
Endothelial NOX5 Obliterates the Reno-Protective Effect of Nox4 Deletion by Promoting Renal Fibrosis via Activation of EMT and ROS-Sensitive Pathways in Diabetes


 , ,
, , 
Abstract
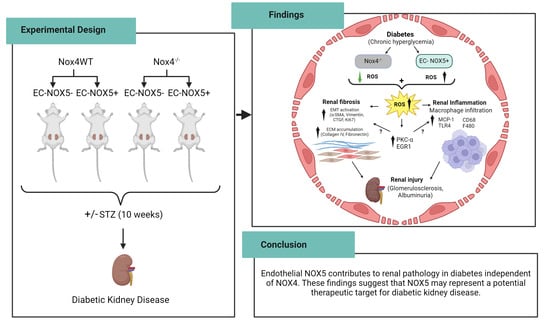
:
1. Introduction
2. Materials and Methods
2.1. Experimental Design
2.2. Assessment of Metabolic Parameters and Renal Function
2.3. Quantitative RT-PCR
2.4. Renal Histology and Immunohistochemistry
2.5. Protein Expression of Renal MCP-1 by ELISA
2.6. Renal Protein Expression via Western Blot
2.7. Statistical Analysis
3. Results
3.1. Metabolic Variables and Renal Function
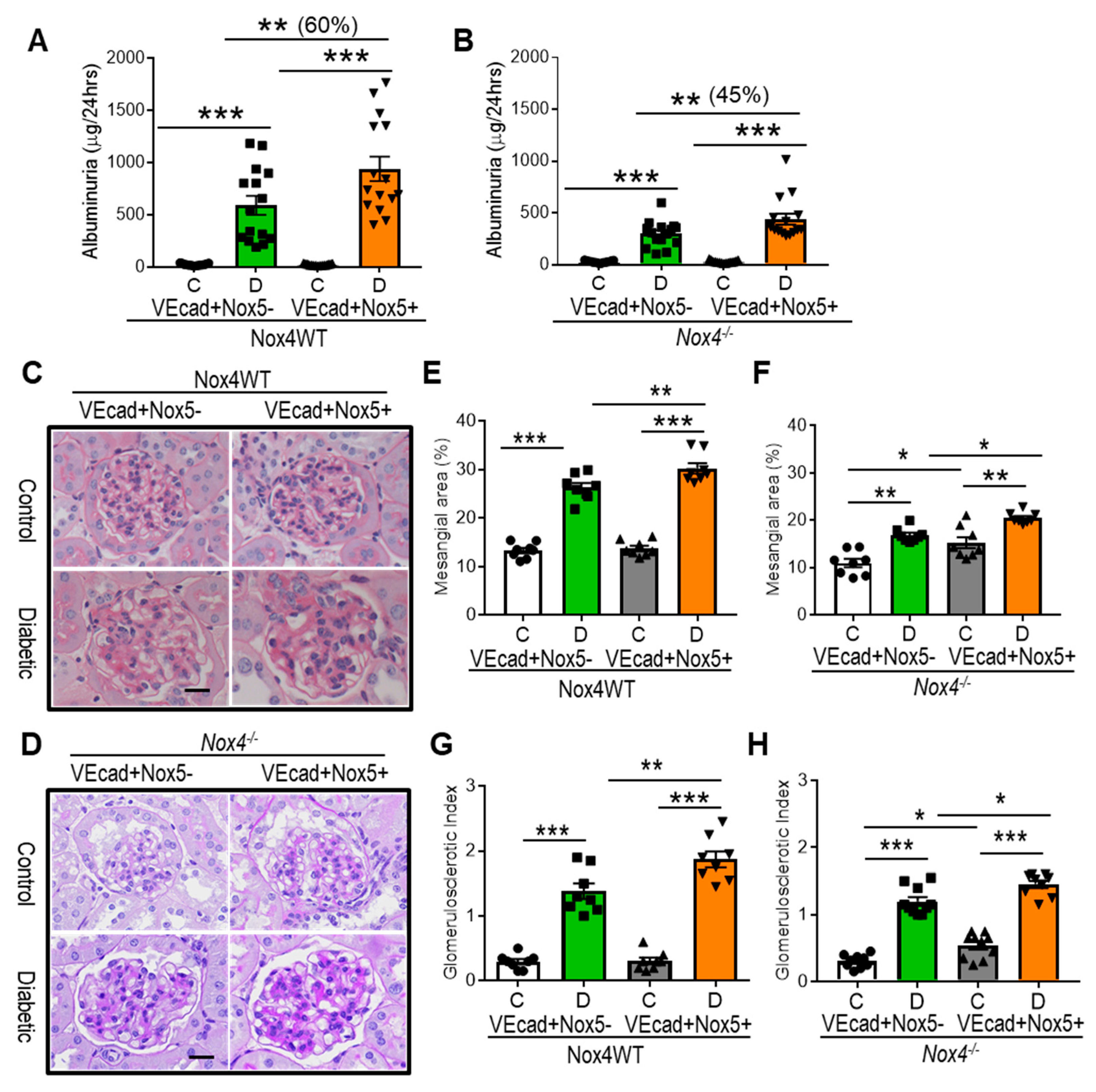
3.2. EC-NOX5 Increases Albuminuria and Renal Injury in WT and Nox4KO Mice in Diabetes
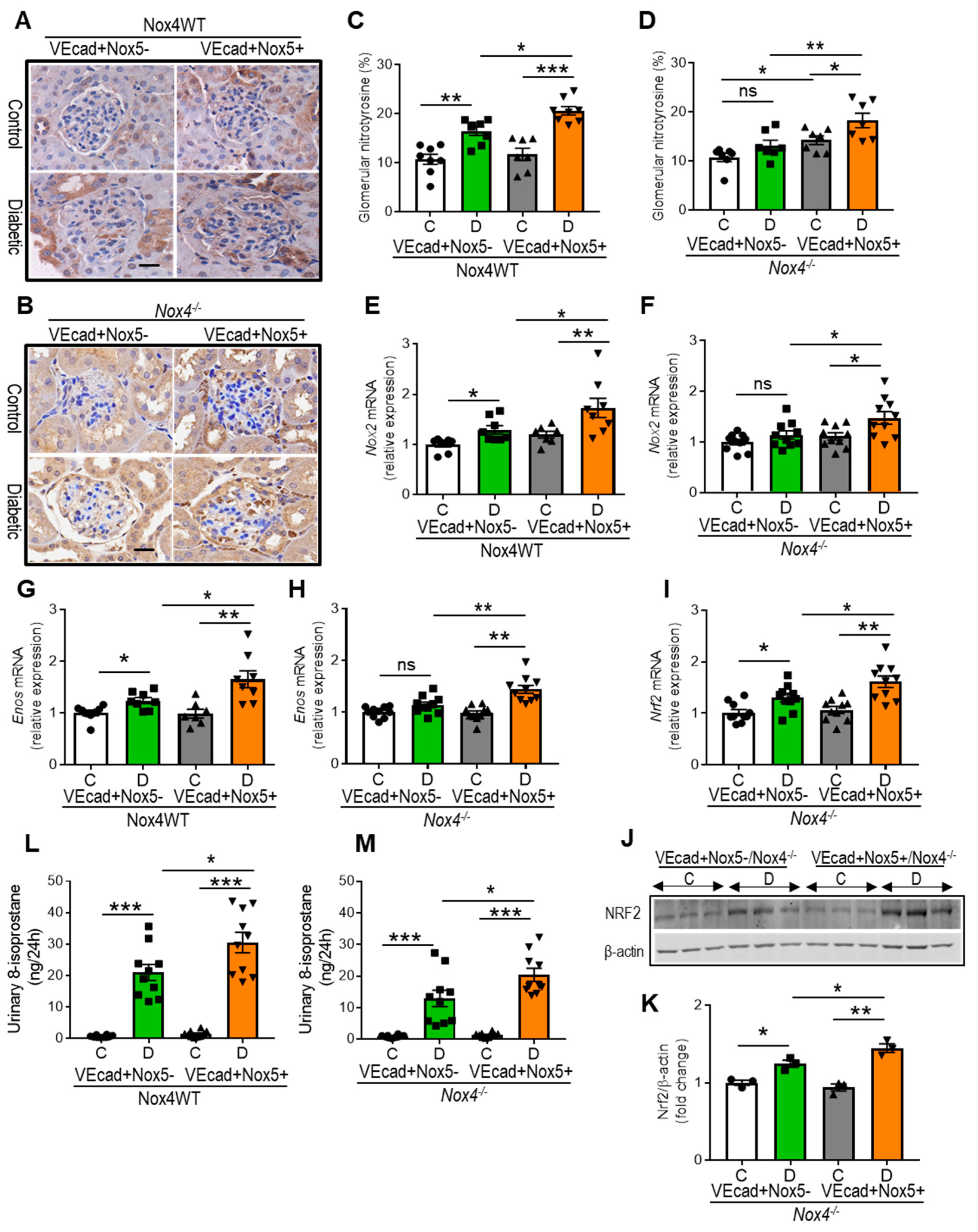
3.3. EC-NOX5 Enhances Renal ROS Formation in WT and Nox4KO Mice in Diabetes
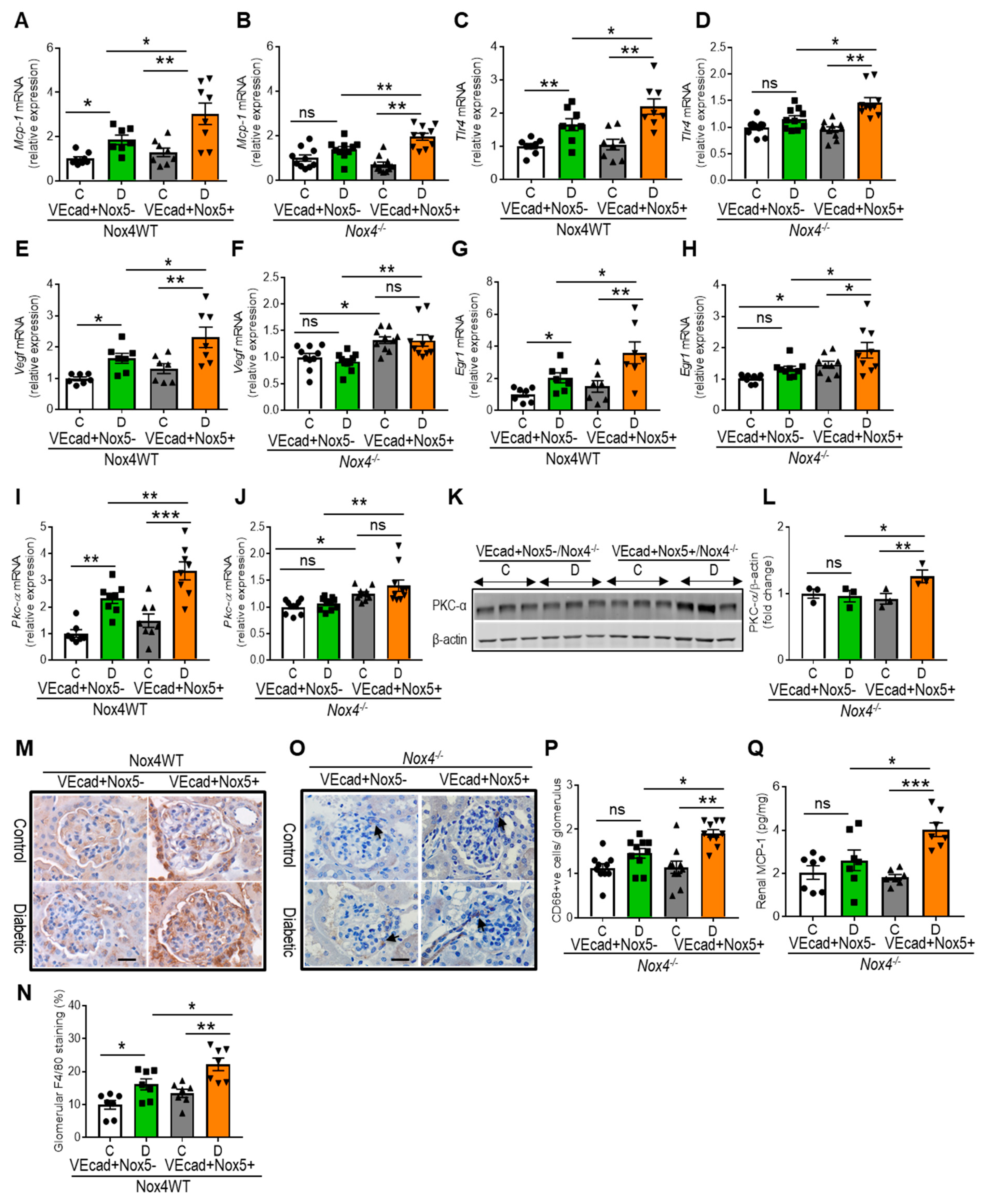
3.4. EC-NOX5 Upregulates Markers of Inflammation and ROS-Sensitive Factors in WT and Nox4KO Mice in Diabetes
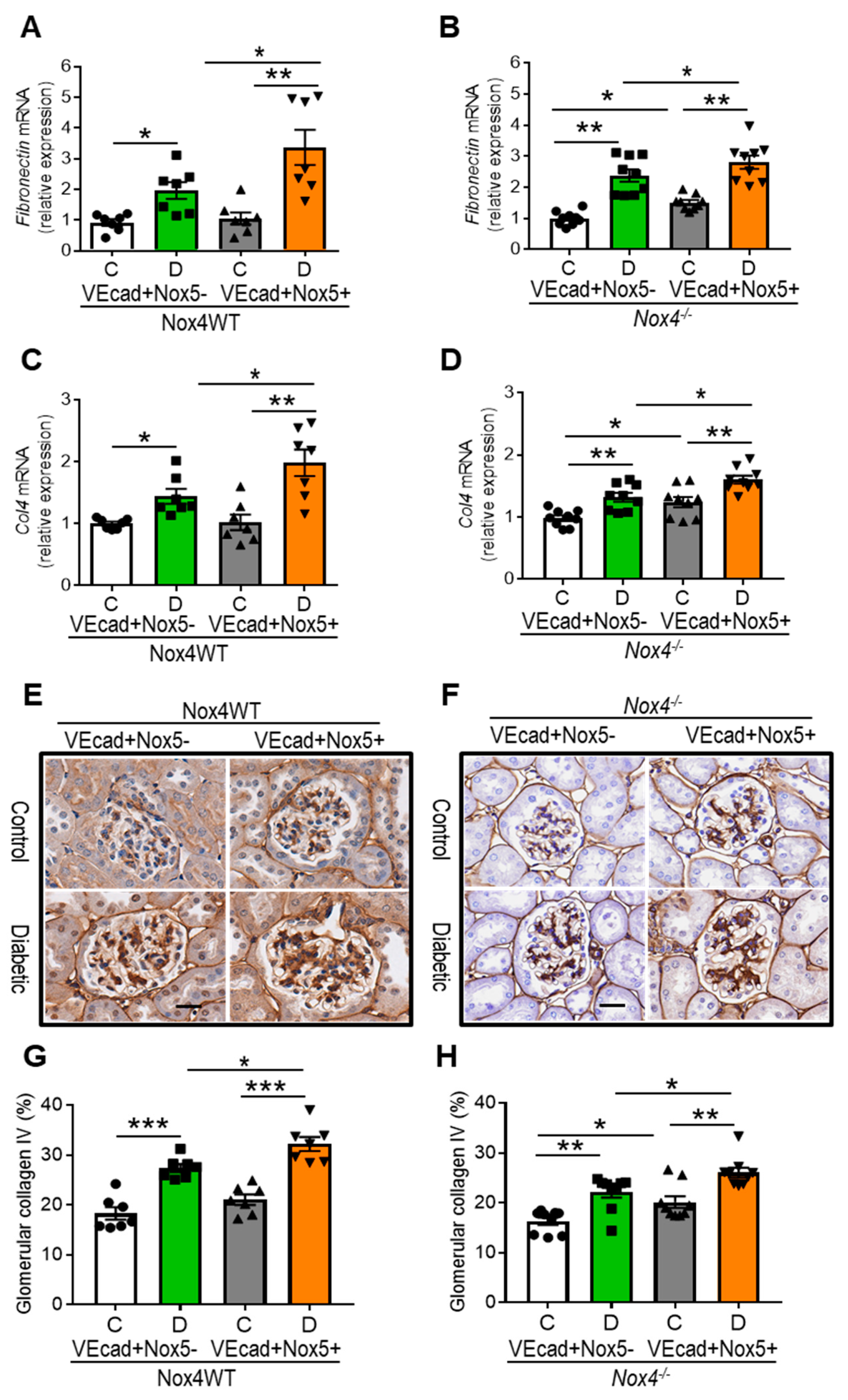
3.5. EC-NOX5 Upsurges Renal ECM Accumulation and Fibrosis in WT and Nox4KO Mice
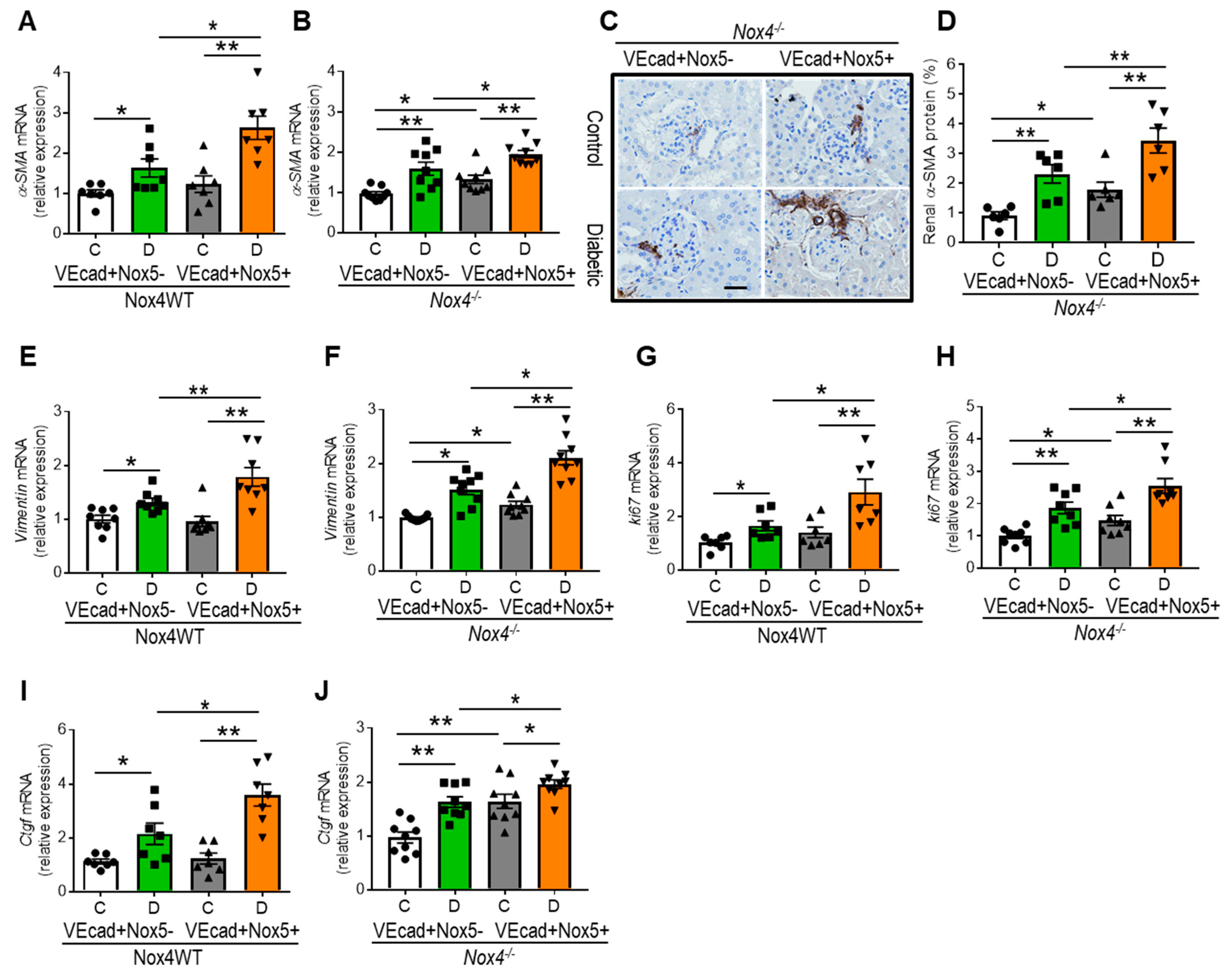
3.6. EC-NOX5 Promotes Renal Fibrosis by Activating EMT-Related Factors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, N.H.; Seo, M.H.; Jung, J.H.; Han, K.D.; Kim, M.K.; Kim, N.H.; Diabetic Kidney Disease Research Group of the Korean Diabetes Association. 2023 Diabetic Kidney Disease Fact Sheet in Korea. Diabetes Metab. J. 2024. [Google Scholar] [CrossRef] [PubMed]
- Sukkar, L.; Kang, A.; Hockham, C.; Young, T.; Jun, M.; Foote, C.; Pecoits-Filho, R.; Neuen, B.; Rogers, K.; Pollock, C.; et al. Incidence and Associations of Chronic Kidney Disease in Community Participants with Diabetes: A 5-Year Prospective Analysis of the EXTEND45 Study. Diabetes Care 2020, 43, 982–990. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Rogers, K.; Sukkar, L.; Jun, M.; Kang, A.; Young, T.; Campain, A.; Cass, A.; Chow, C.K.; Comino, E.; et al. Prevalence, incidence and risk factors of diabetes in Australian adults aged >/=45 years: A cohort study using linked routinely-collected data. J. Clin. Transl. Endocrinol. 2020, 22, 100240. [Google Scholar] [CrossRef] [PubMed]
- Dalla Vestra, M.; Simioni, N.; Masiero, A. Renal effects of dual renin-angiotensin-aldosterone system blockade in patients with diabetic nephropathy. Int. Urol. Nephrol. 2009, 41, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Fioretto, P.; Zambon, A.; Rossato, M.; Busetto, L.; Vettor, R. SGLT2 Inhibitors and the Diabetic Kidney. Diabetes Care 2016, 39 (Suppl. 2), S165–S171. [Google Scholar] [CrossRef] [PubMed]
- Morino, J.; Hirai, K.; Kaneko, S.; Minato, S.; Yanai, K.; Mutsuyoshi, Y.; Ishii, H.; Matsuyama, M.; Kitano, T.; Shindo, M.; et al. Two cases of advanced stage rapidly progressive diabetic nephropathy effectively treated with combination therapy including RAS blocker, GLP-1 receptor agonist and SGLT-2 inhibitor. CEN Case Rep. 2019, 8, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Rossing, K.; Christensen, P.K.; Jensen, B.R.; Parving, H.H. Dual blockade of the renin-angiotensin system in diabetic nephropathy: A randomized double-blind crossover study. Diabetes Care 2002, 25, 95–100. [Google Scholar] [CrossRef]
- Tommerdahl, K.L.; Kendrick, J.; Bjornstad, P. The Role of Glucagon-Like Peptide 1 (GLP-1) Receptor Agonists in the Prevention and Treatment of Diabetic Kidney Disease: Insights from the AMPLITUDE-O Trial. Clin. J. Am. Soc. Nephrol. 2022, 17, 905–907. [Google Scholar] [CrossRef] [PubMed]
- Asaba, K.; Tojo, A.; Onozato, M.L.; Goto, A.; Quinn, M.T.; Fujita, T.; Wilcox, C.S. Effects of NADPH oxidase inhibitor in diabetic nephropathy. Kidney Int. 2005, 67, 1890–1898. [Google Scholar] [CrossRef]
- Block, K.; Gorin, Y.; Abboud, H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390. [Google Scholar] [CrossRef]
- Bondi, C.D.; Manickam, N.; Lee, D.Y.; Block, K.; Gorin, Y.; Abboud, H.E.; Barnes, J.L. NAD(P)H oxidase mediates TGF-β1-induced activation of kidney myofibroblasts. J. Am. Soc. Nephrol. 2010, 21, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Etoh, T.; Inoguchi, T.; Kakimoto, M.; Sonoda, N.; Kobayashi, K.; Kuroda, J.; Sumimoto, H.; Nawata, H. Increased expression of NAD(P)H oxidase subunits, NOX4 and p22phox, in the kidney of streptozotocin-induced diabetic rats and its reversibity by interventive insulin treatment. Diabetologia 2003, 46, 1428–1437. [Google Scholar] [CrossRef] [PubMed]
- Gorin, Y.; Block, K. Nox4 and diabetic nephropathy: With a friend like this, who needs enemies? Free Radic. Biol. Med. 2013, 61C, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Gorin, Y.; Block, K.; Hernandez, J.; Bhandari, B.; Wagner, B.; Barnes, J.L.; Abboud, H.E. Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J. Biol. Chem. 2005, 280, 39616–39626. [Google Scholar] [CrossRef] [PubMed]
- Jha, J.C.; Banal, C.; Chow, B.S.; Cooper, M.E.; Jandeleit-Dahm, K. Diabetes and Kidney Disease: Role of Oxidative Stress. Antioxid. Redox Signal. 2016, 25, 657–684. [Google Scholar] [CrossRef] [PubMed]
- Jha, J.C.; Gray, S.P.; Barit, D.; Okabe, J.; El-Osta, A.; Namikoshi, T.; Thallas-Bonke, V.; Wingler, K.; Szyndralewiez, C.; Heitz, F.; et al. Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. J. Am. Soc. Nephrol. 2014, 25, 1237–1254. [Google Scholar] [CrossRef] [PubMed]
- Jha, J.C.; Thallas-Bonke, V.; Banal, C.; Gray, S.P.; Chow, B.S.; Ramm, G.; Quaggin, S.E.; Cooper, M.E.; Schmidt, H.H.; Jandeleit-Dahm, K.A. Podocyte-specific Nox4 deletion affords renoprotection in a mouse model of diabetic nephropathy. Diabetologia 2016, 59, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Holterman, C.E.; Boisvert, N.C.; Thibodeau, J.F.; Kamto, E.; Novakovic, M.; Abd-Elrahman, K.S.; Ferguson, S.S.G.; Kennedy, C.R.J. Podocyte NADPH Oxidase 5 Promotes Renal Inflammation Regulated by the Toll-Like Receptor Pathway. Antioxid. Redox Signal. 2019, 30, 1817–1830. [Google Scholar] [CrossRef] [PubMed]
- Holterman, C.E.; Thibodeau, J.F.; Towaij, C.; Gutsol, A.; Montezano, A.C.; Parks, R.J.; Cooper, M.E.; Touyz, R.M.; Kennedy, C.R. Nephropathy and elevated BP in mice with podocyte-specific NADPH oxidase 5 expression. J. Am. Soc. Nephrol. 2014, 25, 784–797. [Google Scholar] [CrossRef]
- Jha, J.C.; Banal, C.; Okabe, J.; Gray, S.P.; Hettige, T.; Chow, B.S.M.; Thallas-Bonke, V.; De Vos, L.; Holterman, C.E.; Coughlan, M.T.; et al. NADPH Oxidase Nox5 Accelerates Renal Injury in Diabetic Nephropathy. Diabetes 2017, 66, 2691–2703. [Google Scholar] [CrossRef]
- Jha, J.C.; Dai, A.; Garzarella, J.; Charlton, A.; Urner, S.; Ostergaard, J.A.; Okabe, J.; Holterman, C.E.; Skene, A.; Power, D.A.; et al. Independent of Renox, NOX5 Promotes Renal Inflammation and Fibrosis in Diabetes by Activating ROS-Sensitive Pathways. Diabetes 2022, 71, 1282–1298. [Google Scholar] [CrossRef] [PubMed]
- Jha, J.C.; Dai, A.; Holterman, C.E.; Cooper, M.E.; Touyz, R.M.; Kennedy, C.R.; Jandeleit-Dahm, K.A.M. Endothelial or vascular smooth muscle cell-specific expression of human NOX5 exacerbates renal inflammation, fibrosis and albuminuria in the Akita mouse. Diabetologia 2019, 62, 1712–1726. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Chen, W.; Gongora, M.C.; Guzik, B.; Lob, H.E.; Mangalat, D.; Hoch, N.; Dikalov, S.; Rudzinski, P.; Kapelak, B.; et al. Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J. Am. Coll. Cardiol. 2008, 52, 1803–1809. [Google Scholar] [CrossRef] [PubMed]
- Marzaioli, V.; Hurtado-Nedelec, M.; Pintard, C.; Tlili, A.; Marie, J.C.; Monteiro, R.C.; Gougerot-Pocidalo, M.A.; Dang, P.M.; El-Benna, J. NOX5 and p22phox are 2 novel regulators of human monocytic differentiation into dendritic cells. Blood 2017, 130, 1734–1745. [Google Scholar] [CrossRef] [PubMed]
- Deliyanti, D.; Alrashdi, S.F.; Touyz, R.M.; Kennedy, C.R.; Jha, J.C.; Cooper, M.E.; Jandeleit-Dahm, K.A.; Wilkinson-Berka, J.L. Nox (NADPH Oxidase) 1, Nox4, and Nox5 Promote Vascular Permeability and Neovascularization in Retinopathy. Hypertension 2020, 75, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Han, W.; Villar, V.A.; Yang, Y.; Lu, Q.; Lee, H.; Li, F.; Quinn, M.T.; Gildea, J.J.; Felder, R.A.; et al. Unique role of NADPH oxidase 5 in oxidative stress in human renal proximal tubule cells. Redox Biol. 2014, 2, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Kumar, S.; Yu, Y.; Aggarwal, S.; Gross, C.; Wang, Y.; Chakraborty, T.; Verin, A.D.; Catravas, J.D.; Lucas, R.; et al. PKC-dependent phosphorylation of eNOS at T495 regulates eNOS coupling and endothelial barrier function in response to G+-toxins. PLoS ONE 2014, 9, e99823. [Google Scholar] [CrossRef]
- Chen, F.; Yu, Y.; Haigh, S.; Johnson, J.; Lucas, R.; Stepp, D.W.; Fulton, D.J. Regulation of NADPH oxidase 5 by protein kinase C isoforms. PLoS ONE 2014, 9, e88405. [Google Scholar] [CrossRef]
- Fulton, D.J. Nox5 and the regulation of cellular function. Antioxid. Redox Signal. 2009, 11, 2443–2452. [Google Scholar] [CrossRef]
- Touyz, R.M.; Anagnostopoulou, A.; Rios, F.; Montezano, A.C.; Camargo, L.L. NOX5: Molecular biology and pathophysiology. Exp. Physiol. 2019, 104, 605–616. [Google Scholar] [CrossRef]
- Kleinschnitz, C.; Grund, H.; Wingler, K.; Armitage, M.E.; Jones, E.; Mittal, M.; Barit, D.; Schwarz, T.; Geis, C.; Kraft, P.; et al. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 2010, 8, e1000479. [Google Scholar] [CrossRef] [PubMed]
- Eid, A.A.; Gorin, Y.; Fagg, B.M.; Maalouf, R.; Barnes, J.L.; Block, K.; Abboud, H.E. Mechanisms of podocyte injury in diabetes: Role of cytochrome P450 and NADPH oxidases. Diabetes 2009, 58, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Genkyotex. 2015. Available online: https://www.businesswire.com/news/home/20150909005080/en/Genkyotex-Announces-Top-Line-Results-of-Phase-2-Clinical-Program (accessed on 9 September 2015).
- Reutens, A.T.; Jandeleit-Dahm, K.; Thomas, M.; Salim, A.; De Livera, A.M.; Bach, L.A.; Colman, P.G.; Davis, T.M.E.; Ekinci, E.I.; Fulcher, G.; et al. A physician-initiated double-blind, randomised, placebo-controlled, phase 2 study evaluating the efficacy and safety of inhibition of NADPH oxidase with the first-in-class Nox-1/4 inhibitor, GKT137831, in adults with type 1 diabetes and persistently elevated urinary albumin excretion: Protocol and statistical considerations. Contemp. Clin. Trials 2020, 90, 105892. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, E.; Gray, S.P.; Kennedy, K.; Szyndralewiez, C.; Lyle, A.N.; Lassegue, B.; Griendling, K.K.; Cooper, M.E.; Schmidt, H.; Jandeleit-Dahm, K.A.M. NOX4-derived reactive oxygen species limit fibrosis and inhibit proliferation of vascular smooth muscle cells in diabetic atherosclerosis. Free Radic. Biol. Med. 2016, 97, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Haigh, S.; Yu, Y.; Benson, T.; Wang, Y.; Li, X.; Dou, H.; Bagi, Z.; Verin, A.D.; Stepp, D.W.; et al. Nox5 stability and superoxide production is regulated by C-terminal binding of Hsp90 and CO-chaperones. Free Radic. Biol. Med. 2015, 89, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Chiarelli, F.; Cipollone, F.; Mohn, A.; Marini, M.; Iezzi, A.; Fazia, M.; Tumini, S.; De Cesare, D.; Pomilio, M.; Pierdomenico, S.D.; et al. Circulating monocyte chemoattractant protein-1 and early development of nephropathy in type 1 diabetes. Diabetes Care 2002, 25, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Reddy, Y.S.; Kiranmayi, V.S.; Bitla, A.R.; Krishna, G.S.; Rao, P.V.; Sivakumar, V. Nitric oxide status in patients with chronic kidney disease. Indian J. Nephrol. 2015, 25, 287–291. [Google Scholar] [CrossRef]
- Bondi, C.D.; Hartman, H.L.; Tan, R.J. NRF2 in kidney physiology and disease. Physiol. Rep. 2024, 12, e15961. [Google Scholar] [CrossRef] [PubMed]
- Nezu, M.; Suzuki, N. Nrf2 activation for kidney disease treatment-a mixed blessing? Kidney Int. 2021, 99, 20–22. [Google Scholar] [CrossRef]
- Zhao, S.; Lo, C.S.; Miyata, K.N.; Ghosh, A.; Zhao, X.P.; Chenier, I.; Cailhier, J.F.; Ethier, J.; Lattouf, J.B.; Filep, J.G.; et al. Overexpression of Nrf2 in Renal Proximal Tubular Cells Stimulates Sodium-Glucose Cotransporter 2 Expression and Exacerbates Dysglycemia and Kidney Injury in Diabetic Mice. Diabetes 2021, 70, 1388–1403. [Google Scholar] [CrossRef]
- Huang, M.J.; Wei, R.B.; Zhao, J.; Su, T.Y.; Li, Q.P.; Yang, X.; Chen, X.M. Albuminuria and Endothelial Dysfunction in Patients with Non-Diabetic Chronic Kidney Disease. Med. Sci. Monit. 2017, 23, 4447–4453. [Google Scholar] [CrossRef]
- Lassen, E.; Daehn, I.S. Molecular Mechanisms in Early Diabetic Kidney Disease: Glomerular Endothelial Cell Dysfunction. Int. J. Mol. Sci. 2020, 21, 9456. [Google Scholar] [CrossRef]
- D’Amico, G.; Bazzi, C. Pathophysiology of proteinuria. Kidney Int. 2003, 63, 809–825. [Google Scholar] [CrossRef]
- Myers, B.D. Pathophysiology of proteinuria in diabetic glomerular disease. J. Hypertens. Suppl. 1990, 8, S41–S46. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Smyth, B. From Proteinuria to Fibrosis: An Update on Pathophysiology and Treatment Options. Kidney Blood Press. Res. 2021, 46, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Meier, M.; Park, J.K.; Overheu, D.; Kirsch, T.; Lindschau, C.; Gueler, F.; Leitges, M.; Menne, J.; Haller, H. Deletion of protein kinase C-β isoform in vivo reduces renal hypertrophy but not albuminuria in the streptozotocin-induced diabetic mouse model. Diabetes 2007, 56, 346–354. [Google Scholar] [CrossRef]
- Menne, J.; Park, J.K.; Boehne, M.; Elger, M.; Lindschau, C.; Kirsch, T.; Meier, M.; Gueler, F.; Fiebeler, A.; Bahlmann, F.H.; et al. Diminished loss of proteoglycans and lack of albuminuria in protein kinase C-α-deficient diabetic mice. Diabetes 2004, 53, 2101–2109. [Google Scholar] [CrossRef]
- Menne, J.; Shushakova, N.; Bartels, J.; Kiyan, Y.; Laudeley, R.; Haller, H.; Park, J.K.; Meier, M. Dual inhibition of classical protein kinase C-α and protein kinase C-β isoforms protects against experimental murine diabetic nephropathy. Diabetes 2013, 62, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.J.; Hyun, Y.Y.; Lee, M.H.; Kim, J.E.; Nam, D.H.; Song, H.K.; Kang, Y.S.; Lee, J.E.; Kim, H.W.; Han, J.Y.; et al. Renal protective effects of toll-like receptor 4 signaling blockade in type 2 diabetic mice. Endocrinology 2013, 154, 2144–2155. [Google Scholar] [CrossRef]
- Chow, F.Y.; Nikolic-Paterson, D.J.; Ozols, E.; Atkins, R.C.; Rollin, B.J.; Tesch, G.H. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 2006, 69, 73–80. [Google Scholar] [CrossRef]
- Szabo, Z.; Alachkar, N.; Xia, J.; Mathews, W.B.; Rabb, H. Molecular imaging of the kidneys. Semin. Nucl. Med. 2011, 41, 20–28. [Google Scholar] [CrossRef]
- Daehn, I.S. Glomerular Endothelial Cell Stress and Cross-Talk With Podocytes in Early [corrected] Diabetic Kidney Disease. Front. Med. 2018, 5, 76. [Google Scholar] [CrossRef]
- Ho, L.C.; Sung, J.M.; Shen, Y.T.; Jheng, H.F.; Chen, S.H.; Tsai, P.J.; Tsai, Y.S. Egr-1 deficiency protects from renal inflammation and fibrosis. J. Mol. Med. 2016, 94, 933–942. [Google Scholar] [CrossRef]
- Hu, F.; Xue, M.; Li, Y.; Jia, Y.J.; Zheng, Z.J.; Yang, Y.L.; Guan, M.P.; Sun, L.; Xue, Y.M. Early Growth Response 1 (Egr1) Is a Transcriptional Activator of NOX4 in Oxidative Stress of Diabetic Kidney Disease. J. Diabetes Res. 2018, 2018, 3405695. [Google Scholar] [CrossRef]
- Hu, F.; Xue, R.; Wei, X.; Wang, Z.; Luo, S.; Lin, J.; Yan, Z.; Sun, L. Egr1 Knockdown Combined with an ACE Inhibitor Ameliorates Diabetic Kidney Disease in Mice: Blockade of Compensatory Renin Increase. Diabetes Metab. Syndr. Obes. 2020, 13, 1005–1013. [Google Scholar] [CrossRef]
- Lim, A.K.; Tesch, G.H. Inflammation in diabetic nephropathy. Mediat. Inflamm. 2012, 2012, 146154. [Google Scholar] [CrossRef]
- Aaltonen, P.; Luimula, P.; Astrom, E.; Palmen, T.; Gronholm, T.; Palojoki, E.; Jaakkola, I.; Ahola, H.; Tikkanen, I.; Holthofer, H. Changes in the expression of nephrin gene and protein in experimental diabetic nephropathy. Lab. Investig. 2001, 81, 1185–1190. [Google Scholar] [CrossRef]
- Lei, C.T.; Wei, Y.H.; Tang, H.; Wen, Q.; Ye, C.; Zhang, C.; Su, H. PKC-α Triggers EGFR Ubiquitination, Endocytosis and ERK Activation in Podocytes Stimulated with High Glucose. Cell. Physiol. Biochem. 2017, 42, 281–294. [Google Scholar] [CrossRef]
- Thallas-Bonke, V.; Jha, J.C.; Gray, S.P.; Barit, D.; Haller, H.; Schmidt, H.H.; Coughlan, M.T.; Cooper, M.E.; Forbes, J.M.; Jandeleit-Dahm, K.A. Nox-4 deletion reduces oxidative stress and injury by PKC-α-associated mechanisms in diabetic nephropathy. Physiol. Rep. 2014, 2, e12192. [Google Scholar] [CrossRef] [PubMed]
- Tossidou, I.; Starker, G.; Kruger, J.; Meier, M.; Leitges, M.; Haller, H.; Schiffer, M. PKC-alpha modulates TGF-β signaling and impairs podocyte survival. Cell. Physiol. Biochem. 2009, 24, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, T.; Paik, Y.H.; Watanabe, S.; Laleu, B.; Gaggini, F.; Fioraso-Cartier, L.; Molango, S.; Heitz, F.; Merlot, C.; Szyndralewiez, C.; et al. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology 2012, 56, 2316–2327. [Google Scholar] [CrossRef] [PubMed]
- Dimke, H.; Sparks, M.A.; Thomson, B.R.; Frische, S.; Coffman, T.M.; Quaggin, S.E. Tubulovascular cross-talk by vascular endothelial growth factor a maintains peritubular microvasculature in kidney. J. Am. Soc. Nephrol. 2015, 26, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Kern, T.S.; Engerman, R.L. Kidney morphology in experimental hyperglycemia. Diabetes 1987, 36, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Kiritoshi, S.; Nishikawa, T.; Sonoda, K.; Kukidome, D.; Senokuchi, T.; Matsuo, T.; Matsumura, T.; Tokunaga, H.; Brownlee, M.; Araki, E. Reactive oxygen species from mitochondria induce cyclooxygenase-2 gene expression in human mesangial cells: Potential role in diabetic nephropathy. Diabetes 2003, 52, 2570–2577. [Google Scholar] [CrossRef]
- Lee, S.R.; Lee, H.E.; Yoo, J.Y.; An, E.J.; Song, S.J.; Han, K.H.; Cha, D.R.; Bae, Y.S. Nox4-SH3YL1 complex is involved in diabetic nephropathy. iScience 2024, 27, 108868. [Google Scholar] [CrossRef]
- Loeffler, I.; Wolf, G. Epithelial-to-Mesenchymal Transition in Diabetic Nephropathy: Fact or Fiction? Cells 2015, 4, 631–652. [Google Scholar] [CrossRef]
- Mason, R.M.; Wahab, N.A. Extracellular matrix metabolism in diabetic nephropathy. J. Am. Soc. Nephrol. 2003, 14, 1358–1373. [Google Scholar] [CrossRef]
- McLennan, S.V.; Wang, X.Y.; Moreno, V.; Yue, D.K.; Twigg, S.M. Connective tissue growth factor mediates high glucose effects on matrix degradation through tissue inhibitor of matrix metalloproteinase type 1: Implications for diabetic nephropathy. Endocrinology 2004, 145, 5646–5655. [Google Scholar] [CrossRef]
- Nlandu Khodo, S.; Dizin, E.; Sossauer, G.; Szanto, I.; Martin, P.Y.; Feraille, E.; Krause, K.H.; de Seigneux, S. NADPH-oxidase 4 protects against kidney fibrosis during chronic renal injury. J. Am. Soc. Nephrol. 2012, 23, 1967–1976. [Google Scholar] [CrossRef]
- Shakour, N.; Karami, S.; Iranshahi, M.; Butler, A.E.; Sahebkar, A. Antifibrotic effects of sodium-glucose cotransporter-2 inhibitors: A comprehensive review. Diabetes Metab. Syndr. 2024, 18, 102934. [Google Scholar] [CrossRef]
- Acheva, A.; Haghdoost, S.; Sollazzo, A.; Launonen, V.; Kamarainen, M. Presence of Stromal Cells Enhances Epithelial-to-Mesenchymal Transition (EMT) Induction in Lung Bronchial Epithelium after Protracted Exposure to Oxidative Stress of Gamma Radiation. Oxid. Med. Cell. Longev. 2019, 2019, 4120379. [Google Scholar] [CrossRef] [PubMed]
- Burns, W.C.; Twigg, S.M.; Forbes, J.M.; Pete, J.; Tikellis, C.; Thallas-Bonke, V.; Thomas, M.C.; Cooper, M.E.; Kantharidis, P. Connective tissue growth factor plays an important role in advanced glycation end product-induced tubular epithelial-to-mesenchymal transition: Implications for diabetic renal disease. J. Am. Soc. Nephrol. 2006, 17, 2484–2494. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, E.; Parri, M.; Chiarugi, P. EMT and oxidative stress: A bidirectional interplay affecting tumor malignancy. Antioxid. Redox Signal. 2012, 16, 1248–1263. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Wan, R.; Chen, Z.; Huo, L.; Zhu, M.; Yang, Y.; Chen, Q.; Zhang, X.; Wang, X. Triptolide Alleviates Podocyte Epithelial-Mesenchymal Transition via Kindlin-2 and EMT-Related TGF-β/Smad Signaling Pathway in Diabetic Kidney Disease. Appl. Biochem. Biotechnol. 2022, 194, 1000–1012. [Google Scholar] [CrossRef] [PubMed]
- Gonlusen, G.; Ergin, M.; Paydas, S.; Tunali, N. The expression of cytoskeletal proteins (α-SMA, vimentin, desmin) in kidney tissue: A comparison of fetal, normal kidneys, and glomerulonephritis. Int. Urol. Nephrol. 2001, 33, 299–305. [Google Scholar] [CrossRef]
- Zhang, S.; Huang, Q.; Cai, X.; Jiang, S.; Xu, N.; Zhou, Q.; Cao, X.; Hultstrom, M.; Tian, J.; Lai, E.Y. Osthole Ameliorates Renal Fibrosis in Mice by Suppressing Fibroblast Activation and Epithelial-Mesenchymal Transition. Front. Physiol. 2018, 9, 1650. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VEcad+Nox5−/Nox4WT | VEcad+Nox5+/Nox4WT | |||
|---|---|---|---|---|
| Control | Diabetes | Control | Diabetes | |
| Plasma glucose (mmol/L) | 12.2 ± 0.6 | 33.1 ± 1.3 ** | 12.1 ± 0.4 | 33.8 ± 0.6 ^^ |
| Glycated hemoglobin (%) | 4.1 ± 0.1 | 10.3 ± 0.4 * | 4.2 ± 0.1 | 10.2 ± 0.3 ^ |
| Body weight (g) | 31 ± 0.5 | 28 ± 0.9 * | 30 ± 0.7 | 27 ± 0.5 ^ |
| Kidney weight/Body weight (mg/g) | 13.1 ± 0.3 | 21.1 ± 1.3 * | 13.6 ± 0.5 | 23.1 ± 1.2 ^ |
| Systolic BP (mmHg) | 107 ± 1 | 111 ± 2 | 110 ± 2 | 109 ± 1 |
| Plasma cystatin C (ng/mL) | 450 ± 74 | 272 ± 36 * | 428 ± 61 | 234 ± 27 ^ |
| VEcad+Nox5−/Nox4−/− | VEcad+Nox5+/Nox4−/− | |||
|---|---|---|---|---|
| Control | Diabetes | Control | Diabetes | |
| Plasma glucose (mmol/L) | 13.2 ±0.6 | 33.3 ± 0.1 $$ | 12.7 ± 0.8 | 29.1 ± 1.7 ## |
| Glycated hemoglobin (%) | 4.1 ± 0.1 | 11.1 ± 0.3 $ | 4.1 ± 0.1 | 10.2 ± 0.6 # |
| Body weight (g) | 40 ± 1.2 | 32 ± 0.7 $ | 36 ± 0.7 | 30 ± 0.9 # |
| Kidney weight /Body weight (mg/g) | 10.5 ± 0.3 | 18.4 ± 0.6 $ | 11.6 ± 0.4 | 18.9 ± 0.9 # |
| Systolic BP (mmHg) | 120 ± 8 | 119 ± 7 | 110 ± 6 | 131 ± 8 # |
| Plasma cystatin C (ng/mL) | 417 ± 64 | 159 ± 16 $ | 406 ± 41 | 177 ± 31 # |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jandeleit-Dahm, K.A.M.; Kankanamalage, H.R.; Dai, A.; Meister, J.; Lopez-Trevino, S.; Cooper, M.E.; Touyz, R.M.; Kennedy, C.R.J.; Jha, J.C. Endothelial NOX5 Obliterates the Reno-Protective Effect of Nox4 Deletion by Promoting Renal Fibrosis via Activation of EMT and ROS-Sensitive Pathways in Diabetes. Antioxidants 2024, 13, 396. https://doi.org/10.3390/antiox13040396
Jandeleit-Dahm KAM, Kankanamalage HR, Dai A, Meister J, Lopez-Trevino S, Cooper ME, Touyz RM, Kennedy CRJ, Jha JC. Endothelial NOX5 Obliterates the Reno-Protective Effect of Nox4 Deletion by Promoting Renal Fibrosis via Activation of EMT and ROS-Sensitive Pathways in Diabetes. Antioxidants. 2024; 13(4):396. https://doi.org/10.3390/antiox13040396
Chicago/Turabian StyleJandeleit-Dahm, Karin A. M., Haritha R. Kankanamalage, Aozhi Dai, Jaroslawna Meister, Sara Lopez-Trevino, Mark E. Cooper, Rhian M. Touyz, Christopher R. J. Kennedy, and Jay C. Jha. 2024. "Endothelial NOX5 Obliterates the Reno-Protective Effect of Nox4 Deletion by Promoting Renal Fibrosis via Activation of EMT and ROS-Sensitive Pathways in Diabetes" Antioxidants 13, no. 4: 396. https://doi.org/10.3390/antiox13040396
APA StyleJandeleit-Dahm, K. A. M., Kankanamalage, H. R., Dai, A., Meister, J., Lopez-Trevino, S., Cooper, M. E., Touyz, R. M., Kennedy, C. R. J., & Jha, J. C. (2024). Endothelial NOX5 Obliterates the Reno-Protective Effect of Nox4 Deletion by Promoting Renal Fibrosis via Activation of EMT and ROS-Sensitive Pathways in Diabetes. Antioxidants, 13(4), 396. https://doi.org/10.3390/antiox13040396