Heat Sepsis Precedes Heat Toxicity in the Pathophysiology of Heat Stroke—A New Paradigm on an Ancient Disease

{kind=link}
Abstract
:1. Introduction
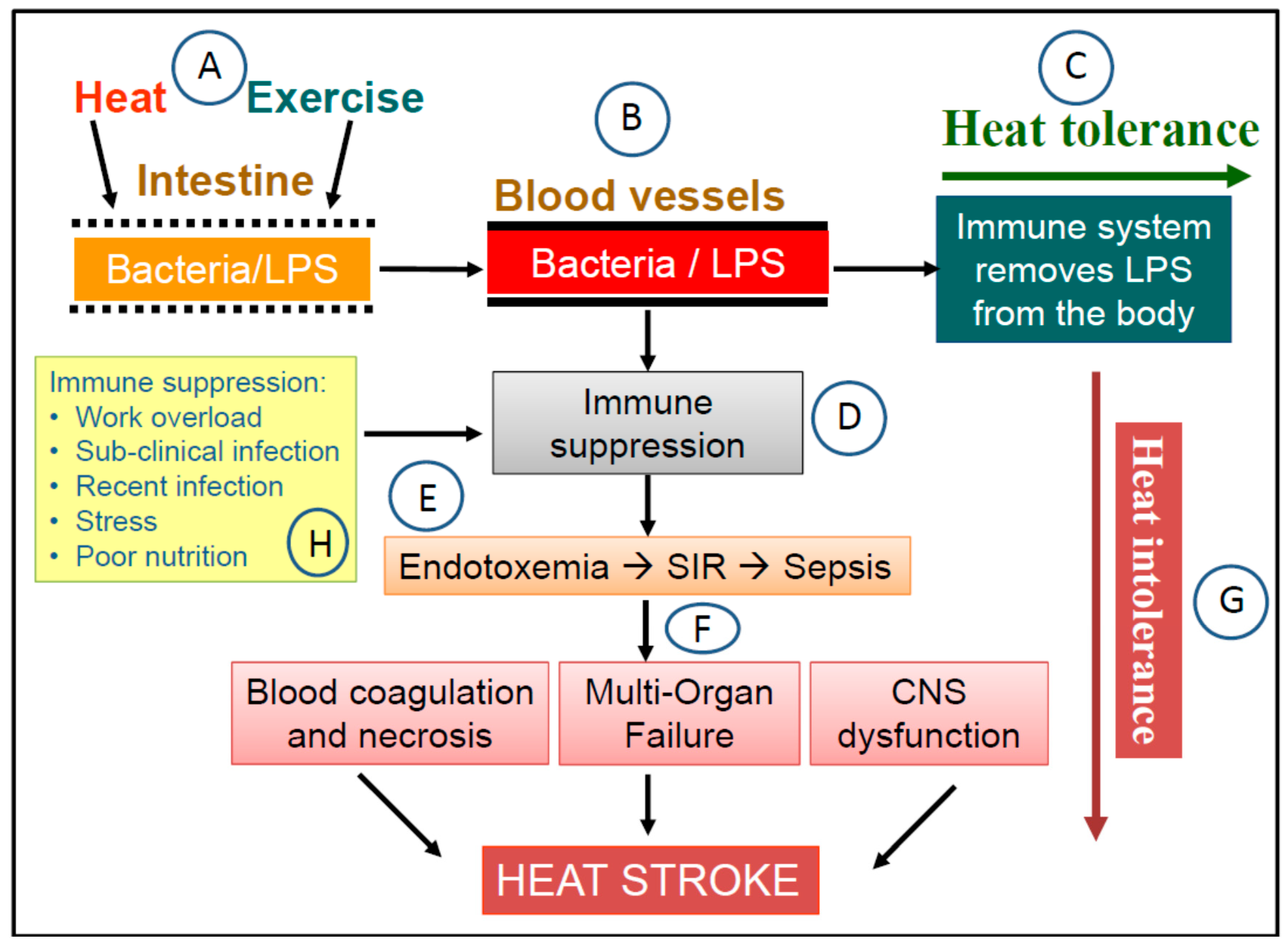
2. The Endotoxemia Model of Heat Stroke
3. Evidence Supporting the Endotoxemia Models of Heat Stroke
3.1. Pathological Reports and Clinical Data
3.2. Animal Studies
3.3. Human Studies
4. The Dual Pathway Model of Heat Stroke
Potential Mitigation of Heat Sepsis
5. Future Research
6. Conclusions
Funding
Conflicts of Interest
References
- Osler, W. The Principles and Practice of Medicine; D. Appleton and Co.: New York, NY, USA, 1893; pp. 1017–1018. [Google Scholar]
- Knochel, J.P. Dog days and siriasis. JAMA 1975, 233, 513–515. [Google Scholar] [CrossRef]
- Eichler, A.C.; McFee, A.S.; Root, H.D. Heat Stroke. Am. J. Surg. 1969, 118, 855–863. [Google Scholar] [CrossRef]
- Shibolet, S.; Lancaster, M.C.; Danon, Y. Heatstroke: A review. Aviat. Space Environ. Med. 1976, 47, 280–301. [Google Scholar] [PubMed]
- Jarco, S.A. Roman experience with heat stroke in 24 B.C. Bull. N. Y. Acad. Med. 1967, 43, 767. [Google Scholar]
- Bouchama, A. Heatstroke: A new look at an ancient disease. Intens. Care Med. 1995, 21, 623–625. [Google Scholar] [CrossRef]
- Prawer, J. A History of the Latin Kingdom of Jerusalem; Vol 1: The Crusaders and the First Kingdom; Bialic Institute: Jerusalem, Israel, 1984; pp. 526–561. [Google Scholar]
- Shapiro, Y.; Seidman, D.S. Field and clinical observations of exertional heat stroke patients. Med. Sci. Sports Exerc. 1990, 22, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Abriat, A.; Brosset, C.; Bregigeon, M.; Sagui, E. Report of 182 cases of exertional heat stroke in the French Armed Forces. Mil. Med. 2014, 179, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Demartini, J.K.; Case, D.J.; Stearns, R.; Belval, L.; Crago, A.E.; Davis, R.; Jardine, J. Effectiveness of cold water immersion in the treatment of exertional heat stroke at the Falmouth Road Race. Med. Sci. Sports Exerc. 2015, 47, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Shibolet, S.; Coll, R.; Gilat, T.; Sohar, E. Heat stroke: Its clinical picture and mechanism in 36 cases. Q. J. Med. 1967, 36, 525–547. [Google Scholar] [PubMed]
- Divine, J.G.; Daggy, M.W.; Dixon, E.E.; LeBlanc, D.P.; Okragley, R.A.; Hesselfeld, K.A. Case series of exertional heat stroke in runners during early spring: 2014–2016 Cincinnati Flying Pig Marathon. Curr. Sports Med. Rep. 2018, 17, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Adolf, E.F.; Fulton, F.B. The effects of exposure to high temperature upon the circulation in man. Am. J. Physiol. 1924, 67, 573–588. [Google Scholar] [CrossRef]
- Adolf, E.F. Man in the Desert; Interscience Publishers Inc.: New York, NY, USA, 1947; pp. 1–357. [Google Scholar]
- Malamud, N.; Haymaker, W.; Custer, R. Heat stroke: A clinico-pathologic study of 125 fatal cases. Mil. Surg. 1946, 99, 397–449. [Google Scholar] [PubMed]
- O’Connor, F.; Casa, D.G.; Bergeron, M.F.; Carter, R., III; Deuster, P.A.; Heled, Y.; Leon, L.; McDermott, B.; O’Brien, K.; Roberts, W.O. American College of Sports Medicine rountable on exertional heat stroke—Return to duty/return to play: Conference proceedings. Curr. Sports Med. Rep. 2010, 9, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, E.G.; Hall, W.W. Heat injuries. JAMA 1927, 89, 92. [Google Scholar] [CrossRef]
- Levick, J.J. Remarks on sunstroke. Am. J. Med. Sci. 1859, 73, 40–55. [Google Scholar] [CrossRef]
- Watts, J., Jr. Remarks on the supposed effects of drinking cold water. Med. Surg. Reg. 1818, 9, 377. [Google Scholar]
- Shanks, N.J.; Papworth, G. Environmental factors and heatstroke. Occup. Med. (Lond.) 2001, 51, 45–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coris, E.E.; Ramirez, A.M.; Van Durme, D.J. Heat illness in athletes. Sports Med. 2004, 34, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Simon, H.B. Hyperthermia. N. Engl. J. Med. 1993, 329, 483–487. [Google Scholar] [PubMed]
- Barrow, M.W.; Clark, K.A. Heat-related illnesses. Am. Fam. Phys. 1998, 58, 749–756. [Google Scholar]
- American College of Sports Medicine. Position stand: Heat and cold illnesses during distance running. Med. Sci. Sports Exerc. 1996, 28, i–x. [Google Scholar]
- National Athlete Trainer, A. National Athlete Trainer Association’s Position Statement: Exertional Heat Illness. J. Athl. Train. 2002, 37, 329–343. [Google Scholar]
- Aarseth, H.P.; Eide, I.; Skeie, B.; Thaulow, E. Heat stroke in endurance exercise. Acta Med. Scand. 1986, 220, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Hanson, P.G.; Zimmermann, S.W. Exertional heat stroke in novice runners. JAMA 1979, 242, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.W.; Nio, A.Q.X.; Lim, C.L.; Teo, Y.N.E.; Byrne, C. Thermoregulation, pacing and fluid balance during mass participation distance running in a warm and humid environment. Eur. J. Appl. Physiol. 2010, 109, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Hughson, R.L.; Green, H.J.; Houston, M.E.; Thomson, J.A.; MacLean, R.D.; Sutton, J.R. Heat injuries in Canadian mass participation runs. CMAJ 1980, 122, 1141–1144. [Google Scholar]
- Nielsen, B. Heat stress and acclimation. Ergonomics 1994, 137, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Moseley, P.L. Heat shock proteins and heat adaptation of the whole organism. J. Appl. Physiol. 1997, 83, 1413–1417. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, Y.; McLellan, T.M.; Shephard, R.J. Effects of 6 versus 12 days of heat acclimation on heat tolerance in lightly exercising men wearing protective clothing. Eur. J. Appl. Physiol. 1995, 71, 187–196. [Google Scholar] [CrossRef]
- Lim, C.L.; Ng, K.K.C.; Lee, L.K.H. The effects of prolonged passive heat exposure and Basic Military Training on thermoregulatory and cardiovascular responses in recruits from a tropical country. Mil. Med. 1997, 162, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Epstein, Y. Heat intolerance: Predisposing factors or residual injury? Med. Sci. Sports Exerc. 1990, 22, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Charan, N.B.; Robinson, W.A.; Mathew, M. Heat stroke, disseminated intravascular coagulation and death in a long distance runner. J. Assoc. Phys. India 1975, 23, 917–919. [Google Scholar]
- Dickinson, J.G. Heat illness in the services. J. R. Army Med. Corps 1994, 140, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Epstein, Y.; Moran, D.S.; Shapiro, Y.; Sohar, E.; Shemer, J. Exertional heat stroke: A case series. Med. Sci. Sports Exerc. 1999, 31, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.P.; Bishop, G.F.; Ashton, C.M. Severe heat stroke in an experienced athlete. Med. J. Aust. 1990, 153, 100–104. [Google Scholar] [PubMed]
- Hart, L.E.; Egier, B.P.; Shimizu, A.G.; Tandan, P.J.; Sutton, J.R. Exertional heat stroke: The runner’s nemesis. Can. Med. Assoc. J. 1980, 122, 1144–1150. [Google Scholar] [PubMed]
- Byrne, C.; Lee, J.; Chew, S.A.N.; Lim, C.L.; Tan, Y.M. Continuous thermoregulatory responses to mass-participation distance running in heat. Med. Sci. Sports Exerc. 2006, 38, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Pugh, L.G.C.E.; Corbett, J.L.; Johnson, R.H. Rectal temperatures, weight losses and sweat rates in marathon running. J. Appl. Physiol. 1967, 23, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Maron, M.B.; Wagner, J.A.; Horvath, S.M. Thermoregulatory responses during competitive marathon running. J. Appl. Physiol. 1977, 42, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Moseley, P.; Gisolfi, C.V. New frontiers in thermoregulation and exercise. Sports Med. 1993, 16, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Gathiram, P.; Wells, M.T.; Brock Utne, J.G.; Gaffin, S.L. Antilipopolysaccharide improves survival in primates subjected to heat stroke. Circ. Shock 1987, 23, 157–164. [Google Scholar] [PubMed]
- Lim, C.L.; Wilson, G.; Brown, L.; Coombes, J.S.; Mackinnon, L.T. Pre-existing inflammatory state compromises heat tolerance in rats exposed to heat stress. Am. J. Physiol. 2007, 292, R186–R194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bynum, G.D.; Brown, J.; Dubose, D.A.; Marsili, M.; Leav, I.; Pistole, T.G.; Hamlet, M.; Lemarie, M.; Caleb, B. Increased survival in experimental dog heatstroke after reduction of gut flora. Aviat. Space Environ. Med. 1979, 50, 816–819. [Google Scholar] [PubMed]
- Chao, T.C.; Sinniah, R.; Pakiam, J.E. Acute heat stroke deaths. Pathology 1981, 13, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.K.C.; Pin, C.H. Obesity and the occurrence of heat disorders. Mil. Med. 1996, 161, 739–742. [Google Scholar]
- Camus, G.; Poortman, J.R.; Nys, M.; Deby-Dupont, G.; Duchateau, J.; Deby, C.; Lamy, M. Mild endotoxemia and the inflammatory response induced by a marathon race. Clin. Sci. 1997, 92, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Jeukendrup, A.E.; Vet-Joop, K.; Sturk, A.; Stegen, J.H.J.C.; Senden, J.; Saris, W.H.M.; Wagenmakers, A.J. Relationship between gastro-intestinal complaints and endotoxemia, cytokine release and the acute-phase reaction during and after a long-distance triathlon in highly trained men. Clin. Sci. 2000, 98, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Nakaji, S.; Yamada, M.; Totsuka, M.; Sato, K.; Sugawara, K. Systemic inflammatory response to exhaustive exercise: Cytokine kinetics. Exerc. Immunol. Rev. 2002, 8, 6–48. [Google Scholar] [PubMed]
- Bouchama, A. Features and outcomes of classic heat stroke. Ann. Intern. Med. 1999, 130, 613. [Google Scholar] [CrossRef] [PubMed]
- Hart, G.R.; Anderson, R.J.; Crumpler, C.P.; Shulkin, A.; Reed, G.; Knochel, J.P. Epidemic classical heat stroke: Clinical characteristics and course of 28 patients. Medicine 1982, 61, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Lambert, G.P. Role of gastrointestinal permeability in exertional heatstroke. Exerc. Sport Sci. Rev. 2004, 32, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Lambert, G.P.; Gisolfi, C.V.; Berg, D.J.; Moseley, P.L.; Oberley, L.W.; Kregel, K.C. Hyperthermia-induced intestinal permeability and role of oxidative and nitrosative stress. J. Appl. Physiol. 2002, 92, 1750–1761. [Google Scholar] [CrossRef] [PubMed]
- Bouchama, A.; Bridey, F.; Hammami, M.M.; Lacombe, C.; al Shail, E.; al Ohali, Y.; Combe, F.; al Sedairy, S.; de Prost, D. Activation of coagulation and fibrinolysis in heatstroke. Thromb. Haemost. 1996, 76, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Hales, R.J.S.; Hubbard, R.W.; Gaffin, S.L. Limitation of heat tolerance. In Hand Book of Physiology: Environmental Physiology; Fregly, M.J., Blatteis, C.M., Eds.; Oxford University Press: New York, NY, USA, 1996; Volume 1, pp. 285–287, 295–312. [Google Scholar]
- Bouchama, A.; Knochel, J.P. Heat Stroke. N. Engl. J. Med. 2002, 346, 1978–1988. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.J.; Law, L.Y.L.; Lim, C.L. Gastrointestinal response and endotoxemia during intense exercise in hot and cool environments. Eur. J. Appl. Physiol. 2013, 113, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Dokladny, K.; Zuhl, M.N.; Moseley, P.L. Intestinal epithelial barrier function and tight junction proteins with heat and exercise. J. Appl. Physiol. 2016, 120, 692–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doran, J.E. Biological Effects of Endotoxin. In Gut-Derived Infectious Toxic Shock. Current Studies in Hematology and Blood Transfusion; Kraft, C.H., Ed.; Karger: Basel, Switzerland, 1992; Volume 59, pp. 66–99. [Google Scholar]
- Williams, K.L. Endotoxin Structure, Function and Activity. In Endotoxins: Pyrogens, LAL Testing, and Depyrogenisation, 3rd ed.; Williams, K.L., Ed.; Informa Healthcare USA Inc.: New York, NY, USA, 2007; pp. 47–90. [Google Scholar]
- Greisman, S.E. Induction of endotoxin tolerance. In Beneficial Effects of Endotoxins; Nowotny, A., Ed.; Plenum Press: New York, NY, USA, 1983; pp. 149–196. [Google Scholar]
- Lim, C.L.; Mackinnon, L.T. The role of exercise-induced immune system disturbances in the pathology of heat stroke: The dual pathway model of heat stroke. Sports Med. 2006, 36, 39–64. [Google Scholar] [CrossRef] [PubMed]
- Gaffin, S.L.; Brock Utne, J.G.; Zanotti, A.; Wells, M.T. Hypoxia-induced endotoxemia in primates: Role of reticuloendothelial system function and anti-lipopolysaccharide plasma. Aviat. Space Environ. Med. 1986, 57, 1044–1049. [Google Scholar] [PubMed]
- Dubose, D.A.; McCreary, J.; Sowders, L.; Goode, L. Relationship between rat heat stress mortality and alterations in reticuloendothelial carbon clearance function. Aviat. Space Environ. Med. 1983, 54, 1090–1095. [Google Scholar] [PubMed]
- Clow, A.; Hucklebridge, F. The impact of psychological stress on immune function in the athlete population. Exerc. Immunol. Rev. 2001, 7, 5–17. [Google Scholar] [PubMed]
- Eichner, E.R. Gastrointestinal bleeding in athletes. Phys. Sportsmed. 1989, 17, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Moses, F.M. Gastrointestinal bleeding and the athlete. Am. J. Gastroenterol. 1993, 88, 1157–1159. [Google Scholar] [PubMed]
- Sullivan, S.N.; Wong, C. Runners’ Diarrhea: Different pattern and associated factors. J. Clin. Gastroenterol. 1992, 14, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Butcher, J.D. Runner’s diarrhea and other intestinal problems of athletes. Am. Fam. Phys. 1993, 48, 623–625. [Google Scholar]
- Buckman, M.T. Gastrointestinal bleeding in long-distance runners. Ann. Intern. Med. 1984, 101, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Clausen, J.P. Effect of physical training on cardiovascular adjustment to exercise in man. Physiol. Rev. 1977, 57, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.N.; Richardson, P.D.; Kvietys, P.R.; Mortillaro, N.A. Intestinal blood flow. Gastroenterology 1980, 78, 837–863. [Google Scholar] [PubMed]
- Hall, D.M.; Baumgardner, K.R.; Oberley, T.D.; Gisolfi, C.V. Splanchnic tissue undergo hypoxic stress during whole body hyperthermia. Am. J. Physiol. 1999, 276, G1195–G1203. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.M.; Buettner, G.R.; Oberley, L.W.; Xu, L.; Matthes, R.D.; Gisolfi, C.V. Mechanisms of circulatory and intestinal barrier dysfunction during whole body hyperthermia. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H509–H521. [Google Scholar] [CrossRef] [PubMed]
- Pals, K.L.; Chang, R.T.; Ryan, A.L.; Gisolfi, C.V. Effect of running intensity on intestinal permeability. J. Appl. Physiol. 1997, 82, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Shing, C.M.; Peake, J.; Lim, C.L.; Briskey, D.; Walsh, N.P.; Fortes, M.B.; Ahuja, K.D.K.; Vitetta, L. Effects of probiotics supplementation on gastrointestinal permeability, inflammation and exercise performance in the heat. Eur. J. Appl. Physiol. 2014, 114, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Nieman, D.C. Exercise, Infection, and Immunity. Int. J. Sports Med. 1994, 15, S131–S141. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.L.; Suzuki, K. The pattern recognition receptors and lipopolysaccharides (LPS)-induced systemic inflammation. Int. J. Res. Stud. Med. Health Sci. 2017, 2, 1–7. [Google Scholar]
- Erridge, C.; Attina, T.; Spickett, M.; Webb, D.J. A high-fat meal induces low-grade endotoxemia: Evidence of a novelmechanism of postprandial inflammation. Am. J. Clin. Nutr. 2007, 86, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, L.E.; Lee, E.C.; Armstrong, E.M. Interactions of gut microbiota, endotoxemia, immune function, and diet in exertional heatstroke. J. Sports Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bouchama, A.; Al-sedairy, S.; Siddiqui, S.; Shail, E.; Razeig, M. Elevated pyrogenic cytokines in heatstroke. Chest 1993, 104, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Bouchama, A.; Parhar, S.; El-Yagi, A.; Sheth, K.; Al-Sedairy, S. Endotoxemia and the release of tumor necrosis factor and interleukin1-alpha in acute heat stroke. J. Appl. Physiol. 1991, 70, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- Gathiram, P.; Gaffin, S.L.; Brock Utne, J.G.; Wells, M.T. Prophylactic corticosteroid suppresses endotoxemia in heat-stressed primates. Aviat. Space Environ. Med. 1988, 59, 142–145. [Google Scholar] [PubMed]
- Gathiram, P.; Wells, M.T.; Brock Utne, J.G.; Wessels, B.C.; Gaffin, S.L. Prevention of endotoxaemia by non-absorbable antibiotics in heat stress. J. Clin. Pathol. 1987, 40, 1364–1368. [Google Scholar] [CrossRef] [PubMed]
- Sakurada, S.; Hales, R.J.S. A role for gastrointestinal endotoxins in enhancement of heat tolerance by physical fitness. J. Appl. Physiol. 1998, 84, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Brock Utne, J.G. Incidence of gram-negative bacteraemia in sepsis syndrome. Anaesthesia 1995, 50, 267–268. [Google Scholar] [CrossRef] [PubMed]
- Bosenberg, A.T.; Brock-Utne, J.G.; Gaffin, S.L.; Wells, M.T.; Blake, G.T.W. Strenuous exercise causes systemic endotoxemia. J. Appl. Physiol. 1988, 65, 106–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, Q.Y.; Lee, K.W.; Byrne, C.; Ho, T.F.; Lim, C.L. Plasma endotoxin and immune responses during a 21-km road racen under a warm and humid environment. Ann. Acad. Med. Singap. 2008, 37, 307–314. [Google Scholar] [PubMed]
- Selkirk, G.A.; McLellan, T.M.; Wright, H.E.; Rhind, S.G. Mild endotoxemia, NF-translocation, and cytokine increase during exertional heat stress in trained and untrained individuals. Am. J. Physiol. 2008, 295, R611–R623. [Google Scholar] [CrossRef] [PubMed]
- Epstein, Y.; Shani, Y.; Moran, D.S.; Shapiro, Y. Exertional heat stroke—The prevention of a medical emergency. J. Basic Clin. Physiol. Pharmacol. 2000, 11, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Nieman, D.C. Exercise effects on systemic immunity. Immunol. Cell Biol. 2000, 78, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Rohde, T.; Zacho, M. Immunity in athletes. J. Sports Med. Phys. Fit. 1996, 36, 236–245. [Google Scholar]
- Burger, F.J.; Fuhrman, F.A. Evidence of injury by heat in mammilian tissues. Am. J. Physiol. 1964, 206, 1057–1061. [Google Scholar] [PubMed]
- Frankel, H.M. Effect of restraint on rat exposed to high temperature. J. Appl. Physiol. 1959, 14, 997. [Google Scholar] [CrossRef] [PubMed]
- Nadel, E.R.; Mack, G.W.; Nose, H.; Tripathi, A. Tolerance to severe heat and exercise: Peropheral vascular responses to body fluid changes. In Heat Stress: Physical Exertion and Environment; Hales, R.J.S., Richards, D.A.B., Eds.; Elsevier Science Publisher: Amsterdam, The Netherlands, 1987; pp. 117–131. [Google Scholar]
- Lock, M.; Noble, E.G. Stress Proteins: The Exercise Response. Can. J. Appl. Physiol. 1995, 20, 155–167. [Google Scholar] [CrossRef]
- Craig, E.A. The heat shock response. Crit. Rev. Biochem. 1984, 18, 239–280. [Google Scholar] [CrossRef]
- Bouchama, A.; Hammami, M.M.; Haq, A.; Jackson, J.; al Sedairy, S. Evidence for endothelial cell activation/injury in heatstroke. Crit. Care Med. 1996, 24, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Bowers, W.D.; Hubbard, R.W.; Leav, I.; Daum, R.; Conlon, M.; Hamlet, M.; Mager, M.; Brandt, P. Histological and ultrastructural alterations of rat liver subsequent to heat overload. Arch. Pathol. Lab. Med. 1978, 102, 154–157. [Google Scholar] [PubMed]
- Ferries, E.B., Jr.; Blankenhorn, M.A.; Robinson, H.W.; Cullen, G.E. Heat stroke: Clinical and chemical observations in 44 cases. J. Clin. Investig. 1938, 17, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, V.H. Critical thermal maxima in salamanders. Physiol. Zool. 1961, 34, 92–125. [Google Scholar] [CrossRef]
- Bynum, G.D.; Pandolf, K.B.; Schuette, W.H.; Goldman, R.F.; Lees, D.E.; Whang-Peng, J.; Atkinson, E.R.; Bull, J.M. Induced hyperthermia in sedated humans and the concept of critical thermal maximum. Am. J. Physiol. 1978, 235, R228–R236. [Google Scholar] [CrossRef] [PubMed]
- Pettigrew, R.T.; Galt, J.M.; Ludgate, C.M.; Horn, D.B.; Smith, A.N. Circulatory and biochemic al effects of whole body hyperthermia. Br. J. Surg. 1974, 61, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Pettigrew, R.T.; Galt, J.M.; Ludgate, C.M.; Smith, A.N. Clinical effects of whole-body hyperthermia in advanced malignancy. Br. Med. J. 1974, 4, 479–682. [Google Scholar] [CrossRef]
- Stafanini, M.; Spicer, D.D. Hemostatic breakdown, fibrinolysis and hemolytic anemia in patient with heat stroke: Pathogenetic mechanisms. Am. J. Clin. Pathol. 1971, 55, 180–186. [Google Scholar] [CrossRef]
- Al-Mashhadani, S.A.; Gader, A.G.M.A.; Al Harthi, S.S.; Kangav, D. The coagulopathy of heat stroke: Alterations in coagulation and fibrinolysis in heat stroke patients during the pilgrimage (Haj) to Makkah. Blood Coagul. Fibrinol. 1994, 5, 731–736. [Google Scholar] [CrossRef]
- Ang, C.; Dawes, J. The effects of hyperthermia on human endothelial monolayers: Modulation of thrombotic potential and permeability. Blood Coagul. Fibrinol. 1994, 5, 193–199. [Google Scholar] [CrossRef]
- Smith, L.L. Overtraining, excessive exercise, and altered immunity. Sports Med. 2003, 33, 347–364. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Toft, A.D. Effects of exercise on lymphocytes and cytokines. Br. J. Sports Med. 2000, 34, 246–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieman, D.C. Is infection risk linked to exercise workload? Med. Sci. Sports Exerc. 2000, 32, S406–S411. [Google Scholar] [CrossRef] [PubMed]
- Nieman, D.C. Exercise Immunology: Practical Application. Int. J. Sports Med. 1997, 18 (Suppl. 1), S91–S100. [Google Scholar] [CrossRef]
- Nieman, D.C. Exercise, upper respiratory tract infection, and the immune system. Med. Sci. Sports Exerc. 1994, 26, 128–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, B.K. Influence of physical activity on the cellular immune system: Mechanisms of action. Int. J. Sports Med. 1991, 12 (Suppl. 1), S23–S29. [Google Scholar] [CrossRef]
- Shani, Y.; Moran, D.S.; Heled, Y.; Shapiro, Y.; Epstein, Y. Exertional heat illness. Lancet 2000, 355, 1992–1993. [Google Scholar] [CrossRef]
- Lim, C.L.; Suzuki, K. Systemic inflammation mediates the effects of endotoxemia in the mechanisms of heat stroke. Biol. Med. 2016, 9, 1–3. [Google Scholar] [CrossRef]
- Gathiram, P.; Wells, M.T.; Brock Utne, J.G.; Wessels, B.C.; Gaffin, S.L. Oral administered nonabsorbable antibiotics prevent endotoxemia in primates following intestinal ischemia. J. Surg. Res. 1988, 45, 187–193. [Google Scholar] [CrossRef]
- Guy, J.H.; Vincent, G.E. Nutrition and Supplementation Considerations to Limit Endotoxemia When Exercising in the Heat. Sports 2018, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Taioli, E. Use of permitted drugs in Italian professional soccer players. Br. J. Sports Med. 2007, 41, 439–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alaranta, A.; Alaranta, H.; Heliovaara, M.; Airaksinen, M.; Helenius, I. Ample use of physician-prescribed medications in Finnish elite athletes. Int. J. Sports Med. 2006, 27, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Garcin, M.; Mille-Hamard, L.; Billat, V.; Imbenotte, M.; Humbert, L.; Lhermitte, Z. Use of acetaminophen in young subelite athletes. J. Sports Med. Phys. Fit. 2005, 45, 604–607. [Google Scholar]
- Gorski, T.; Cadore, E.L.; Pinto, S.S.; da Silva, E.M.; Correa, C.S.; Beltrami, F.G.; Kruel, L.F. Use of NSAIDs in triathletes: Prevalence, level of awareness and reasons for use. Br. J. Sports Med. 2011, 45, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Camus, G.; Nys, M.; Poortmans, J.R.; Venneman, I.; Monfils, T.; Deby-Dupont, G.; Juchmes-Ferir, A.; Deby, C.; Lamy, M.; Duchateau, J. Endotoxemia, production of tumor necrosis factor alpha and polymorphonuclear neutrophil activation following strenuous exercise in humans. Eur. J. Appl. Physiol. 1998, 79, 62–68. [Google Scholar] [CrossRef]
- Van Wijck, K.; Lenaerts, K.; Van Bijnen, A.A.; Boonen, B.; Van Loon, L.; Dejong, C.; Buurman, W.A. Aggravation of exercise-induced intestinal injury by ibuprofen in athletes. Med. Sci. Sports Exerc. 2012, 44, 2257–2262. [Google Scholar] [CrossRef] [PubMed]
- Audet, G.N.; Dineen, S.M.; Stewart, D.A.; Plamper, M.L.; Pathmasiri, W.W.; McRitchie, S.L.; Sumner, S.J.; Leon, L.R. Pretreatment with indomethacin results in increased heat stroke severity during recovery in a rodent model of heat stroke. J. Appl. Physiol. 2017, 123, 544–557. [Google Scholar] [CrossRef] [PubMed]
- Nieman, D.C.; Henson, D.A.; Dumke, C.L.; Oley, K.; McNaulty, S.R.; Davis, J.M.; Murphy, E.A.; Utter, A.C.; Lind, R.H.; McNaulty, L.S.; et al. Ibuprofen use, endotoxemia, inflammation, and plasma cytokines during ultramarathon competitions. Brain Behav. Immun. 2006, 20, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Peppelenbosch, M.P.; Ferreira, C.V. Immunology of pre- and probiotic supplementation. Br. J. Nutr. 2009, 101, 2–4. [Google Scholar] [CrossRef] [PubMed]
- West, N.P.; Pyne, D.B.; Peake, J.M.; Cripps, A.W. Probiotics, immunity and exercise: A review. Exerc. Immunol. Rev. 2009, 15, 107–126. [Google Scholar] [PubMed]
- Mack, D.R.; Ahrne, S.; Hyde, L.; Wei, S.; Hollingsworth, M.A. Extracellular MUC3 mucin secretion follows adherence of Lactobacillus strains to intestinal epithelial cells in vitro. Gut 2003, 52, 827–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mack, D.R.; Michail, S.; Wei, S.; McDougall, L.; Hollingsworth, M.A. Probiotics inhibit enteropathogenic E. coli adherence in vitro by inducing intestinal mucin gene expression. Am. J. Physiol. 1999, 276, G941–G950. [Google Scholar] [PubMed]
- Shing, C.M.; Hunter, D.C.; Stevenson, L.M. Bovine colostrum supplementation and exercise performance: Potential mechanisms. Sports Med. 2009, 39, 1033–1054. [Google Scholar] [CrossRef] [PubMed]
- Playford, R.J.; Floyd, D.N.; Macdonald, C.E.; Calnan, D.P.; Adenekan, R.O.; Johnson, W.; Goodlad, R.A.; Marchbank, T. Bovine colostrum is a health food supplement which prevents NSAID induced gut damage. Gut 1999, 44, 653–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Playford, R.J.; MacDonald, C.E.; Calnan, D.P.; Floyd, D.N.; Podas, T.; Johnson, W.; Wicks, A.C.; Bashir, O.; Marchbank, T. Co-administration of the health food supplement, bovine colostrum, reduces the acute non-steroidal anti-inflammatory drug-induced increase in intestinal permeability. Clin. Sci. (Lond.) 2001, 100, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Jeon, W.K.; Kim, E.J. Combined effects of bovine colostrum and glutamine in diclofenac-induced bacterial translocation in rat. Clin. Nutr. 2005, 24, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Marchbank, T.; Davison, G.; Oakes, J.R.; Ghatei, M.A.; Patterson, M.; Moyer, M.P.; Playford, R.J. The nutraceutical bovine colostrum truncates the increase in gut permeability caused by heavy intense exercise in athletes. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G477–G484. [Google Scholar] [CrossRef] [PubMed]
- Brinkworth, G.D.; Buckley, J.D. Concentrated bovine colostrum protein supplementation reduces the incidence of self-reported symptoms of upper respiratory tract infection in adult males. Eur. J. Nutr. 2003, 42, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Crooks, C.V.; Wall, C.R.; Cross, M.L.; Rutherfurd-Markwick, K.J. The effect of bovine colostrum supplementation on salivary IgA in distance runners. Int. J. Sport Nutr. Exerc. Metab. 2006, 16, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Buckley, J.D.; Butler, R.N.; Southcott, E.; Brinkworth, G.D. Bovine colostrum supplementation during running training increases intestinal permeability. Nutrients 2009, 1, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.A.; Cheung, S.S.; Cotter, J.D. Bovine colostrum, training status, and gastrointestinal permeability during exercise in the heat: A placebo-controlled double-blind study. Appl. Physiol. Nutr. Metab. 2014, 39, 1070–1082. [Google Scholar] [CrossRef] [PubMed]
- Sithinamsuwan, P.; Piyavechvirathanat, K.; Kitthaweesin, T.; Chusri, W.; Orrawanhanothai, P.; Wongsat, A.; Wattanathum, A.; Chinvarun, Y.; Nidhinandana, S.; Satirapoj, B.; et al. Exertional heat stroke: Early recognition and outcome with agressive combined-cooling—A 12-year experience. Mil. Med. 2009, 174, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Keren, G.; Epstein, Y.; Magazanik, A. Temporary heat intolerance in a heatstroke patient. Aviat. Space Environ. Med. 1981, 52, 116–117. [Google Scholar] [PubMed]
- Ekblom, B.; Ekblom, O.; Malm, C. Infectious episodes befoe and after a marathon race. Scand. J. Med. Sci. Sports 2006, 16, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Matos, N.; Winsley, R.; Williams, C. Prevalence of nonfunctional overeaching/overtraining in young English athletes. Med. Sci. Sports Exerc. 2011, 43, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Brock Utne, J.G.; Gaffin, S.L.; Wells, M.T.; Gathiram, P.; Sohar, E.; James, M.F.; Morrell, D.F.; Norman, R.J. Endotoxaemia in exhausted runners after a long-distance race. S. Afr. Med. J. 1988, 73, 533–536. [Google Scholar] [PubMed]
- Chen, S.H.; Lin, M.T.; Chang, C.P. Ischemic and oxidative damage to the hypothalamus may be responsible for heat stroke. Curr. Neuropharmacol. 2013, 11, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Karemer, W.J.; Armstrong, L.E.; Watson, G. The effects of exertional heatstroke on exercise and exercise-heat acclimation on plasma beta-endorphin concentrations. Aviat. Space Environ. Med. 2003, 74, 758–762. [Google Scholar]
- Gathiram, P.; Wells, M.T.; Brock Utne, J.G.; Gaffin, S.L. Prophylactic corticosteroid increases survival in experimental heat stroke in primates. Aviat. Space Environ. Med. 1988, 59, 352–355. [Google Scholar] [PubMed]
- Liu, C.C.; Chien, C.H.; Lin, M.T. Glucocorticoids reduce interleukin-1 concentration and result in neuroprotective effects in rat heatstroke. J. Physiol. 2000, 527, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Bouchama, A.; Kwaasi, A.; Dehbi, M.; Al Mohanna, F.; Eldali, A.; El-Sayed, R.; Tbakhi, A.; Alzahrani, A.S.; Roberts, A.G. Glucocorticoids do not protect against the lethal effects of experimental heatstroke in baboons. Shock 2007, 27, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.S.; Chou, M.T.; Chao, C.M.; Chang, C.K.; Lin, M.T.; Chang, C.P. Melatonin reduces acute lung inflammation, edema, and hemorrhage in heatstroke rats. Acta Pharmacol. Sin. 2012, 33, 775–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.F.; Lin, C.H.; Hsu, S.F.; Lin, M.T. Melatonin improves outcomes of heatstroke in mice by reducing brain inflammation and oxidative damage and multiple organ dysfunction. Mediat. Inflamm. 2013. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, C.L. Heat Sepsis Precedes Heat Toxicity in the Pathophysiology of Heat Stroke—A New Paradigm on an Ancient Disease. Antioxidants 2018, 7, 149. https://doi.org/10.3390/antiox7110149
Lim CL. Heat Sepsis Precedes Heat Toxicity in the Pathophysiology of Heat Stroke—A New Paradigm on an Ancient Disease. Antioxidants. 2018; 7(11):149. https://doi.org/10.3390/antiox7110149
Chicago/Turabian StyleLim, Chin Leong. 2018. "Heat Sepsis Precedes Heat Toxicity in the Pathophysiology of Heat Stroke—A New Paradigm on an Ancient Disease" Antioxidants 7, no. 11: 149. https://doi.org/10.3390/antiox7110149
APA StyleLim, C. L. (2018). Heat Sepsis Precedes Heat Toxicity in the Pathophysiology of Heat Stroke—A New Paradigm on an Ancient Disease. Antioxidants, 7(11), 149. https://doi.org/10.3390/antiox7110149