Interleukin-1β Protects Neurons against Oxidant-Induced Injury via the Promotion of Astrocyte Glutathione Production

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cell Culture
2.3. IL-1β Treatment
2.4. Measurement of GSH
2.5. Drug Exposure
2.6. Measurement of Cell Death and Viability
2.7. Statistical Analysis
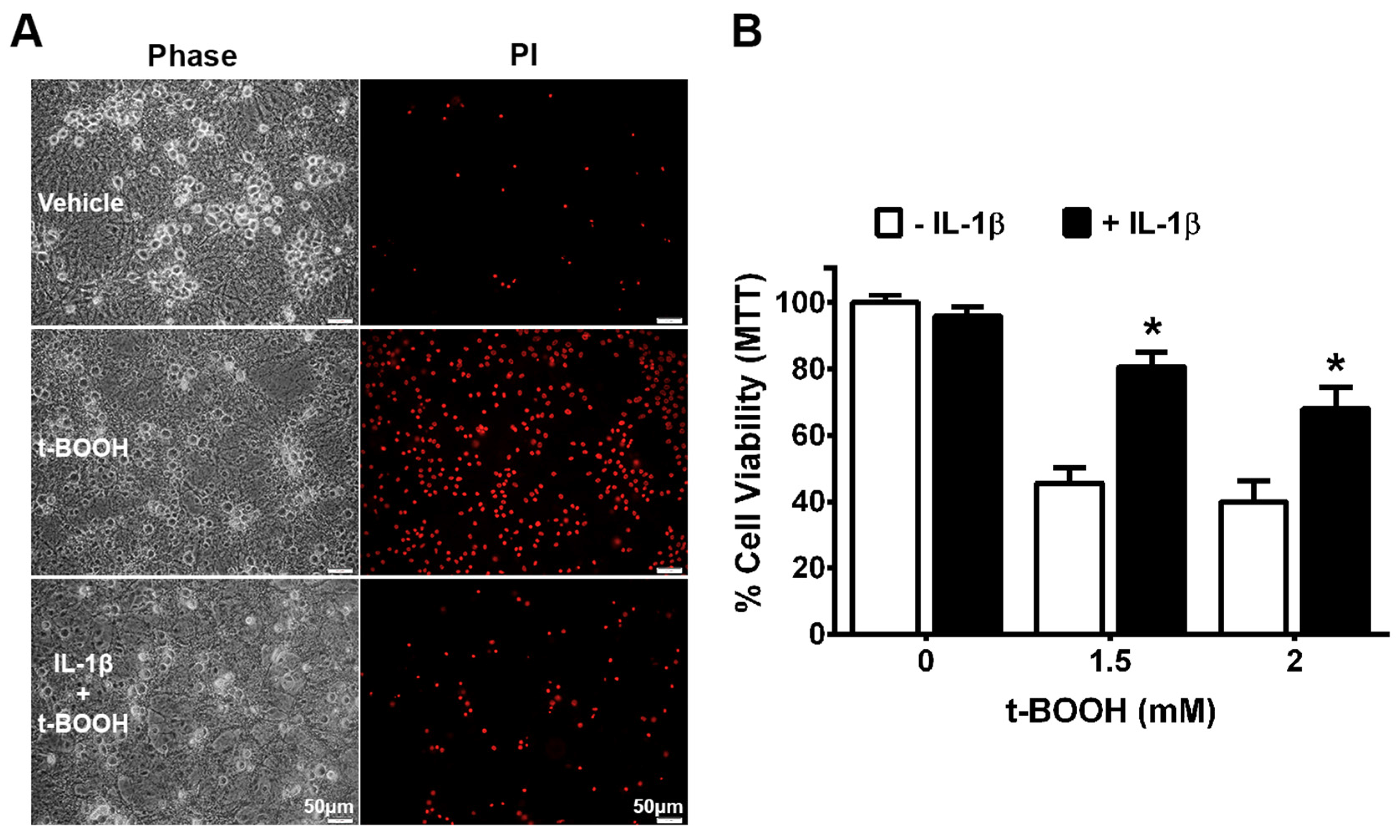
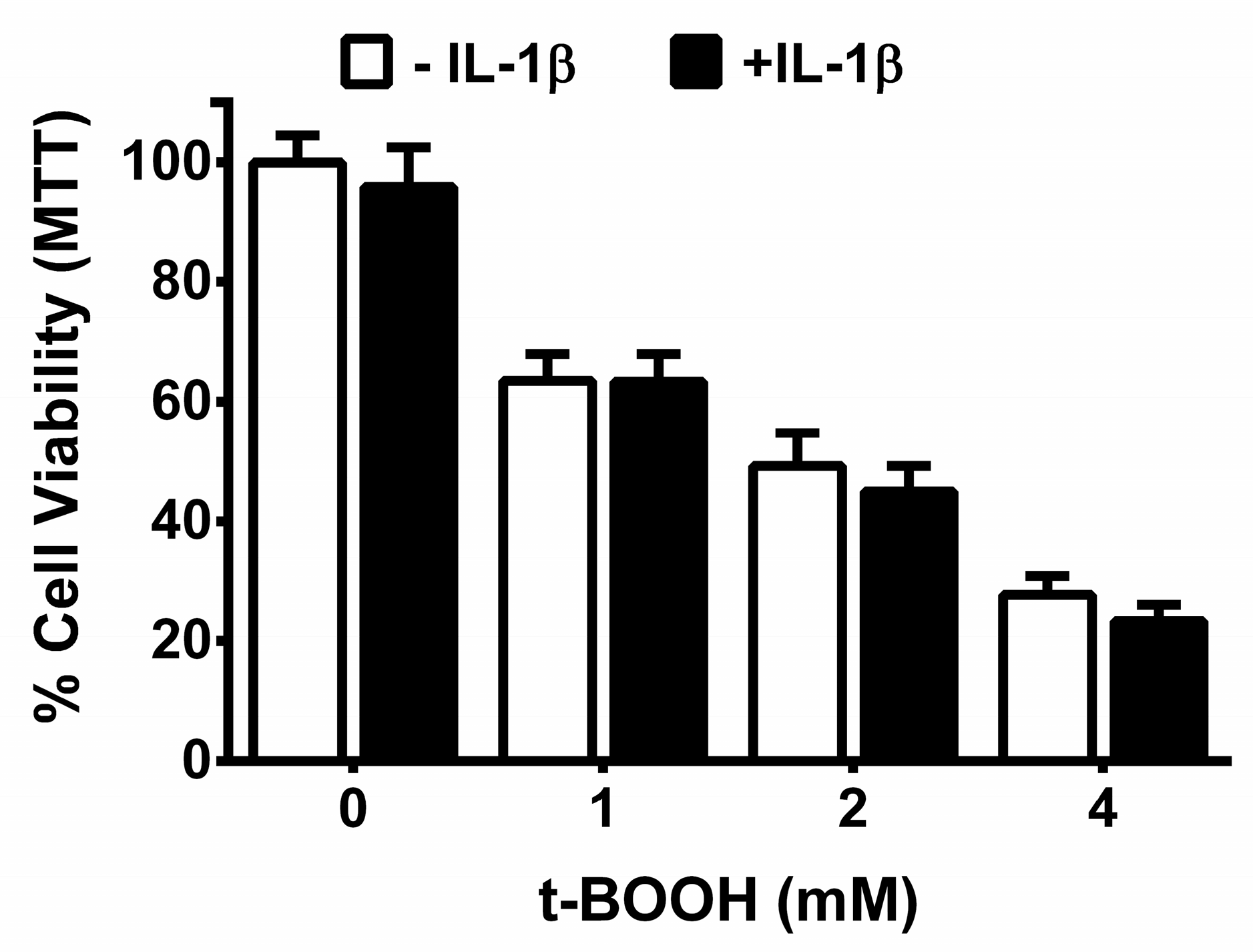
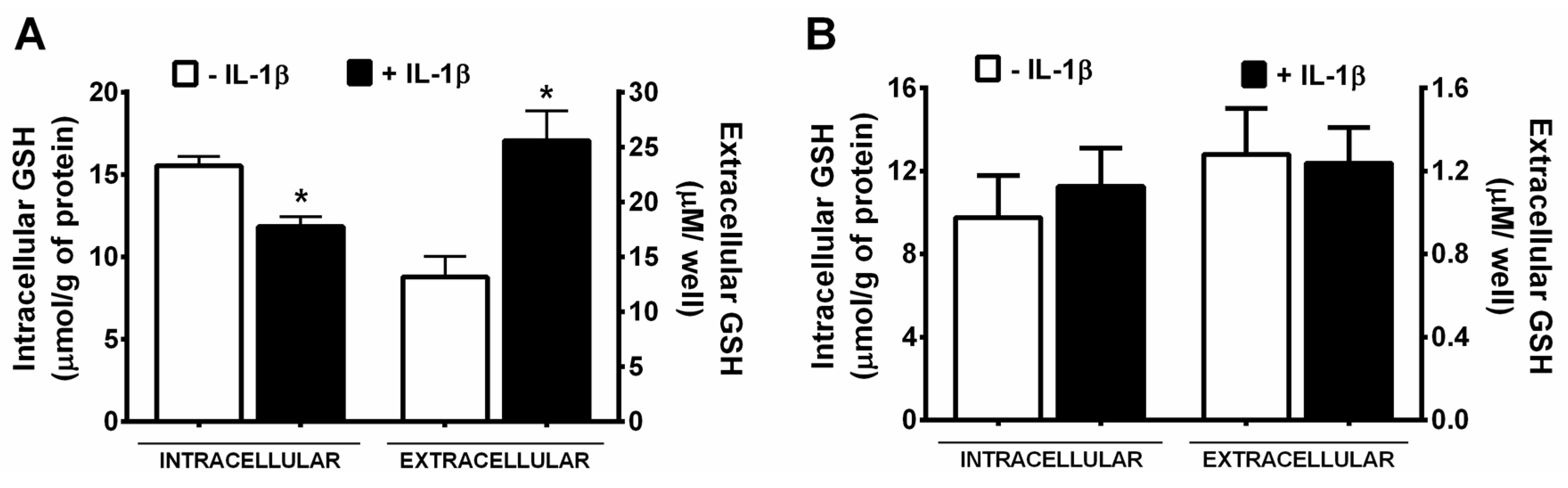
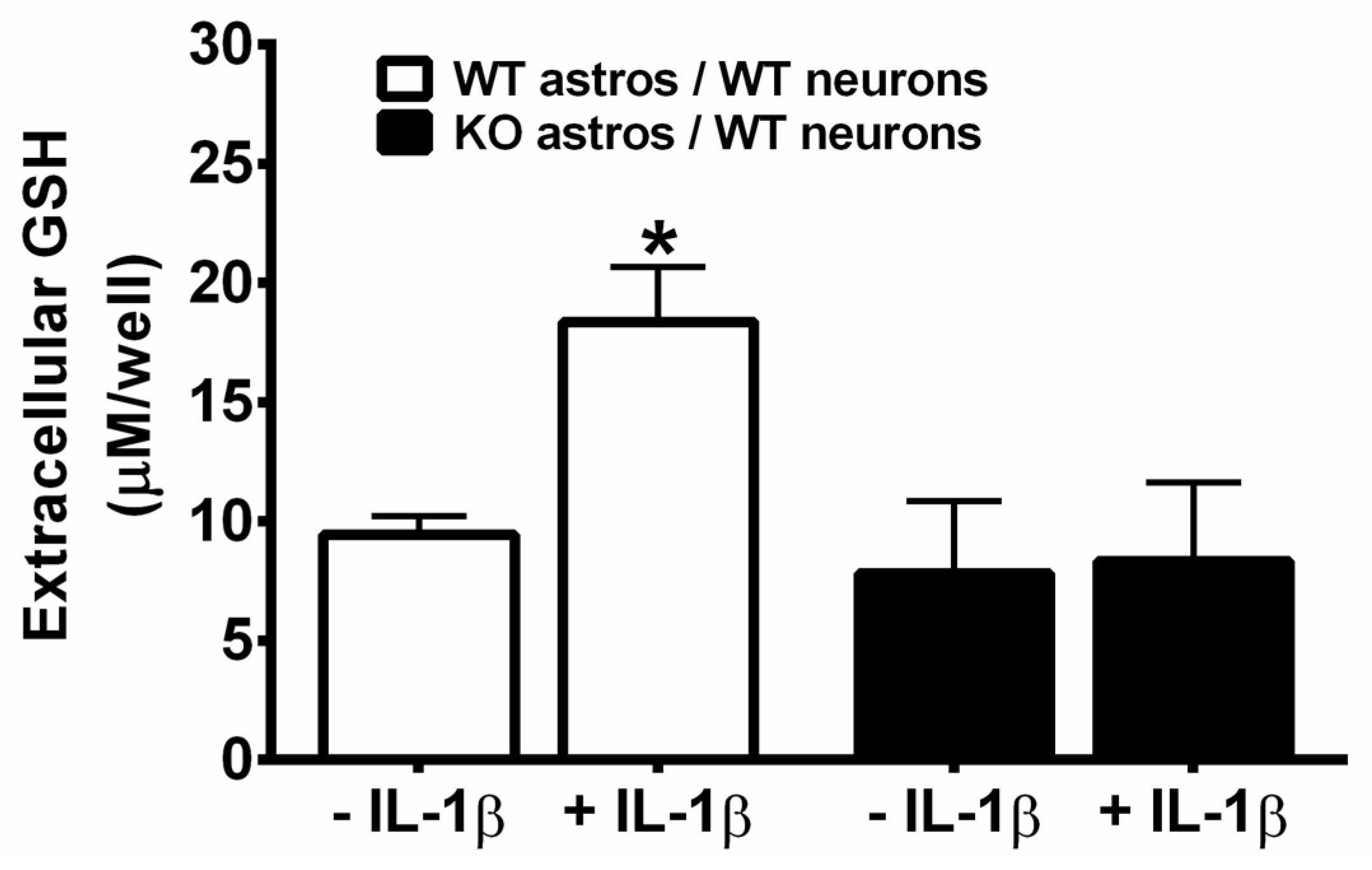
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ross, F.M.; Allan, S.M.; Rothwell, N.J.; Verkhratsky, A. A dual role for interleukin-1 in LTP in mouse hippocampal slices. J. Neuroimmunol. 2003, 144, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.; Pitossi, F.; Balschun, D.; Wagner, A.; del Rey, A.; Besedovsky, H.O. A neuromodulatory role of interleukin-1β in the hippocampus. Proc. Natl. Acad. Sci. USA 1998, 95, 7778–7783. [Google Scholar] [CrossRef] [PubMed]
- Avital, A.; Goshen, I.; Kamsler, A.; Segal, M.; Iverfeldt, K.; Richter-Levin, G.; Yirmiya, R. Impaired interleukin-1 signaling is associated with deficits in hippocampal memory processes and neural plasticity. Hippocampus 2003, 13, 826–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Wang, Y.; Krueger, J.M. Effects of interleukin-1 beta on sleep are mediated by the type I receptor. Am. J. Physiol. 1998, 274, R655–R660. [Google Scholar] [PubMed]
- Krueger, J.M.; Fang, J.; Taishi, P.; Chen, Z.; Kushikata, T.; Gardi, J. Sleep. A physiologic role for IL-1 beta and TNF-alpha. Ann. N. Y. Acad. Sci. 1998, 856, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.M.; Linz, B.; Diekelmann, S.; Besedovsky, L.; Lange, T.; Born, J. Effects of an interleukin-1 receptor antagonist on human sleep, sleep-associated memory consolidation, and blood monocytes. Brain Behav. Immun. 2015, 47, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Hewett, S.J.; Jackman, N.A.; Claycomb, R.J. Interleukin-1β in central nervous system injury and repair. Eur. J. Neurodegener. Dis. 2012, 1, 195–211. [Google Scholar] [PubMed]
- Jackman, N.A.; Uliasz, T.F.; Hewett, J.A.; Hewett, S.J. Regulation of system xc− activity and expression in astrocytes by interleukin-1beta: Implications for hypoxic neuronal injury. Glia 2010, 58, 1806–1815. [Google Scholar] [CrossRef] [PubMed]
- Fogal, B.; Li, J.; Lobner, D.; McCullough, L.D.; Hewett, S.J. System xc− activity and astrocytes are necessary for interleukin-1beta-mediated hypoxic neuronal injury. J. Neurosci. 2007, 27, 10094–10105. [Google Scholar] [CrossRef] [PubMed]
- Fogal, B.; Hewett, J.A.; Hewett, S.J. Interleukin-1beta potentiates neuronal injury in a variety of injury models involving energy deprivation. J. Neuroimmunol. 2005, 161, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Jackman, N.A.; Melchior, S.E.; Hewett, J.A.; Hewett, S.J. Non-cell autonomous influence of the astrocyte system xc− on hypoglycaemic neuronal cell death. ASN Neuro 2012, 4, e00074. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Jackman, N.A.; Thorn, T.L.; Vought, V.E.; Hewett, S.J. Interleukin-1beta protects astrocytes against oxidant-induced injury via an nf-kappab-dependent upregulation of glutathione synthesis. Glia 2015, 63, 1568–1580. [Google Scholar] [CrossRef] [PubMed]
- Glaccum, M.B.; Stocking, K.L.; Charrier, K.; Smith, J.L.; Willis, C.R.; Maliszewski, C.; Livingston, D.J.; Peschon, J.J.; Morrissey, P.J. Phenotypic and functional characterization of mice that lack the type I receptor for IL-1. J. Immunol. 1997, 159, 3364–3371. [Google Scholar] [PubMed]
- Hamby, M.E.; Uliasz, T.F.; Hewett, S.J.; Hewett, J.A. Characterization of an improved procedure for the removal of microglia from confluent monolayers of primary astrocytes. J. Neurosci. Methods 2006, 150, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Trackey, J.L.; Uliasz, T.F.; Hewett, S.J. Sin-1-induced cytotoxicity in mixed cortical cell culture: Peroxynitrite-dependent and -independent induction of excitotoxic cell death. J. Neurochem. 2001, 79, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Kussmaul, L.; Hamprecht, B. Rapid clearance of tertiary butyl hydroperoxide by cultured astroglial cells via oxidation of glutathione. Glia 1998, 23, 139–145. [Google Scholar] [CrossRef]
- Hirrlinger, J.; Schulz, J.B.; Dringen, R. Glutathione release from cultured brain cells: Multidrug resistance protein 1 mediates the release of GSH from rat astroglial cells. J. Neurosci. Res. 2002, 69, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Borlongan, C.V.; Koutouzis, T.K.; Freeman, T.B.; Cahill, D.W.; Sanberg, P.R. Behavioral pathology induced by repeated systemic injections of 3-nitropropionic acid mimics the motoric symptoms of huntington’s disease. Brain Res. 1995, 697, 254–257. [Google Scholar] [CrossRef]
- Schulz, J.B.; Henshaw, D.R.; MacGarvey, U.; Beal, M.F. Involvement of oxidative stress in 3-nitropropionic acid neurotoxicity. Neurochem. Int. 1996, 29, 167–171. [Google Scholar] [CrossRef]
- Uliasz, T.F.; Hewett, S.J. A microtiter trypan blue absorbance assay for the quantitative determination of excitotoxic neuronal injury in cell culture. J. Neurosci. Methods 2000, 100, 157–163. [Google Scholar] [CrossRef]
- Lobner, D. Comparison of the LDH and mtt assays for quantifying cell death: Validity for neuronal apoptosis? J. Neurosci. Methods 2000, 96, 147–152. [Google Scholar] [CrossRef]
- He, Y.; Akumuo, R.C.; Yang, Y.; Hewett, S.J. Mice deficient in l-12/15 lipoxygenase show increased vulnerability to 3-nitropropionic acid neurotoxicity. Neurosci. Lett. 2017, 643, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.Y.; Imbeault, S.; Barakauskas, V.; Erb, H.; Jiang, L.; Li, P.; Murphy, T.H. Induction of the Nrf2-driven antioxidant response confers neuroprotection during mitochondrial stress in vivo. J. Biol. Chem. 2005, 280, 22925–22936. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Bankiewicz, K.S.; Plunkett, R.J.; Oldfield, E.H. Intrastriatal implantation of interleukin-1. Reduction of parkinsonism in rats by enhancing neuronal sprouting from residual dopaminergic neurons in the ventral tegmental area of the midbrain. J. Neurosurg. 1994, 80, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Fagan, A.M.; Gage, F.H. Cholinergic sprouting in the hippocampus: A proposed role for IL-1. Exp. Neurol. 1990, 110, 105–120. [Google Scholar] [CrossRef]
- Temporin, K.; Tanaka, H.; Kuroda, Y.; Okada, K.; Yachi, K.; Moritomo, H.; Murase, T.; Yoshikawa, H. IL-1beta promotes neurite outgrowth by deactivating RhoA via p38 mapk pathway. Biochem. Biophys. Res. Commun. 2008, 365, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, T.; Ruetzler, C.A.; Tasaki, K.; Hallenbeck, J.M. Interleukin-1 mediates induction of tolerance to global ischemia in gerbil hippocampal CA1 neurons. J. Cereb. Blood Flow Metab. 1996, 16, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Pringle, A.K.; Niyadurupola, N.; Johns, P.; Anthony, D.C.; Iannotti, F. Interleukin-1beta exacerbates hypoxia-induced neuronal damage, but attenuates toxicity produced by simulated ischaemia and excitotoxicity in rat organotypic hippocampal slice cultures. Neurosci. Lett. 2001, 305, 29–32. [Google Scholar] [CrossRef]
- Carlson, N.G.; Wieggel, W.A.; Chen, J.; Bacchi, A.; Rogers, S.W.; Gahring, L.C. Inflammatory cytokines IL-1 alpha, IL-1 beta, IL-6, and TNF-alpha impart neuroprotection to an excitotoxin through distinct pathways. J. Immunol. 1999, 163, 3963–3968. [Google Scholar] [PubMed]
- Strijbos, P.J.; Rothwell, N.J. Interleukin-1 beta attenuates excitatory amino acid-induced neurodegeneration in vitro: Involvement of nerve growth factor. J. Neurosci. 1995, 15, 3468–3474. [Google Scholar] [CrossRef] [PubMed]
- Shaftel, S.S.; Kyrkanides, S.; Olschowka, J.A.; Miller, J.N.; Johnson, R.E.; O’Banion, M.K. Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates alzheimer plaque pathology. J. Clin. Investig. 2007, 117, 1595–1604. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.H.; Wu, M.; Shaftel, S.S.; Graham, K.A.; O’Banion, M.K. Sustained expression of interleukin-1beta in mouse hippocampus impairs spatial memory. Neuroscience 2009, 164, 1484–1495. [Google Scholar] [CrossRef] [PubMed]
- Matousek, S.B.; Hein, A.M.; Shaftel, S.S.; Olschowka, J.A.; Kyrkanides, S.; O’Banion, M.K. Cyclooxygenase-1 mediates prostaglandin E2 elevation and contextual memory impairment in a model of sustained hippocampal interleukin-1beta expression. J. Neurochem. 2010, 114, 247–258. [Google Scholar] [PubMed]
- Hassel, B.; Sonnewald, U. Selective inhibition of the tricarboxylic acid cycle of gabaergic neurons with 3-nitropropionic acid in vivo. J. Neurochem. 1995, 65, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Brouillet, E.; Conde, F.; Beal, M.F.; Hantraye, P. Replicating huntington’s disease phenotype in experimental animals. Prog. Neurobiol. 1999, 59, 427–468. [Google Scholar] [CrossRef]
- Beal, M.F.; Brouillet, E.; Jenkins, B.G.; Ferrante, R.J.; Kowall, N.W.; Miller, J.M.; Storey, E.; Srivastava, R.; Rosen, B.R.; Hyman, B.T. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J. Neurosci. 1993, 13, 4181–4192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouillet, E.; Jacquard, C.; Bizat, N.; Blum, D. 3-nitropropionic acid: A mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in huntington’s disease. J. Neurochem. 2005, 95, 1521–1540. [Google Scholar] [CrossRef] [PubMed]
- Palfi, S.; Ferrante, R.J.; Brouillet, E.; Beal, M.F.; Dolan, R.; Guyot, M.C.; Peschanski, M.; Hantraye, P. Chronic 3-nitropropionic acid treatment in baboons replicates the cognitive and motor deficits of huntington’s disease. J. Neurosci. 1996, 16, 3019–3025. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, D.S.; Carter, R.J.; Morton, A.J. Dopamine modulates the susceptibility of striatal neurons to 3-nitropropionic acid in the rat model of huntington’s disease. J. Neurosci. 1998, 18, 10116–10127. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann. Neurol. 1995, 38, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Ban, E.M. Interleukin-1 receptors in the brain: Characterization by quantitative in situ autoradiography. Immunomethods 1994, 5, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Ban, E.M.; Sarlieve, L.L.; Haour, F.G. Interleukin-1 binding sites on astrocytes. Neuroscience 1993, 52, 725–733. [Google Scholar] [CrossRef]
- Choi, S.; Friedman, W.J. Inflammatory cytokines IL-1beta and TNF-alpha regulate p75NTR expression in CNS neurons and astrocytes by distinct cell-type-specific signalling mechanisms. ASN Neuro 2009, 1, e00010. [Google Scholar] [CrossRef] [PubMed]
- Friedman, W.J. Cytokines regulate expression of the type 1 interleukin-1 receptor in rat hippocampal neurons and glia. Exp. Neurol. 2001, 168, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Tomozawa, Y.; Inoue, T.; Satoh, M. Expression of type I interleukin-1 receptor mrna and its regulation in cultured astrocytes. Neurosci. Lett. 1995, 195, 57–60. [Google Scholar] [CrossRef]
- Srinivasan, D.; Yen, J.H.; Joseph, D.J.; Friedman, W. Cell type-specific interleukin-1beta signaling in the CNS. J. Neurosci. 2004, 24, 6482–6488. [Google Scholar] [CrossRef] [PubMed]
- Sagara, J.I.; Miura, K.; Bannai, S. Maintenance of neuronal glutathione by glial cells. J. Neurochem. 1993, 61, 1672–1676. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Shih, A.Y.; Johannssen, H.C.; Erb, H.; Li, P.; Murphy, T.H. Two-photon imaging of glutathione levels in intact brain indicates enhanced redox buffering in developing neurons and cells at the cerebrospinal fluid and blood-brain interface. J. Biol. Chem. 2006, 281, 17420–17431. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef] [PubMed]
- Keelan, J.; Allen, N.J.; Antcliffe, D.; Pal, S.; Duchen, M.R. Quantitative imaging of glutathione in hippocampal neurons and glia in culture using monochlorobimane. J. Neurosci. Res. 2001, 66, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Raps, S.P.; Lai, J.C.; Hertz, L.; Cooper, A.J. Glutathione is present in high concentrations in cultured astrocytes but not in cultured neurons. Brain Res. 1989, 493, 398–401. [Google Scholar] [CrossRef]
- Jakel, R.J.; Townsend, J.A.; Kraft, A.D.; Johnson, J.A. Nrf2-mediated protection against 6-hydroxydopamine. Brain Res. 2007, 1144, 192–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, J.; Toku, K.; Zhang, B.; Ishihara, K.; Sakanaka, M.; Maeda, N. Astrocytes protect neuronal death induced by reactive oxygen and nitrogen species. Glia 1999, 28, 85–96. [Google Scholar] [CrossRef]
- Desagher, S.; Glowinski, J.; Premont, J. Astrocytes protect neurons from hydrogen peroxide toxicity. J. Neurosci. 1996, 16, 2553–2562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makar, T.K.; Nedergaard, M.; Preuss, A.; Gelbard, A.S.; Perumal, A.S.; Cooper, A.J. Vitamin E, ascorbate, glutathione, glutathione disulfide, and enzymes of glutathione metabolism in cultures of chick astrocytes and neurons: Evidence that astrocytes play an important role in antioxidative processes in the brain. J. Neurochem. 1994, 62, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Clark, J.B.; Heales, S.J.R. Co-culture of neurones with glutathione deficient astrocytes leads to increased neuronal susceptibility to nitric oxide and increased glutamate-cysteine ligase activity. Brain Res. 2005, 1036, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Drukarch, B.; Schepens, E.; Jongenelen, C.A.M.; Stoof, J.C.; Langeveld, C.H. Astrocyte-mediated enhancement of neuronal survival is abolished by glutathione deficiency. Brain Res. 1997, 770, 123–130. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J. Neurosci. 2003, 23, 5088–5095. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; He, Y.; Hewett, S.J.; Hewett, J.A. Interleukin 1beta regulation of the system xc− substrate-specific subunit, xCT, in primary mouse astrocytes involves the RNA-binding protein HuR. J. Biol. Chem. 2016, 291, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Gutterer, J.M.; Hirrlinger, J. Glutathione metabolism in brain. Metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur. J. Biochem. 2000, 267, 4912–4916. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Pfeiffer, B.; Hamprecht, B. Synthesis of the antioxidant glutathione in neurons: Supply by astrocytes of CysGly as precursor for neuronal glutathione. J. Neurosci. 1999, 19, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.B.; Lindenau, J.; Seyfried, J.; Dichgans, J. Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem. 2000, 267, 4904–4911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kranich, O.; Hamprecht, B.; Dringen, R. Different preferences in the utilization of amino acids for glutathione synthesis in cultured neurons and astroglial cells derived from rat brain. Neurosci. Lett. 1996, 219, 211–214. [Google Scholar] [CrossRef]
- Stridh, M.H.; Tranberg, M.; Weber, S.G.; Blomstrand, F.; Sandberg, M. Stimulated efflux of amino acids and glutathione from cultured hippocampal slices by omission of extracellular calcium: Likely involvement of connexin hemichannels. J. Biol. Chem. 2008, 283, 10347–10356. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Dringen, R. Gap junction hemichannel-mediated release of glutathione from cultured rat astrocytes. Neurosci. Lett. 2007, 415, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Hanigan, M.H.; Ricketts, W.A. Extracellular glutathione is a source of cysteine for cells that express gamma-glutamyl transpeptidase. Biochemistry 1993, 32, 6302–6306. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.K.; Binkley, F. Metabolism of glutathione. III. Enzymatic hydrolysis of cysteinylglycine. J. Biol. Chem. 1950, 186, 731–735. [Google Scholar] [PubMed]
- Dringen, R.; Gutterer, J.M.; Gros, C.; Hirrlinger, J. Aminopeptidase n mediates the utilization of the GSH precursor CysGly by cultured neurons. J. Neurosci. Res. 2001, 66, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Babior, B.M. Oxygen-dependent microbial killing by phagocytes (second of two parts). N. Engl. J. Med. 1978, 298, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Babior, B.M. Oxygen-dependent microbial killing by phagocytes (first of two parts). N. Engl. J. Med. 1978, 298, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Carreras, M.C.; Pargament, G.A.; Catz, S.D.; Poderoso, J.J.; Boveris, A. Kinetics of nitric oxide and hydrogen peroxide production and formation of peroxynitrite during the respiratory burst of human neutrophils. FEBS Lett. 1994, 341, 65–68. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Hatter, A.; Liu, B. Manganese chloride stimulates rat microglia to release hydrogen peroxide. Toxicol. Lett. 2007, 173, 88–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banati, R.B.; Gehrmann, J.; Schubert, P.; Kreutzberg, G.W. Cytotoxicity of microglia. Glia 1993, 7, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Henzler, T.; Steudle, E. Transport and metabolic degradation of hydrogen peroxide in chara corallina: Model calculations and measurements with the pressure probe suggest transport of H2O2 across water channels. J. Exp. Bot. 2000, 51, 2053–2066. [Google Scholar] [CrossRef] [PubMed]
- Badaut, J.; Lasbennes, F.; Magistretti, P.J.; Regli, L. Aquaporins in brain: Distribution, physiology, and pathophysiology. J. Cereb. Blood Flow Metab. 2002, 22, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Badaut, J.; Fukuda, A.M.; Jullienne, A.; Petry, K.G. Aquaporin and brain diseases. Biochim. Biophys. Acta 2014, 1840, 1554–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, H.; Tamba, M.; Ishii, T.; Bannai, S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem. 1999, 274, 11455–11458. [Google Scholar] [CrossRef] [PubMed]
- Bassi, M.T.; Gasol, E.; Manzoni, M.; Pineda, M.; Riboni, M.; Martin, R.; Zorzano, A.; Borsani, G.; Palacin, M. Identification and characterisation of human xCT that co-expresses, with 4F2 heavy chain, the amino acid transport activity system xc. Pflugers Archiv Eur. J. Physiol. 2001, 442, 286–296. [Google Scholar] [CrossRef]
- Bridges, C.C.; Kekuda, R.; Wang, H.; Prasad, P.D.; Mehta, P.; Huang, W.; Smith, S.B.; Ganapathy, V. Structure, function, and regulation of human cystine/glutamate transporter in retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2001, 42, 47–54. [Google Scholar]
- Deneke, S.M.; Fanburg, B.L. Regulation of cellular glutathione. Am. J. Physiol. 1989, 257, L163–L173. [Google Scholar] [CrossRef] [PubMed]
- Gennuso, F.; Fernetti, C.; Tirolo, C.; Testa, N.; L’Episcopo, F.; Caniglia, S.; Morale, M.C.; Ostrow, J.D.; Pascolo, L.; Tiribelli, C.; et al. Bilirubin protects astrocytes from its own toxicity by inducing up-regulation and translocation of multidrug resistance-associated protein 1 (Mrp1). Proc. Natl. Acad. Sci. USA 2004, 101, 2470–2475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, B.; Shen, H.; Zhang, J.; Zhu, Y.G.; Ransom, B.R.; Chen, X.C.; Ye, Z.C. Dual pathways mediate beta-amyloid stimulated glutathione release from astrocytes. Glia 2015, 63, 2208–2219. [Google Scholar] [CrossRef] [PubMed]
- Ballatori, N.; Hammond, C.L.; Cunningham, J.B.; Krance, S.M.; Marchan, R. Molecular mechanisms of reduced glutathione transport: Role of the MRP/CFTR/ABCC and OATP/SLC21A families of membrane proteins. Toxicol. Appl. Pharmacol. 2005, 204, 238–255. [Google Scholar] [CrossRef] [PubMed]
- Becher, B.; Spath, S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2017, 17, 49–59. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chowdhury, T.; Allen, M.F.; Thorn, T.L.; He, Y.; Hewett, S.J. Interleukin-1β Protects Neurons against Oxidant-Induced Injury via the Promotion of Astrocyte Glutathione Production. Antioxidants 2018, 7, 100. https://doi.org/10.3390/antiox7080100
Chowdhury T, Allen MF, Thorn TL, He Y, Hewett SJ. Interleukin-1β Protects Neurons against Oxidant-Induced Injury via the Promotion of Astrocyte Glutathione Production. Antioxidants. 2018; 7(8):100. https://doi.org/10.3390/antiox7080100
Chicago/Turabian StyleChowdhury, Twinkle, Matthew F. Allen, Trista L. Thorn, Yan He, and Sandra J. Hewett. 2018. "Interleukin-1β Protects Neurons against Oxidant-Induced Injury via the Promotion of Astrocyte Glutathione Production" Antioxidants 7, no. 8: 100. https://doi.org/10.3390/antiox7080100
APA StyleChowdhury, T., Allen, M. F., Thorn, T. L., He, Y., & Hewett, S. J. (2018). Interleukin-1β Protects Neurons against Oxidant-Induced Injury via the Promotion of Astrocyte Glutathione Production. Antioxidants, 7(8), 100. https://doi.org/10.3390/antiox7080100