Assessing the Efficacy of Dietary Selenomethionine Supplementation in the Setting of Cardiac Ischemia/Reperfusion Injury




Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents and Quantification of Hypohalous Acids
2.2. Cell Culture
2.3. In Vitro SeMet Supplementation and Oxidative Insult
2.4. Animals and Diets
2.5. In Vivo Cardiac Ischemia/Reperfusion
2.6. Flow Cytometric Analysis
2.7. Glutathione Peroxidase Activity Assay
2.8. Cell Proliferation Assay
2.9. Lactate Dehydrogenase (LDH) Release Assay
2.10. Quantification of Cellular Thiols
2.11. Quantification of GSH
2.12. Echocardiography
2.13. Tissue Collection
2.14. Quantification of Selenium Using Inductively Coupled Plasma Mass Spectrometry (ICP-MS)
2.15. Triphenyl Tetrazolium Chloride (TTC) Staining
2.16. Histological Studies
2.17. qPCR
2.18. Statistical Analysis
3. Results
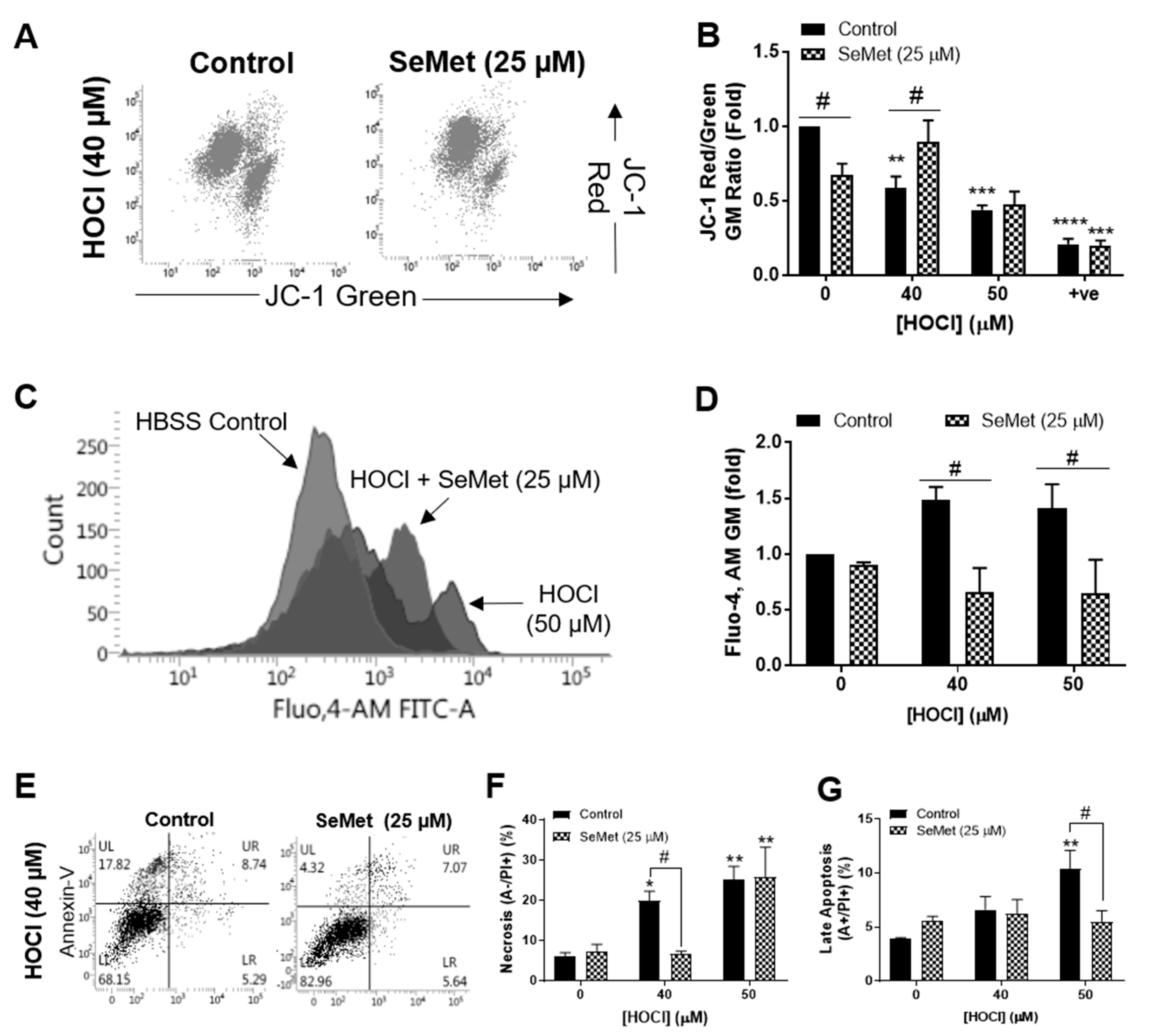
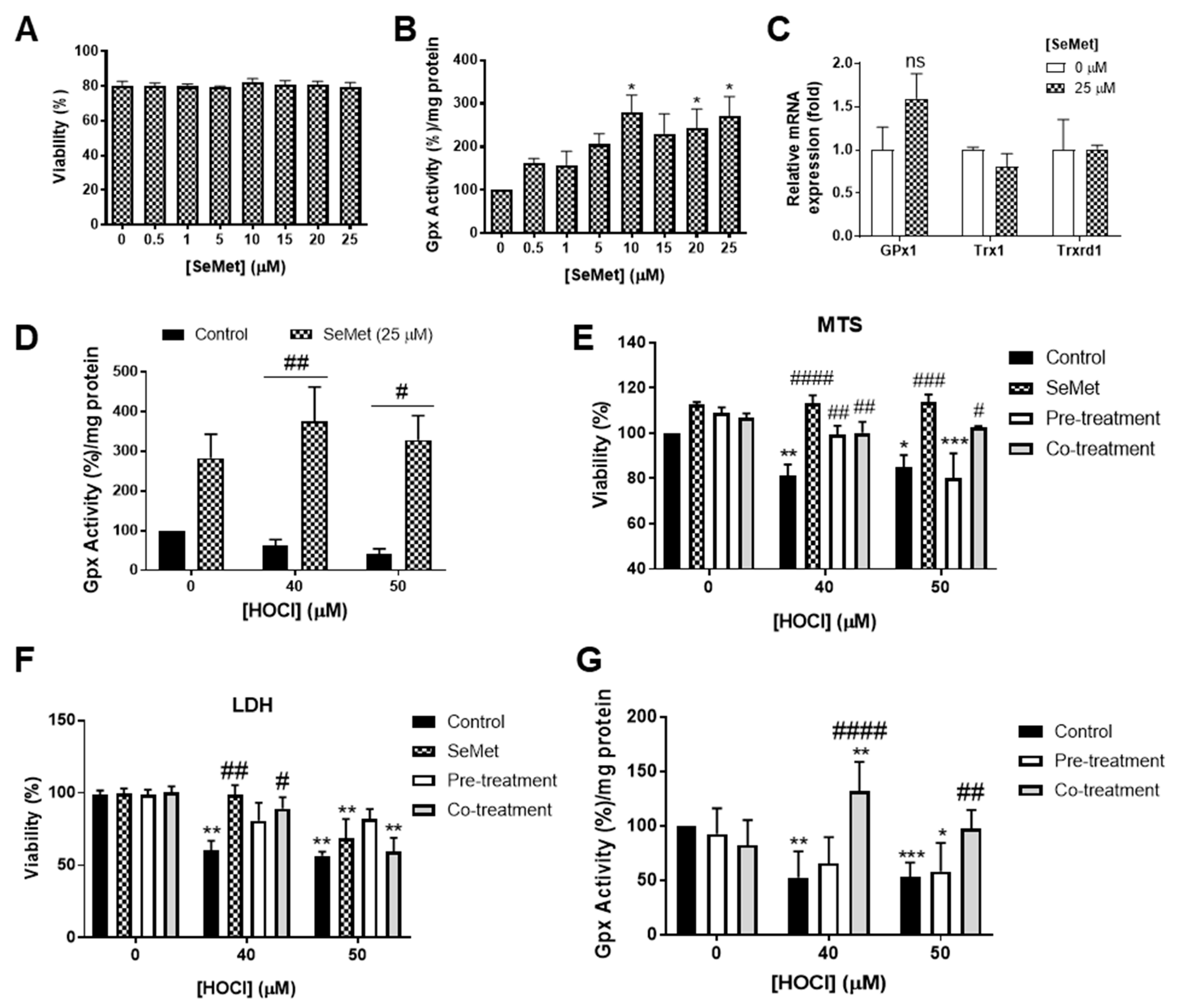
3.1. SeMet Protects Against Cellular Damage Elicited by HOCl in H9c2 Cells
3.2. Redox Status of H9c2 Cells Exposed to HOCl is Altered with SeMet Leading to Protection
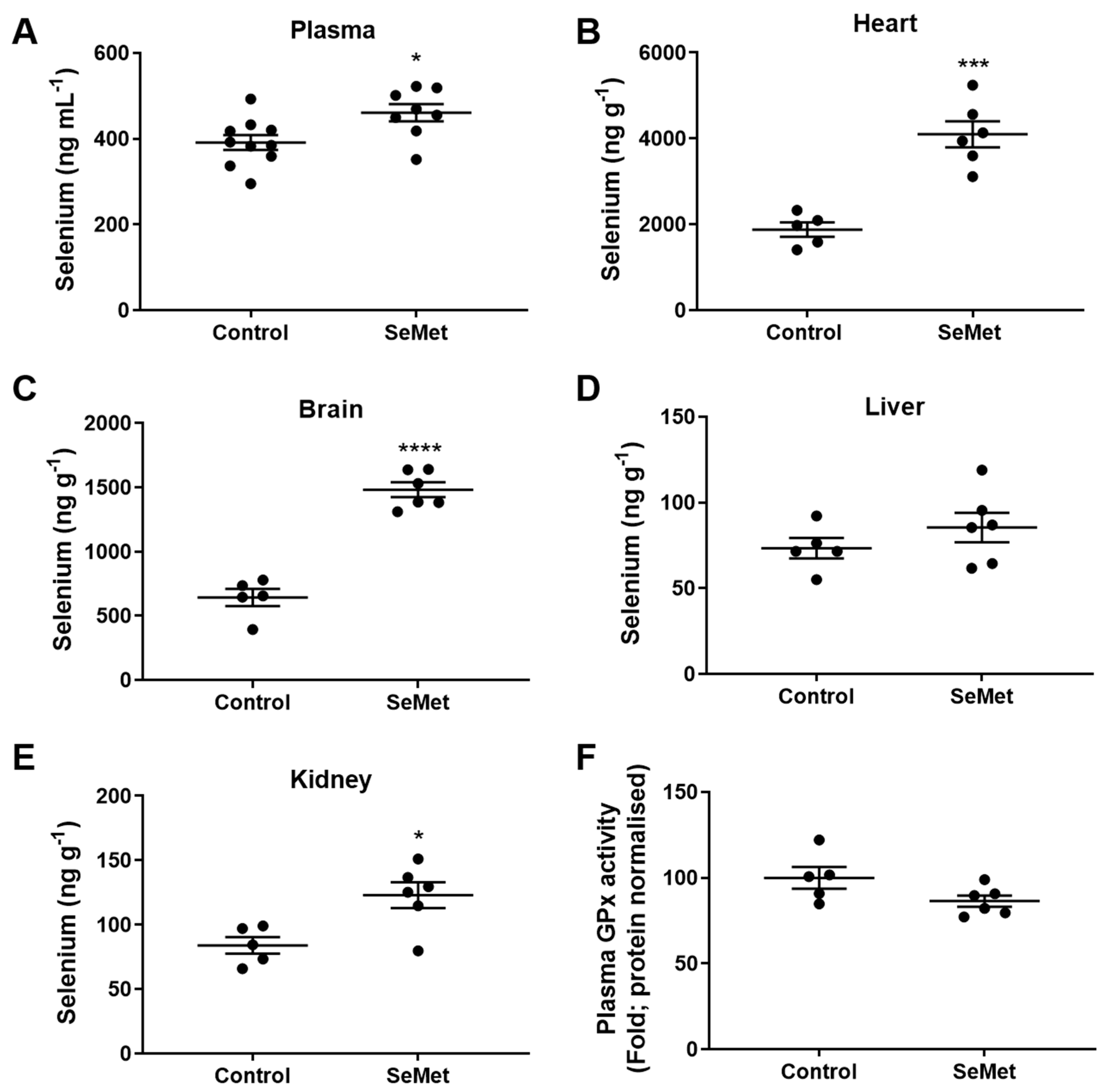
3.3. Profile of Selenium Content in Tissues of SeMet-Supplemented Rats
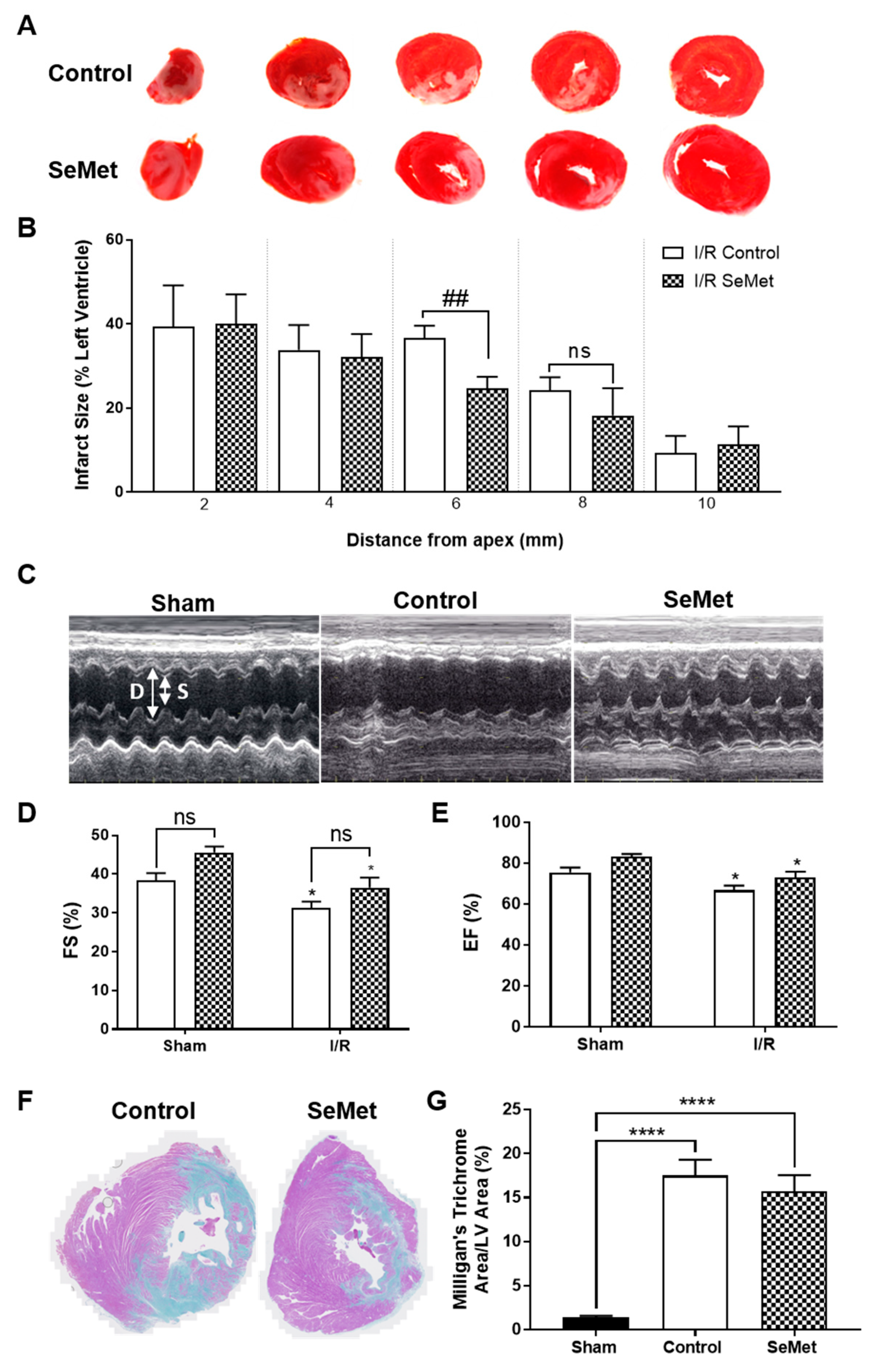
3.4. Effect of SeMet Supplementation Following I/R Injury
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Frangogiannis, N.G.; Dewald, O.; Xia, Y.; Ren, G.; Haudek, S.; Leucker, T.; Kraemer, D.; Taffet, G.; Rollins, B.J.; Entman, M.L. Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation 2007, 115, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Hess, M.L.; Rowe, G.T.; Caplan, M.; Romson, J.L.; Lucchesi, B. Identification of hydrogen peroxide and hydroxyl radicals as mediators of leukocyte-induced myocardial dysfunction. Limitation of infarct size with neutrophil inhibition and depletion. Adv. Myocardiol. 1985, 5, 159–175. [Google Scholar] [PubMed]
- Liao, Y.H.; Xia, N.; Zhou, S.F.; Tang, T.T.; Yan, X.X.; Lv, B.J.; Nie, S.F.; Wang, J.; Iwakura, Y.; Xiao, H.; et al. Interleukin-17A contributes to myocardial ischemia/reperfusion injury by regulating cardiomyocyte apoptosis and neutrophil infiltration. J. Am. Coll. Cardiol. 2012, 59, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, N.; Wada, H.; Kanda, T.; Niwa, T.; Yamada, Y.; Saito, K.; Fujiwara, H.; Sekikawa, K.; Seishima, M. Improved myocardial ischemia/reperfusion injury in mice lacking tumor necrosis factor-alpha. J. Am. Coll. Cardiol. 2002, 39, 1229–1235. [Google Scholar] [CrossRef]
- Duilio, C.; Ambrosio, G.; Kuppusamy, P.; DiPaula, A.; Becker, L.C.; Zweier, J.L. Neutrophils are primary source of O2 radicals during reperfusion after prolonged myocardial ischemia. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2649–H2657. [Google Scholar] [CrossRef]
- Bae, S.; Park, M.; Kang, C.; Dilmen, S.; Kang, T.H.; Kang, D.G.; Ke, Q.; Lee, S.U.; Lee, D.; Kang, P.M. Hydrogen Peroxide-Responsive Nanoparticle Reduces Myocardial Ischemia/Reperfusion Injury. J. Am. Heart Assoc. 2016, 5, e003697. [Google Scholar] [CrossRef]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria: Central role of complex III. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef]
- Kaminishi, T.; Kako, K.J. Sensitivity to oxidants of mitochondrial and sarcoplasmic reticular calcium uptake in saponin-treated cardiac myocytes. Basic Res. Cardiol. 1989, 84, 282–290. [Google Scholar] [CrossRef]
- Kaminishi, T.; Matsuoka, T.; Yanagishita, T.; Kako, K.J. Increase vs. decrease of calcium uptake by isolated heart cells induced by H2O2 vs. HOCl. Am. J. Physiol. 1989, 256, C598–C607. [Google Scholar] [CrossRef]
- Ali, M.A.; Eid, R.; Hanafi, M.Y. Vitamin C and E chronic supplementation differentially affect hepatic insulin signaling in rats. Life Sci. 2018, 194, 196–204. [Google Scholar] [CrossRef]
- Cheng, Y.C.; Sheen, J.M.; Hu, W.L.; Hung, Y.C. Polyphenols and Oxidative Stress in Atherosclerosis-Related Ischemic Heart Disease and Stroke. Oxidative Med. Cell. Longev. 2017, 2017, 8526438. [Google Scholar] [CrossRef] [PubMed]
- Dubick, M.A.; Omaye, S.T. Evidence for Grape, Wine and Tea Polyphenols as Modulators of Atherosclerosis and Ischemic Heart Disease in Humans. J. Nutraceuticals Funct. Med. Foods 2001, 3, 67–93. [Google Scholar] [CrossRef]
- Kojo, S. Vitamin C: Basic metabolism and its function as an index of oxidative stress. Curr. Med. Chem. 2004, 11, 1041–1064. [Google Scholar] [CrossRef] [PubMed]
- Myung, S.K.; Ju, W.; Cho, B.; Oh, S.W.; Park, S.M.; Koo, B.K.; Park, B.J.; Korean Meta-Analysis Study Group. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: Systematic review and meta-analysis of randomised controlled trials. BMJ 2013, 346, f10. [Google Scholar] [CrossRef]
- Weekley, C.M.; Harris, H.H. Which form is that? The importance of selenium speciation and metabolism in the prevention and treatment of disease. Chem. Soc. Rev. 2013, 42, 8870–8894. [Google Scholar] [CrossRef]
- Nagy, P.; Jameson, G.N.; Winterbourn, C.C. Kinetics and mechanisms of the reaction of hypothiocyanous acid with 5-thio-2-nitrobenzoic acid and reduced glutathione. Chem. Res. Toxicol. 2009, 22, 1833–1840. [Google Scholar] [CrossRef]
- Padmaja, S.; Squadrito, G.L.; Lemercier, J.N.; Cueto, R.; Pryor, W.A. Rapid oxidation of DL-selenomethionine by peroxynitrite. Free Radic. Biol. Med. 1996, 21, 317–322. [Google Scholar] [CrossRef]
- Skaff, O.; Pattison, D.I.; Morgan, P.E.; Bachana, R.; Jain, V.K.; Priyadarsini, K.I.; Davies, M.J. Selenium-containing amino acids are targets for myeloperoxidase-derived hypothiocyanous acid: Determination of absolute rate constants and implications for biological damage. Biochem. J. 2012, 441, 305–316. [Google Scholar] [CrossRef]
- Skaff, O.; Pattison, D.I.; Davies, M.J. Hypothiocyanous acid reactivity with low-molecular-mass and protein thiols: Absolute rate constants and assessment of biological relevance. Biochem. J. 2009, 422, 111–117. [Google Scholar] [CrossRef]
- Bordoni, A.; Biagi, P.L.; Angeloni, C.; Leoncini, E.; Muccinelli, I.; Hrelia, S. Selenium supplementation can protect cultured rat cardiomyocytes from hypoxia/reoxygenation damage. J. Agric. Food Chem. 2003, 51, 1736–1740. [Google Scholar] [CrossRef]
- Demirci, S.; Kutluhan, S.; Naziroglu, M.; Uguz, A.C.; Yurekli, V.A.; Demirci, K. Effects of selenium and topiramate on cytosolic Ca2+ influx and oxidative stress in neuronal PC12 cells. Neurochem. Res. 2013, 38, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Hazane-Puch, F.; Champelovier, P.; Arnaud, J.; Garrel, C.; Ballester, B.; Faure, P.; Laporte, F. Long-term selenium supplementation in HaCaT cells: Importance of chemical form for antagonist (protective versus toxic) activities. Biol. Trace Elem. Res. 2013, 154, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Hazane-Puch, F.; Champelovier, P.; Arnaud, J.; Trocme, C.; Garrel, C.; Faure, P.; Laporte, F. Six-day selenium supplementation led to either UVA-photoprotection or toxic effects in human fibroblasts depending on the chemical form and dose of Se. Met. Integr. Biometal Sci. 2014, 6, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Jornot, L.; Junod, A.F. Differential regulation of glutathione peroxidase by selenomethionine and hyperoxia in endothelial cells. Biochem. J. 1995, 306, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.; Vanderlelie, J.J.; Perkins, A.V. Selenium supplementation protects trophoblast cells from mitochondrial oxidative stress. Placenta 2013, 34, 594–598. [Google Scholar] [CrossRef]
- Watson, M.; van Leer, L.; Vanderlelie, J.J.; Perkins, A.V. Selenium supplementation protects trophoblast cells from oxidative stress. Placenta 2012, 33, 1012–1019. [Google Scholar] [CrossRef]
- Carroll, L.; Pattison, D.I.; Fu, S.; Schiesser, C.H.; Davies, M.J.; Hawkins, C.L. Catalytic oxidant scavenging by selenium-containing compounds: Reduction of selenoxides and N-chloramines by thiols and redox enzymes. Redox Biol. 2017, 12, 872–882. [Google Scholar] [CrossRef]
- Suryo Rahmanto, A.; Davies, M.J. Catalytic activity of selenomethionine in removing amino acid, peptide, and protein hydroperoxides. Free Radic. Biol. Med. 2011, 51, 2288–2299. [Google Scholar] [CrossRef]
- Clausen, J.; Nielsen, S.A. Comparison of whole blood selenium values and erythrocyte glutathione peroxidase activities of normal individuals on supplementation with selenate, selenite, l-selenomethionine, and high selenium yeast. Biol. Trace Elem. Res. 1988, 15, 125–138. [Google Scholar] [CrossRef]
- Tanguy, S.; Morel, S.; Berthonneche, C.; Toufektsian, M.C.; de Lorgeril, M.; Ducros, V.; Tosaki, A.; de Leiris, J.; Boucher, F. Preischemic selenium status as a major determinant of myocardial infarct size in vivo in rats. Antioxid. Redox Signal. 2004, 6, 792–796. [Google Scholar] [CrossRef]
- Wang, S.Q.; Niu, X.L.; Liu, Z.W.; Zhu, Y.H.; Gao, D.F. Selenium deficiency is associated with endoplasmic reticulum stress in a rat model of cardiac malfunction. Biol. Trace Elem. Res. 2013, 156, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Liu, Z.; Yang, G.; Gao, D.; Niu, X. MicroRNA expression profiles in rats with selenium deficiency and the possible role of the Wnt/beta-catenin signaling pathway in cardiac dysfunction. Int. J. Mol. Med. 2015, 35, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Bansal, M.P. Studies on scavenger receptors under experimental hypercholesterolemia: Modulation on selenium supplementation. Biol. Trace Elem. Res. 2011, 143, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.D.; Bansal, M.P. Studies on HDL associated enzymes under experimental hypercholesterolemia: Possible modulation on selenium supplementation. Lipids Health Dis. 2009, 8, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stawiarska-Pieta, B.; Birkner, E.; Szaflarska-Stojko, E.; Stojko, R.; Grucka-Mamczar, E.; Pyrsak, M.; Wyszynska, M. The influence of diet supplementation with methionine on the pathomorphological changes of rabbit organs in experimental atherosclerosis. Arch. Med. Sci. 2008, 4, 371–379. [Google Scholar]
- Vinson, J.A.; Stella, J.M.; Flanagan, T.J. Selenium yeast is an effective in vitro and in vivo antioxidant and hypolipemic agent in normal hamsters. Nutr. Res. 1998, 18, 735–742. [Google Scholar] [CrossRef]
- Treska, V.; Kuntscher, V.; Hasman, D.; Neprasova, P.; Kobr, J.; Racek, J.; Trefil, L.; Hes, O. Importance of selenium for the influence of ischemia-reperfusion syndrome after kidney transplantation from a non-heart beating donor in a pig model. Transplant. Proc. 2002, 34, 3057–3059. [Google Scholar] [CrossRef]
- Zapletal, C.; Heyne, S.; Golling, M.; Kraus, T.; Gebhard, M.M.; Herfarth, C.; Klar, E. Influence of selenium therapy on liver microcirculation after warm ischemia/reperfusion: An intravital microscopy study. Transplant. Proc. 2001, 33, 974–975. [Google Scholar] [CrossRef]
- Flores-Mateo, G.; Navas-Acien, A.; Pastor-Barriuso, R.; Guallar, E. Selenium and coronary heart disease: A meta-analysis. Am. J. Clin. Nutr. 2006, 84, 762–773. [Google Scholar] [CrossRef] [Green Version]
- Lubos, E.; Sinning, C.R.; Schnabel, R.B.; Wild, P.S.; Zeller, T.; Rupprecht, H.J.; Bickel, C.; Lackner, K.J.; Peetz, D.; Loscalzo, J.; et al. Serum selenium and prognosis in cardiovascular disease: Results from the AtheroGene study. Atherosclerosis 2010, 209, 271–277. [Google Scholar] [CrossRef] [Green Version]
- Navarro-Alarcon, M.; Lopez-Garcia de la Serrana, H.; Perez-Valero, V.; Lopez-Martinez, C. Serum and urine selenium concentrations in patients with cardiovascular diseases and relationship to other nutritional indexes. Ann. Nutr. Metab. 1999, 43, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Kalishwaralal, K.; Jeyabharathi, S.; Sundar, K.; Muthukumaran, A. Sodium selenite/selenium nanoparticles (SeNPs) protect cardiomyoblasts and zebrafish embryos against ethanol induced oxidative stress. J. Trace Elem. Med. Biol. Organ Soc. Miner. Trace Elem. 2015, 32, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.W.; Zhu, H.T.; Chen, K.L.; Qiu, C.; Tang, K.F.; Niu, X.L. Selenium attenuates high glucose-induced ROS/TLR-4 involved apoptosis of rat cardiomyocyte. Biol. Trace Elem. Res. 2013, 156, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Okatan, E.N.; Tuncay, E.; Turan, B. Cardioprotective effect of selenium via modulation of cardiac ryanodine receptor calcium release channels in diabetic rat cardiomyocytes through thioredoxin system. J. Nutr. Biochem. 2013, 24, 2110–2118. [Google Scholar] [CrossRef] [PubMed]
- Soncul, H.; Tatlican, O.; Halit, V.; Oz, E.; Sinci, V.; Salman, E.; Gokgoz, L.; Turkozkan, N.; Ersoz, A. The effect of selenium added cardioplegia in guinea pigs. Gen. Pharmacol. 1994, 25, 1493–1497. [Google Scholar] [CrossRef]
- Turan, B.; Saini, H.K.; Zhang, M.; Prajapati, D.; Elimban, V.; Dhalla, N.S. Selenium improves cardiac function by attenuating the activation of NF-kappaB due to ischemia-reperfusion injury. Antioxid. Redox Signal. 2005, 7, 1388–1397. [Google Scholar] [CrossRef]
- Venardos, K.; Harrison, G.; Headrick, J.; Perkins, A. Effects of dietary selenium on glutathione peroxidase and thioredoxin reductase activity and recovery from cardiac ischemia-reperfusion. J. Trace Elem. Med. Biol. Organ Soc. Miner. Trace Elem. 2004, 18, 81–88. [Google Scholar] [CrossRef]
- Beilstein, M.A.; Whanger, P.D. Metabolism of selenomethionine and effects of interacting compounds by mammalian cells in culture. J. Inorg. Biochem. 1987, 29, 137–152. [Google Scholar] [CrossRef]
- Wastney, M.E.; Combs, G.F., Jr.; Canfield, W.K.; Taylor, P.R.; Patterson, K.Y.; Hill, A.D.; Moler, J.E.; Patterson, B.H. A human model of selenium that integrates metabolism from selenite and selenomethionine. J. Nutr. 2011, 141, 708–717. [Google Scholar] [CrossRef] [Green Version]
- Assmann, A.; Briviba, K.; Sies, H. Reduction of methionine selenoxide to selenomethionine by glutathione. Arch. Biochem. Biophys. 1998, 349, 201–203. [Google Scholar] [CrossRef]
- Krause, R.J.; Elfarra, A.A. Reduction of l-methionine selenoxide to seleno-l-methionine by endogenous thiols, ascorbic acid, or methimazole. Biochem. Pharmacol. 2009, 77, 134–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, C.L.; Davies, M.J. Hypochlorite-induced damage to proteins: Formation of nitrogen-centred radicals from lysine residues and their role in protein fragmentation. Biochem. J. 1998, 332, 617–625. [Google Scholar] [CrossRef]
- Hawkins, C.L.; Davies, M.J. Hypochlorite-induced oxidation of proteins in plasma: Formation of chloramines and nitrogen-centred radicals and their role in protein fragmentation. Biochem. J. 1999, 340, 539–548. [Google Scholar] [CrossRef]
- Dunn, B.K.; Richmond, E.S.; Minasian, L.M.; Ryan, A.M.; Ford, L.G. A nutrient approach to prostate cancer prevention: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). Nutr. Cancer 2010, 62, 896–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flohe, L.; Gunzler, W.A. Assays of glutathione peroxidase. Methods Enzymol. 1984, 105, 114–121. [Google Scholar] [PubMed]
- Hawkins, C.L.; Morgan, P.E.; Davies, M.J. Quantification of protein modification by oxidants. Free Radic. Biol. Med. 2009, 46, 965–988. [Google Scholar] [CrossRef]
- Kariya, C.; Chu, H.W.; Huang, J.; Leitner, H.; Martin, R.J.; Day, B.J. Mycoplasma pneumoniae infection and environmental tobacco smoke inhibit lung glutathione adaptive responses and increase oxidative stress. Infect. Immun. 2008, 76, 4455–4462. [Google Scholar] [CrossRef] [Green Version]
- Reyes, L.; Hawkins, C.L.; Rayner, B.S. Characterization of the cellular effects of myeloperoxidase-derived oxidants on H9c2 cardiac myoblasts. Arch. Biochem. Biophys. 2019, 665, 132–142. [Google Scholar] [CrossRef]
- Allan, C.B.; Lacourciere, G.M.; Stadtman, T.C. Responsiveness of selenoproteins to dietary selenium. Annu. Rev. Nutr. 1999, 19, 1–16. [Google Scholar] [CrossRef]
- Cohn, J.N.; Ferrari, R.; Sharpe, N. Cardiac remodeling—Concepts and clinical implications: A consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J. Am. Coll. Cardiol. 2000, 35, 569–582. [Google Scholar] [CrossRef] [Green Version]
- Gyongyosi, M.; Winkler, J.; Ramos, I.; Do, Q.T.; Firat, H.; McDonald, K.; Gonzalez, A.; Thum, T.; Diez, J.; Jaisser, F.; et al. Myocardial fibrosis: Biomedical research from bench to bedside. Eur. J. Heart Fail. 2017, 19, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.; Dong, L.F.; Holland, O.; Vanderlelie, J.; Pasdar, E.A.; Neuzil, J.; Perkins, A.V. Selenium supplementation induces mitochondrial biogenesis in trophoblasts. Placenta 2015, 36, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Valiente-Alandi, I.; Potter, S.J.; Salvador, A.M.; Schafer, A.E.; Schips, T.; Carrillo-Salinas, F.; Gibson, A.M.; Nieman, M.L.; Perkins, C.; Sargent, M.A.; et al. Inhibiting Fibronectin Attenuates Fibrosis and Improves Cardiac Function in a Model of Heart Failure. Circulation 2018, 138, 1236–1252. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Genebank Ref. | Forward Primer | Reverse Primer |
|---|---|---|---|
| Reference | |||
| Nono | NM_001012356 | CCTGATGCGAGAGAACAAGAGA | CTGGACGGTTGAATGCAGGA |
| β-actin | NM_012512.2 | ATCAAGATCATTGCTCCTCCTG | CAGCTCAGTAACAGTCCGCC |
| Seleno-dependent antioxidants | |||
| GPx1 | NM_030826.3 | CAGTCCACCGTGTATGCCTT | TGCCATTCTCCTGATGTCCG |
| Trx1 | NM_053800.3 | GCCCTTCTTTCATTCCCTCTGT | CTCCCCAACCTTTTGACCCTT |
| Trxrd1 | NM_031614.2 | CGTGCCGACGAAAATTGAAC | CATTGATCTTCACGCCCACG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reyes, L.; Bishop, D.P.; Hawkins, C.L.; Rayner, B.S. Assessing the Efficacy of Dietary Selenomethionine Supplementation in the Setting of Cardiac Ischemia/Reperfusion Injury. Antioxidants 2019, 8, 546. https://doi.org/10.3390/antiox8110546
Reyes L, Bishop DP, Hawkins CL, Rayner BS. Assessing the Efficacy of Dietary Selenomethionine Supplementation in the Setting of Cardiac Ischemia/Reperfusion Injury. Antioxidants. 2019; 8(11):546. https://doi.org/10.3390/antiox8110546
Chicago/Turabian StyleReyes, Leila, David P. Bishop, Clare L. Hawkins, and Benjamin S. Rayner. 2019. "Assessing the Efficacy of Dietary Selenomethionine Supplementation in the Setting of Cardiac Ischemia/Reperfusion Injury" Antioxidants 8, no. 11: 546. https://doi.org/10.3390/antiox8110546
APA StyleReyes, L., Bishop, D. P., Hawkins, C. L., & Rayner, B. S. (2019). Assessing the Efficacy of Dietary Selenomethionine Supplementation in the Setting of Cardiac Ischemia/Reperfusion Injury. Antioxidants, 8(11), 546. https://doi.org/10.3390/antiox8110546