Oxidative Imbalance and Kidney Damage: New Study Perspectives from Animal Models to Hospitalized Patients




Abstract
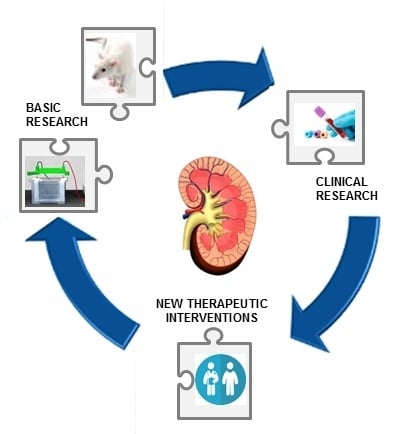
:
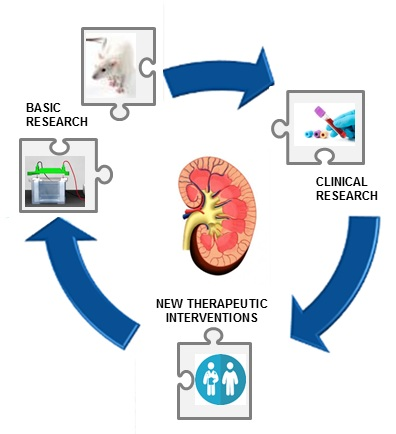{kind=link}
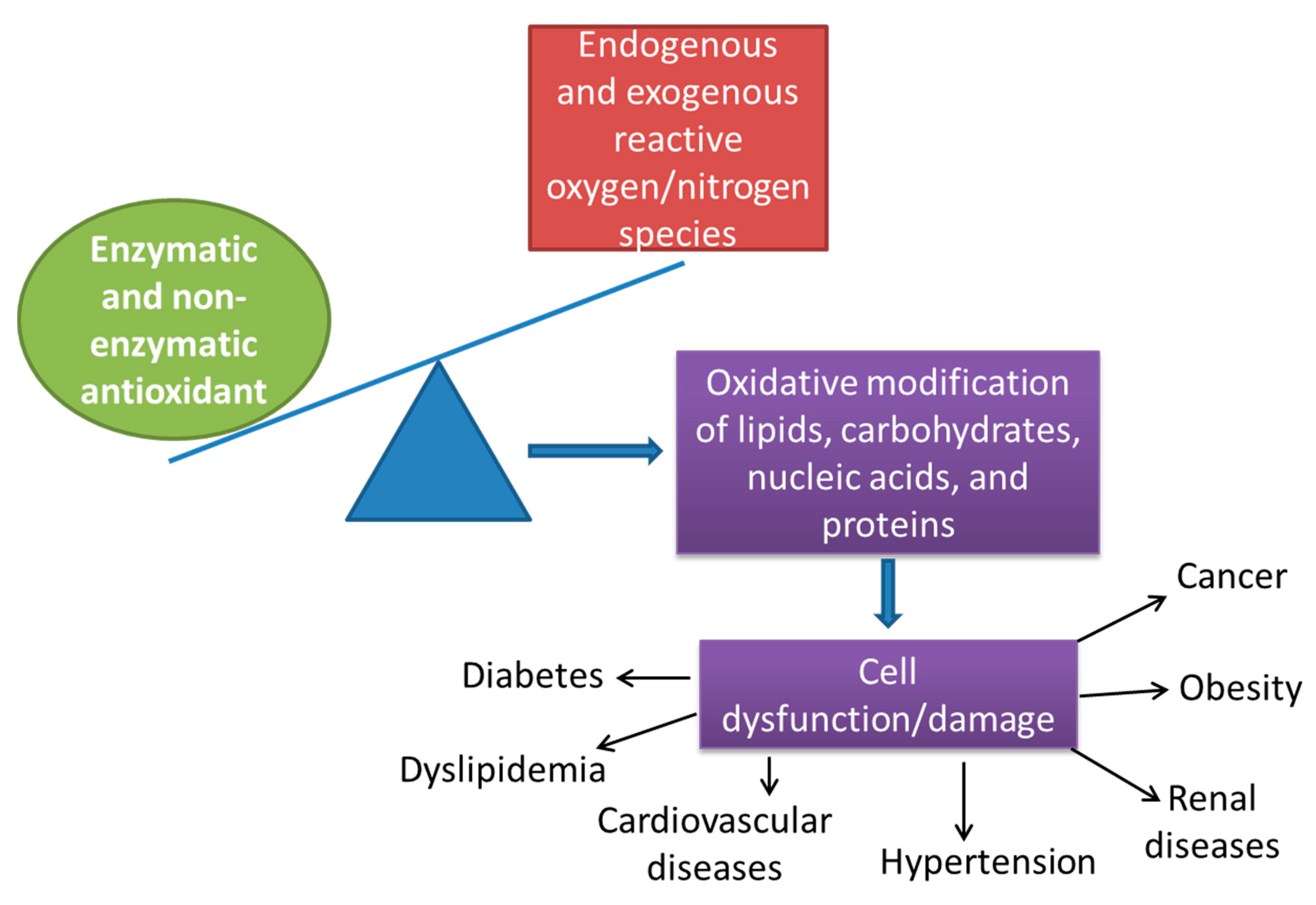{kind=link}
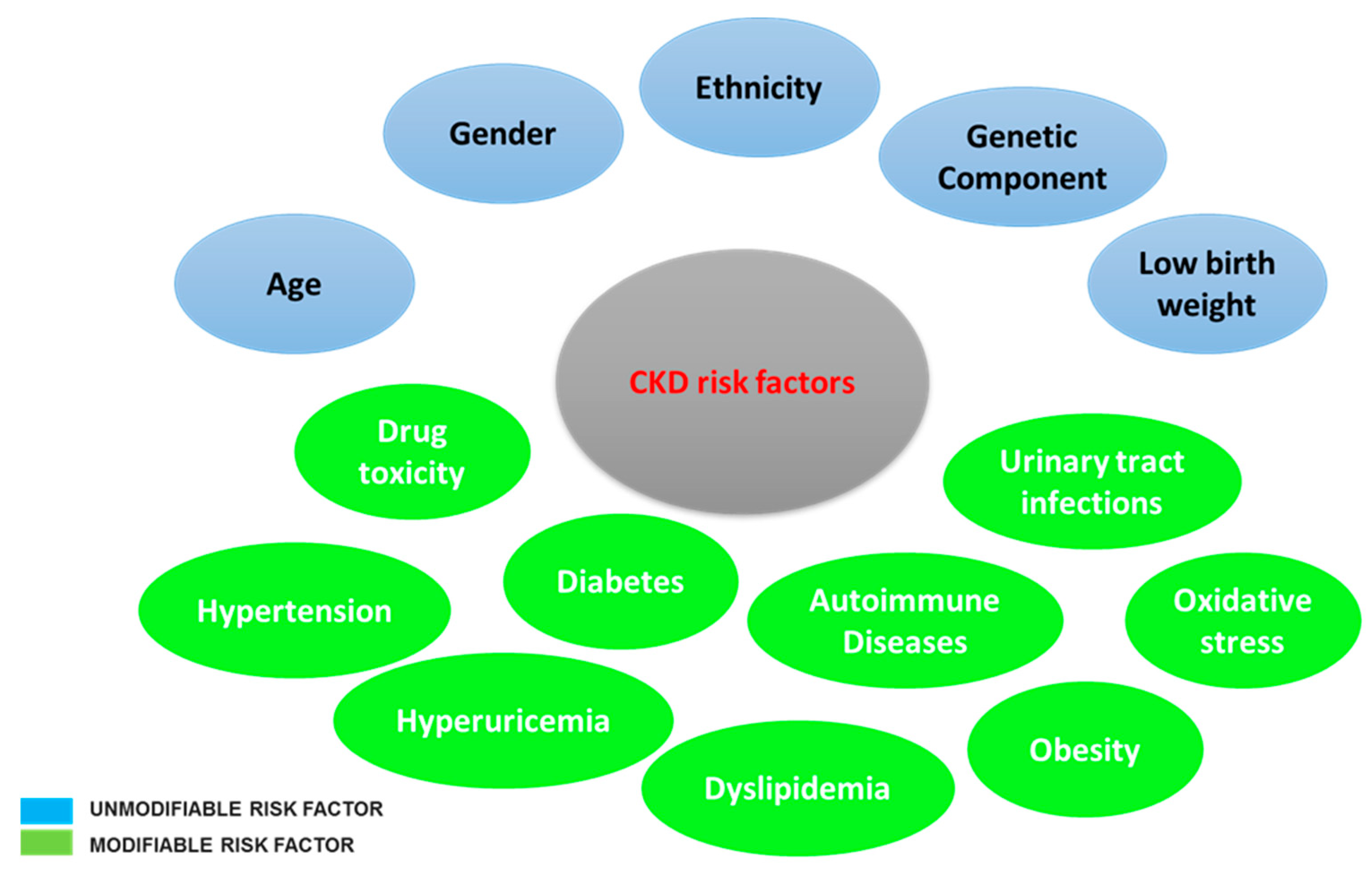{kind=link}
{kind=link}
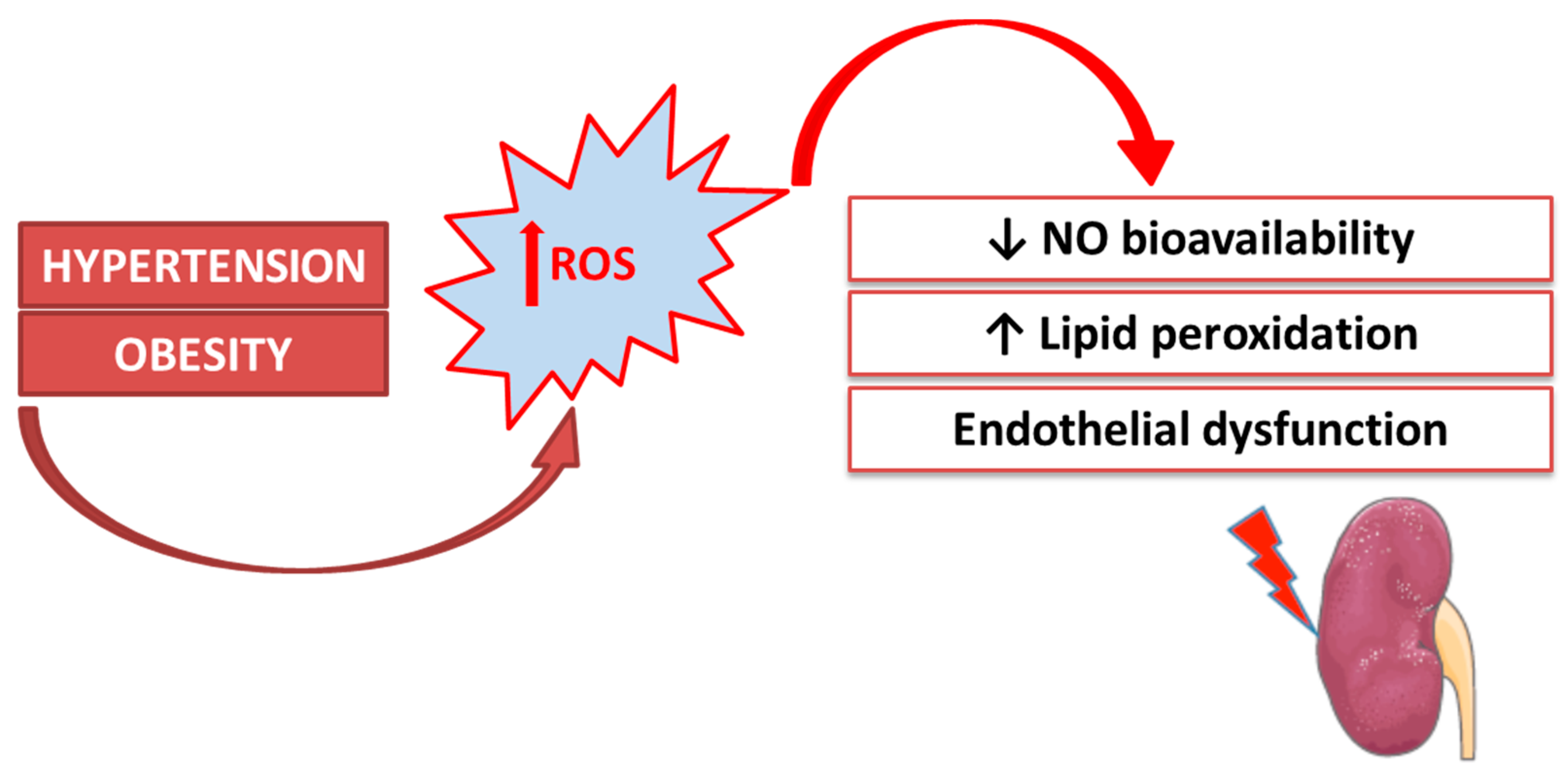
1. Introduction
2. Oxidative Stress and Antioxidant Defense Mechanisms
- (1)
- Lipid peroxidation and loss of membrane fluidity: double bonds in polyunsaturated membrane lipids are vulnerable to attacks by oxygen-free radicals;
- (2)
- Protein cross-linking: free radicals promote sulfhydryl-mediated protein crosslinking, resulting in increased degradation or loss of activity;
- (3)
- DNA fragmentation;
- (4)
- Oxidative damage to carbohydrates impairs the functions of some cellular receptors, including those associated with hormonal and neurotransmitter responses.
3. Pathophysiology of Chronic Kidney Disease
4. Oxidative Stress and Chronic Kidney Disease
5. Conclusions
Funding
Conflicts of Interest
References
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef] [PubMed]
- Che, R.; Yuan, Y.; Huang, S.; Zhang, A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am. J. Physiol. Ren. Physiol. 2014, 306, F367–F378. [Google Scholar] [CrossRef] [PubMed]
- Scholze, A.; Jankowski, J.; Pedraza-Chaverri, J.; Evenepoel, P. Editorial Oxidative Stress in Chronic Kidney Disease. Oxid. Med. Cell. Longev. 2016, 2016, 8375186. [Google Scholar] [CrossRef] [PubMed]
- Salisbury, D.; Bronas, U. Reactive oxygen and nitrogen species: Impact on endothelial dysfunction. Nurs. Res. 2015, 64, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, A.; Idelchik, M.d.P.S.; Melendez, J.A. Redox control of senescence and age-related disease. Redox Biol. 2017, 11, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010, 4, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Ji, L.L.; Kavazis, A.N.; Jackson, M.J. Reactive oxygen species: Impact on skeletal muscle. Compr. Physiol. 2011, 1, 941–969. [Google Scholar]
- Genestra, M. Oxyl radicals, redox-sensitive signalling cascades and antioxidants. Cell. Signal. 2007, 19, 1807–1819. [Google Scholar] [CrossRef]
- Venkataraman, K.; Khurana, S.; Tai, T.C. Oxidative stress in aging-matters of the heart and mind. Int. J. Mol. Sci. 2013, 14, 17897–17925. [Google Scholar] [CrossRef]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free Radicals: Properties, Sources, Targets, and Their Implication in Various Diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef]
- Kulcharyk, P.A.; Heinecke, J.W. Hypochlorous acid produced by the myeloperoxidase system of human phagocytes induces covalent cross-links between DNA and protein. Biochemistry 2001, 40, 3648–3656. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.; Franco, M.C.; Estevez, A.G. Reactive nitrogen species in cellular signaling. Exp. Biol. Med. 2015, 240, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, R.B.; Wardman, P. Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene 2003, 22, 5734–5754. [Google Scholar] [CrossRef] [PubMed]
- Ridnour, L.A.; Thomas, D.D.; Mancardi, D.; Espey, M.G.; Miranda, K.M.; Paolocci, N.; Feelisch, M.; Fukuto, J.; Wink, D.A. The chemistry of nitrosative stress induced by nitric oxide and reactive nitrogen oxide species. Putting perspective on stressful biological situations. Biol. Chem. 2004, 385, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zampelas, A.; Micha, R. Antioxidants in health and disease. Antioxid. Heal. Dis. 2015, 1–302. [Google Scholar]
- Frijhoff, J.; Winyard, P.G.; Zarkovic, N.; Davies, S.S.; Stocker, R.; Cheng, D.; Knight, A.R.; Taylor, E.L.; Oettrich, J.; Ruskovska, T.; et al. Clinical Relevance of Biomarkers of Oxidative Stress. Antioxid. Redox Signal. 2015, 23, 1144–1170. [Google Scholar] [CrossRef] [PubMed]
- Juránek, I.; Bezek, Š. Controversy of free radical hypothesis: Reactive oxygen species-Cause or consequence of tissue injury. Gen. Physiol. Biophys. 2005, 24, 263–278. [Google Scholar] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef]
- Wu, J.Q.; Kosten, T.R.; Zhang, X.Y. Free radicals, antioxidant defense systems, and schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 46, 200–206. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Balasubramanian, S. Progression of chronic kidney disease: Mechanisms and interventions in retardation. Apollo Med. 2013, 10, 19–28. [Google Scholar] [CrossRef]
- Coresh, J.; Selvin, E.; Stevens, L.A.; Manzi, J.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Levey, A.S. Prevalence of chronic kidney disease in the United States. J. Am. Med. Assoc. 2007, 298, 2038–2047. [Google Scholar] [CrossRef] [PubMed]
- Kokubo, Y.; Nakamura, S.; Okamura, T.; Yoshimasa, Y.; Makino, H.; Watanabe, M.; Higashiyama, A.; Kamide, K.; Kawanishi, K.; Okayama, A.; et al. Relationship between blood pressure category and incidence of stroke and myocardial infarction in an Urban Japanese population with and without chronic kidney disease: The suita study. Stroke 2009, 40, 2674–2679. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.R. Global Prevalence of Chronic Kidney Disease–A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef]
- Nitta, K.; Okada, K.; Yanai, M.; Takahashi, S. Aging and chronic kidney disease. Kidney Blood Press. Res. 2014, 38, 109–120. [Google Scholar] [CrossRef]
- Jabarpour, M.; Rashtchizadeh, N.; Argani, H.; Ghorbanihaghjo, A.; Ranjbarzadhag, M.; Sanajou, D.; Panah, F.; Alirezaei, A. The impact of dyslipidemia and oxidative stress on vasoactive mediators in patients with renal dysfunction. Int. Urol. Nephrol. 2019, 51, 2235–2242. [Google Scholar] [CrossRef]
- Tanner, R.M.; Brown, T.M.; Muntner, P. Epidemiology of obesity, the metabolic syndrome, and chronic kidney disease. Curr. Hypertens. Rep. 2012, 14, 152–159. [Google Scholar] [CrossRef]
- Ejaz, A.A.; Johnson, R.J.; Shimada, M.; Mohandas, R.; Alquadan, K.F.; Beaver, T.M.; Lapsia, V.; Dass, B. The Role of Uric Acid in Acute Kidney Injury. Nephron 2019, 142, 275–283. [Google Scholar] [CrossRef]
- Schnaper, H.W. The Tubulointerstitial Pathophysiology of Progressive Kidney Disease. Adv. Chronic Kidney Dis. 2017, 24, 107–116. [Google Scholar] [CrossRef]
- Agarwal, R.; Campbell, R.C.; Warnock, D.G. Oxidative Stress in Hypertension and Chronic Kidney Disease: Role of Angiotensin II. Semin. Nephrol. 2004, 24, 101–114. [Google Scholar] [CrossRef]
- Massy, Z.A.; Nguyen-Khoa, T. Oxidative stress and chronic renal failure: Markers and management. J. Nephrol. 2002, 15, 336–341. [Google Scholar]
- Small, D.M.; Coombes, J.S.; Bennett, N.; Johnson, D.W.; Gobe, G.C. Oxidative stress, anti-oxidant therapies and chronic kidney disease. Nephrology 2012, 17, 311–321. [Google Scholar] [CrossRef]
- Palm, F.; Nordquist, L. Renal oxidative stress, oxygenation, and hypertension. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2011, 301, R1229–R1241. [Google Scholar] [CrossRef]
- Aveles, P.R.; Criminácio, C.R.; Gonçalves, S.; Bignelli, A.T.; Claro, L.M.; Siqueira, S.S.; Nakao, L.S.; Pecoits-Filho, R. Association between biomarkers of carbonyl stress with increased systemic inflammatory response in different stages of chronic kidney disease and after renal transplantation. Nephron Clin. Pract. 2010, 116. [Google Scholar] [CrossRef]
- Poulianiti, K.P.; Kaltsatou, A.; Mitrou, G.I.; Jamurtas, A.Z.; Koutedakis, Y.; Maridaki, M.; Stefanidis, I.; Sakkas, G.K.; Karatzaferi, C. Systemic Redox Imbalance in Chronic Kidney Disease: A Systematic Review. Oxid. Med. Cell. Longev. 2016, 2016, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krata, N.; Zagożdżon, R.; Foroncewicz, B.; Mucha, K. Oxidative Stress in Kidney Diseases: The Cause or the Consequence? Arch. Immunol. Ther. Exp. 2018, 66, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, A.; Pires, M.J.; Oliveira, P.A. Pathophysiological mechanisms of renal fibrosis: A review of animal models and therapeutic strategies. In Vivo 2017, 31, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grynberg, K.; Ma, F.Y.; Nikolic-Paterson, D.J. The JNK signaling pathway in renal fibrosis. Front. Physiol. 2017, 8, 829. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Lin, Q.; Shao, X.; Mou, S.; Gu, L.; Wang, L.; Zhang, Z.; Shen, J.; Zhou, Y.; Qi, C.; et al. NLRP3 inflammasome inhibition attenuates cisplatin-induced renal fibrosis by decreasing oxidative stress and inflammation. Exp. Cell Res. 2019, 383, 111488. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, J.; Kolhe, N.V.; Fluck, R.J.; Elliott, D.; Selby, N.M.; Lawson, N.; McIntyre, C.W.; Packington, R. Defining the Cause of Death in Hospitalised Patients with Acute Kidney Injury. PLoS ONE 2012, 7, e48580. [Google Scholar]
- Di Lullo, L.; House, A.; Gorini, A.; Santoboni, A.; Russo, D.; Ronco, C. Chronic kidney disease and cardiovascular complications. Heart Fail. Rev. 2015, 20, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, M.; Wiebe, N.; Culleton, B.; House, A.; Rabbat, C.; Fok, M.; McAlister, F.; Garg, A.X. Chronic Kidney Disease and Mortality Risk: A Systematic Review. J. Am. Soc. Nephrol. 2006, 17, 2034–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C. Chronic Kidney Disease and the Risks of Death, Cardiovascular Events, and Hospitalization. New Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.X.; Clark, W.F.; Haynes, R.B.; House, A.A. Moderate renal insufficiency and the risk of cardiovascular mortality: Results from the NHANES I. Kidney Int. 2002, 61, 1486–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russa, D.L.; Pellegrino, D.; Montesanto, A.; Gigliotti, P.; Perri, A.; Russa, A.L.; Bonofiglio, R. Oxidative Balance and Inflammation in Hemodialysis Patients: Biomarkers of Cardiovascular Risk? Oxid. Med. Cell. Longev. 2019, 2019, 1–7. [Google Scholar] [CrossRef]
- Dounousi, E.; Papavasiliou, E.; Makedou, A.; Ioannou, K.; Katopodis, K.P.; Tselepis, A.; Siamopoulos, K.C.; Tsakiris, D. Oxidative Stress Is Progressively Enhanced With Advancing Stages of CKD. Am. J. Kidney Dis. 2006, 48, 752–760. [Google Scholar] [CrossRef] [Green Version]
- Recio-Mayoral, A.; Banerjee, D.; Streather, C.; Kaski, J.C. Endothelial dysfunction, inflammation and atherosclerosis in chronic kidney disease—A cross-sectional study of predialysis, dialysis and kidney-transplantation patients. Atherosclerosis 2011, 216, 446–451. [Google Scholar] [CrossRef]
- Nistala, R.; Whaley-Connell, A.; Sowers, J.R. Redox control of renal function and hypertension. Antioxid. Red. Signal. 2008, 10, 2047–2089. [Google Scholar] [CrossRef] [Green Version]
- Liakopoulos, V.; Roumeliotis, S.; Gorny, X.; Eleftheriadis, T.; Mertens, P.R. Oxidative Stress in Patients Undergoing Peritoneal Dialysis: A Current Review of the Literature. Oxid. Med. Cell. Longev. 2017, 2017, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Dursun, E.; Ozben, T.; Süleymanlar, G.; Dursun, B.; Yakupoglu, G. Effect of hemodialysis on the oxidative stress and antioxidants. Clin. Chem. Lab. Med. 2002, 40, 1009–1013. [Google Scholar] [CrossRef]
- Elshamaa, M.F.; Sabry, S.; Nabih, M.; Elghoroury, E.A.; El-Saaid, G.S.; Ismail, A.A.G. Oxidative stress markers and C-reactive protein in pediatric patients on hemodialysis. Ann. Nutr. Metab. 2009, 55, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Kooman, J.P.; van der Sande, F.M.; Leunissen, K.M.L. Kidney disease and aging: A reciprocal relation. Exp. Gerontol. 2017, 87, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Khoa, T.; Massy, Z.A.; Pascal De Bandt, J.; Kebede, M.; Salama, L.; Lambrey, G.; Witko-Sarsat, V.; Drüeke, T.B.; Lacour, B.; Thévenin, M. Oxidative stress and haemodialysis: Role of inflammation and duration of dialysis treatment. Nephrol. Dial. Transplant. 2001, 16, 335–340. [Google Scholar] [CrossRef] [PubMed]
- La Russa, D.; Brunelli, E.; Pellegrino, D. Oxidative imbalance and kidney damage in spontaneously hypertensive rats: Activation of extrinsic apoptotic pathways. Clin. Sci. 2017, 131, 1419–1428. [Google Scholar] [CrossRef] [PubMed]
- La Russa, D.; Giordano, F.; Marrone, A.; Parafati, M.; Janda, E.; Pellegrino, D. Oxidative Imbalance and Kidney Damage in Cafeteria Diet-Induced Rat Model of Metabolic Syndrome: Effect of Bergamot Polyphenolic Fraction. Antioxidants 2019, 8, 66. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Fallahzadeh, M.K.; McCullough, P.A. Aging male spontaneously hypertensive rat as an animal model for the evaluation of the interplay between contrast-induced acute kidney injury and cardiorenal syndrome in humans. Cardiorenal Med. 2016, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Loperena, R.; Harrison, D.G. Oxidative Stress and Hypertensive Diseases. Med. Clin. 2017, 101, 169–193. [Google Scholar] [CrossRef] [Green Version]
- Ward, N.C.; Croft, K.D. Hypertension and oxidative stress. Clin. Exp. Pharmacol. Physiol. 2006, 33, 872–876. [Google Scholar] [CrossRef]
- Rubattu, S.; Pagliaro, B.; Pierelli, G.; Santolamazza, C.; Di Castro, S.; Mennuni, S.; Volpe, M. Pathogenesis of target organ damage in hypertension: Role of mitochondrial oxidative stress. Int. J. Mol. Sci. 2015, 16, 823–839. [Google Scholar] [CrossRef] [Green Version]
- Majzunova, M.; Dovinova, I.; Barancik, M.; Chan, J.Y.H. Redox signaling in pathophysiology of hypertension. J. Biomed. Sci. 2013, 20, 69. [Google Scholar] [CrossRef] [Green Version]
- Ndisang, J.F.; Vannacci, A.; Rastogi, S. Oxidative Stress and Inflammation in Obesity, Diabetes, Hypertension, and Related Cardiometabolic Complications. Oxid. Med. Cell. Longev. 2014, 2014, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, R.; Libuy, M.; Feliú, F.; Hasson, D. Oxidative stress-related biomarkers in essential hypertension and ischemia-reperfusion myocardial damage. Dis. Mark. 2013, 35, 773–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, G.M.; Danaei, G.; Farzadfar, F.; Stevens, G.A.; Woodward, M.; Wormser, D.; Kaptoge, S.; Whitlock, G.; Qiao, Q.; Lewington, S.; et al. The age-specific quantitative effects of metabolic risk factors on cardiovascular diseases and diabetes: A pooled analysis. PLoS ONE 2013, 8, e65174. [Google Scholar] [CrossRef] [PubMed]
- Collaboration, E.R.F.; Wormser, D.; Kaptoge, S.; Di Angelantonio, E.; Wood, A.M.; Pennells, L.; Thompson, A.; Sarwar, N.; Kizer, J.R.; Lawlor, D.A.; et al. Separate and combined associations of body-mass index and abdominal adiposity with cardiovascular disease: Collaborative analysis of 58 prospective studies. Lancet 2011, 377, 1085–1095. [Google Scholar] [CrossRef] [Green Version]
- Gelsomino, L.; Naimo, G.D.; Catalano, S.; Mauro, L.; Andò, S. The Emerging Role of Adiponectin in Female Malignancies. Int. J. Mol. Sci. 2019, 20, 2127. [Google Scholar] [CrossRef] [Green Version]
- Panno, M.L.; Naimo, G.D.; Spina, E.; Andò, S.; Mauro, L. Different molecular signaling sustaining adiponectin action in breast cancer. Curr. Opin. Pharmacol. 2016, 31, 1–7. [Google Scholar] [CrossRef]
- Mauro, L.; Naimo, G.D.; Ricchio, E.; Panno, M.L.; Andò, S. Cross-Talk between Adiponectin and IGF-IR in Breast Cancer. Front. Oncol. 2015, 5, 157. [Google Scholar] [CrossRef] [Green Version]
- Kovesdy, C.P.; Furth, S.; Zoccali, C. Obesity and kidney disease: Hidden consequences of the epidemic. Brunei Int. Med. J. 2017, 13, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Elsayed, E.F.; Sarnak, M.J.; Tighiouart, H.; Griffith, J.L.; Kurth, T.; Salem, D.N.; Levey, A.S.; Weiner, D.E. Waist-to-Hip Ratio, Body Mass Index, and Subsequent Kidney Disease and Death. Am. J. Kidney Dis. 2008, 52, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Tsujimoto, T.; Sairenchi, T.; Iso, H.; Irie, F.; Yamagishi, K.; Watanabe, H.; Tanaka, K.; Muto, T.; Ota, H. The Dose-Response Relationship Between Body Mass Index and the Risk of Incident Stage ≥ 3 Chronic Kidney Disease in a General Japanese Population: The Ibaraki Prefectural Health Study (IPHS). J. Epidemiol. 2014, 24, 444–451. [Google Scholar] [CrossRef] [Green Version]
- Babish, J.G.; Dahlberg, C.J.; Ou, J.J.; Keller, W.J.; Gao, W.; Kaadige, M.R.; Brabazon, H.; Lamb, J.; Soudah, H.C.; Kou, X.; et al. Synergistic in vitro antioxidant activity and observational clinical trial of F105, a phytochemical formulation including Citrus bergamia, in subjects with moderate cardiometabolic risk factors. Can. J. Physiol. Pharmacol. 2016, 186, 1–10. [Google Scholar]
- Postorino, M.; Marino, C.; Tripepi, G.; Zoccali, C. Abdominal Obesity and All-Cause and Cardiovascular Mortality in End-Stage Renal Disease. J. Am. Coll. Cardiol. 2009, 53, 1265–1272. [Google Scholar] [CrossRef] [Green Version]
- Kovesdy, C.P.; Czira, M.E.; Rudas, A.; Ujszaszi, A.; Rosivall, L.; Novak, M.; Kalantar-Zadeh, K.; Molnar, M.Z.; Mucsi, I. Body mass index, waist circumference and mortality in kidney transplant recipients. Am. J. Transplant. 2010, 10, 2644–2651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, M.C.; Hwang, S.J.; Massaro, J.M.; Hoffmann, U.; Deboer, I.H.; Robins, S.J.; Vasan, R.S.; Fox, C.S. Association of subcutaneous and visceral adiposity with albuminuria: The framingham heart study. Obesity 2011, 19, 1284–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity: Implications for metabolic syndrome, diabetes, hypertension, dyslipidemia, atherosclerosis, and cancer. Obes. Res. Clin. Pract. 2013, 7, e330–e341. [Google Scholar] [CrossRef] [PubMed]
- Ramos, L.F.; Shintani, A.; Ikizler, T.A.; Himmelfarb, J. Oxidative Stress and Inflammation Are Associated with Adiposity in Moderate to Severe CKD. J. Am. Soc. Nephrol. 2008, 19, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Parafati, M.; Lascala, A.; Morittu, V.M.; Trimboli, F.; Rizzuto, A.; Brunelli, E.; Coscarelli, F.; Costa, N.; Britti, D.; Ehrlich, J.; et al. Bergamot polyphenol fraction prevents nonalcoholic fatty liver disease via stimulation of lipophagy in cafeteria diet-induced rat model of metabolic syndrome. J. Nutr. Biochem. 2015, 26, 938–948. [Google Scholar] [CrossRef]
- Parafati, M.; Lascala, A.; La Russa, D.; Mignogna, C.; Trimboli, F.; Morittu, V.M.; Riillo, C.; Macirella, R.; Mollace, V.; Brunelli, E.; et al. Bergamot Polyphenols Boost Therapeutic Effects of the Diet on Non-Alcoholic Steatohepatitis (NASH) Induced by “Junk Food”: Evidence for Anti-Inflammatory Activity. Nutrients 2018, 10, 1604. [Google Scholar] [CrossRef] [Green Version]
- Zeeni, N.; Dagher-Hamalian, C.; Dimassi, H.; Faour, W.H. Cafeteria diet-fed mice is a pertinent model of obesity-induced organ damage: A potential role of inflammation. Inflamm. Res. 2015, 64, 501–512. [Google Scholar] [CrossRef]
- Keaney, J.F.; Larson, M.G.; Vasan, R.S.; Wilson, P.W.F.; Lipinska, I.; Corey, D.; Massaro, J.M.; Sutherland, P.; Vita, J.A.; Benjamin, E.J. Obesity and systemic oxidative stress: Clinical correlates of oxidative stress in the Framingham study. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 434–439. [Google Scholar] [CrossRef] [Green Version]
- Brunelli, E.; La Russa, D.; Pellegrino, D. Impaired oxidative status is strongly associated with cardiovascular risk factors. Oxid. Med. Cell. Longev. 2017, 2017, 6480145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrino, D. Antioxidants and Cardiovascular Risk Factors. Diseases 2016, 4, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demerdash, H.M. Role of Oxidative Stress and Associated Alteration in Enzyme Activities in Obesity Comorbidities. Obes. Res. Open J. 2017, 4, 32–43. [Google Scholar] [CrossRef]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef]
- Carillon, J.; Romain, C.; Bardy, G.; Fouret, G.; Feillet-Coudray, C.; Gaillet, S.; Lacan, D.; Cristol, J.P.; Rouanet, J.M. Cafeteria diet induces obesity and insulin resistance associated with oxidative stress but not with inflammation: Improvement by dietary supplementation with a melon superoxide dismutase. Free Radic. Biol. Med. 2013, 65, 254–261. [Google Scholar] [CrossRef]
- Johnson, A.R.; Wilkerson, M.D.; Sampey, B.P.; Troester, M.A.; Hayes, D.N.; Makowski, L. Cafeteria diet-induced obesity causes oxidative damage in white adipose. Biochem. Biophys. Res. Commun. 2016, 473, 545–550. [Google Scholar] [CrossRef] [Green Version]
- Rafieian-Kopaei, M. Medicinal plants for renal injury prevention. J. Ren. Inj. Prev. 2013, 2, 63–65. [Google Scholar]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellegrino, D.; La Russa, D.; Marrone, A. Oxidative Imbalance and Kidney Damage: New Study Perspectives from Animal Models to Hospitalized Patients. Antioxidants 2019, 8, 594. https://doi.org/10.3390/antiox8120594
Pellegrino D, La Russa D, Marrone A. Oxidative Imbalance and Kidney Damage: New Study Perspectives from Animal Models to Hospitalized Patients. Antioxidants. 2019; 8(12):594. https://doi.org/10.3390/antiox8120594
Chicago/Turabian StylePellegrino, Daniela, Daniele La Russa, and Alessandro Marrone. 2019. "Oxidative Imbalance and Kidney Damage: New Study Perspectives from Animal Models to Hospitalized Patients" Antioxidants 8, no. 12: 594. https://doi.org/10.3390/antiox8120594
APA StylePellegrino, D., La Russa, D., & Marrone, A. (2019). Oxidative Imbalance and Kidney Damage: New Study Perspectives from Animal Models to Hospitalized Patients. Antioxidants, 8(12), 594. https://doi.org/10.3390/antiox8120594