Peroxiredoxin6 in Endothelial Signaling



Abstract
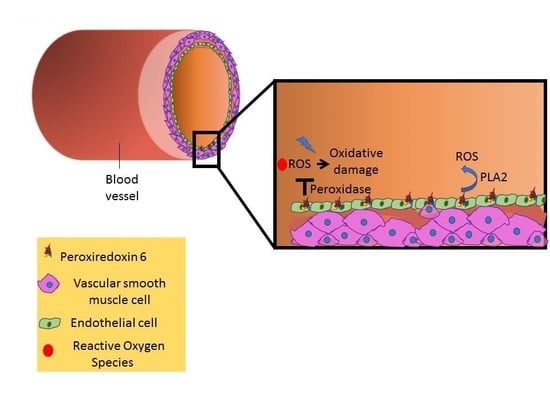
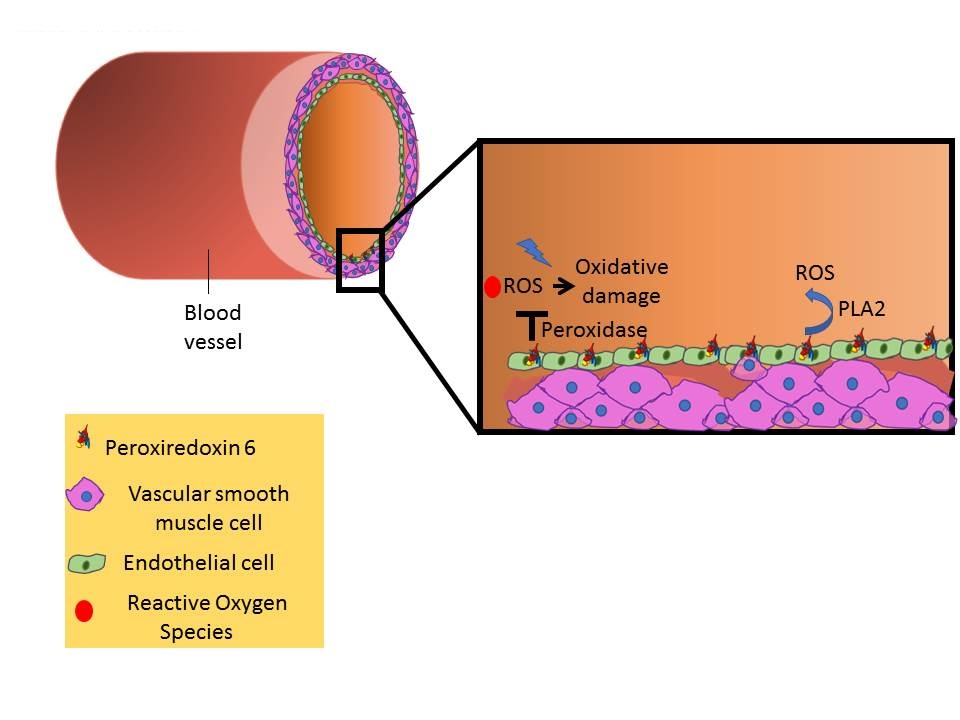
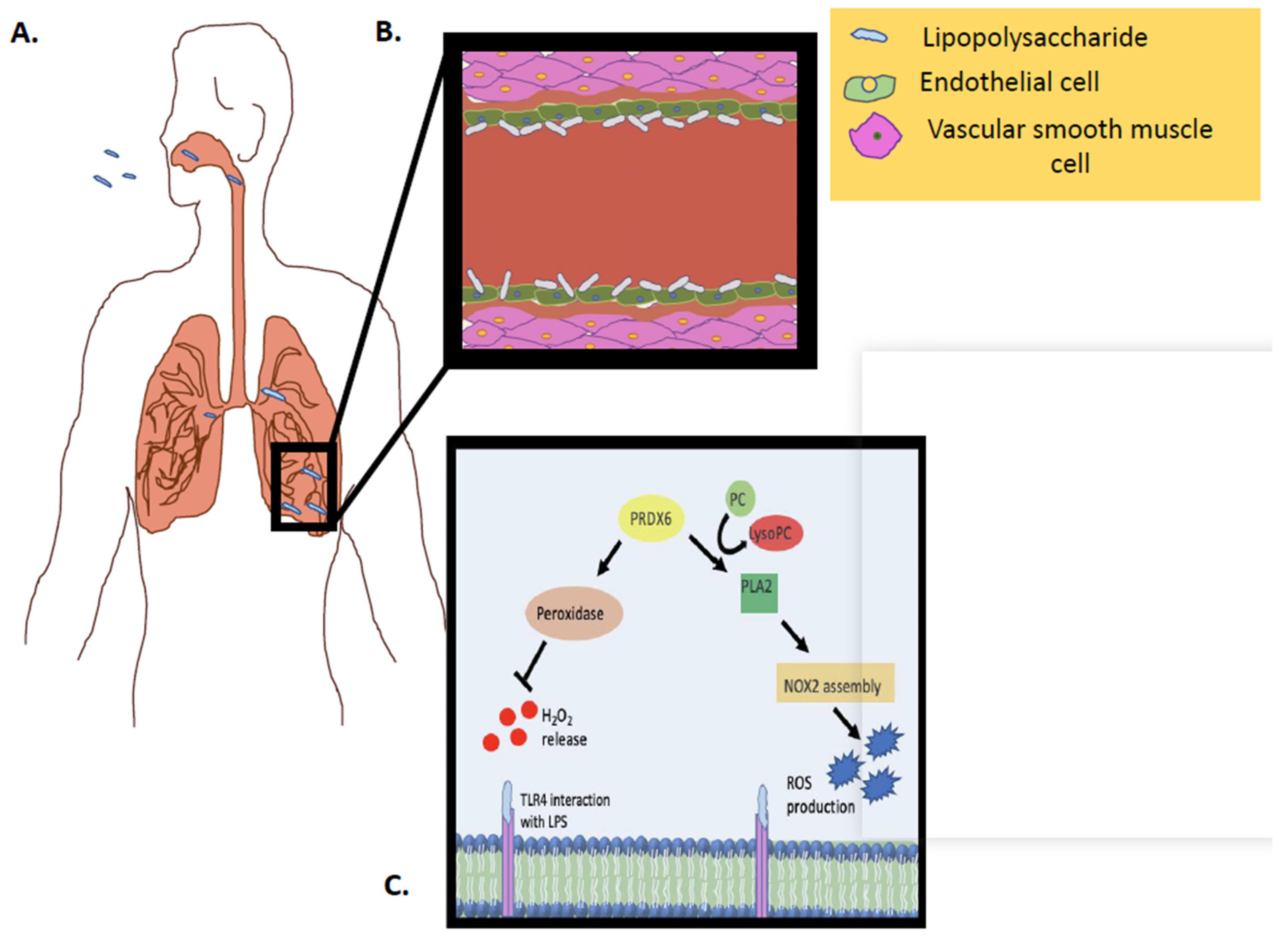
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
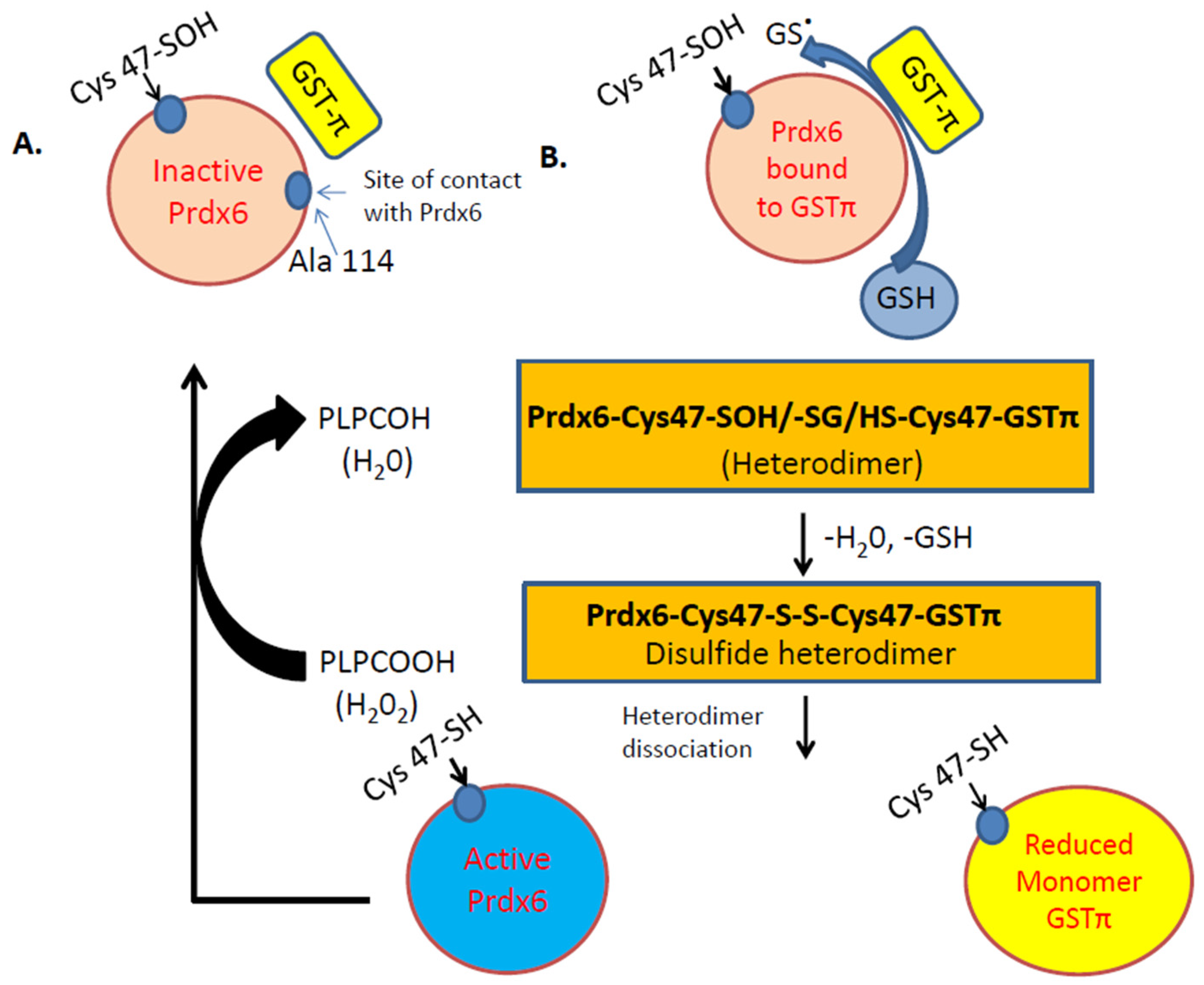
2. Peroxiredoxin 6 in the Endothelium
3. Peroxiredoxin 6 in Endothelial Mechanotransduction
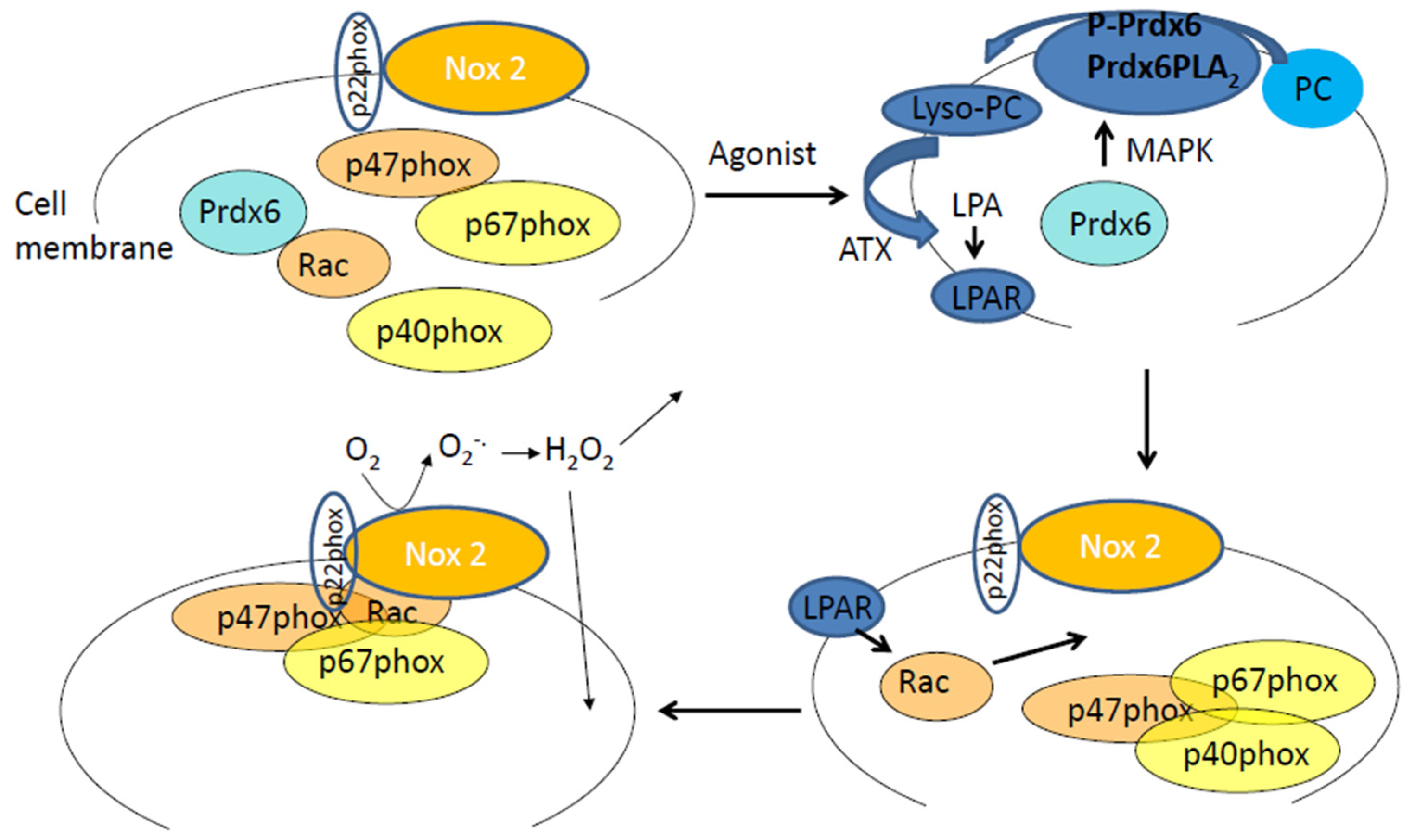
4. Peroxiredoxin 6 in Endothelial Chemotransduction
5. Peroxiredoxin 6 in Inflammatory Response
6. Prdx6 in Wound Repair
7. Prdx6 in the Pathogenesis of Diabetes
8. Conclusions
Funding
Conflicts of Interest
References
- Rhee, S.G.; Chae, H.Z.; Kim, K. Peroxiredoxins: A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic. Biol. Med. 2005, 38, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Kang, S.W.; Chang, T.S.; Jeong, W.; Kim, K. Peroxiredoxin, a novel family of peroxidases. IUBMB Life 2001, 52, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Sorokina, E.M.; Harper, S.; Li, H.; Ralat, L.; Dodia, C.; Speicher, D.W.; Feinstein, S.I.; Fisher, A.B. Peroxiredoxin 6 homodimerization and heterodimerization with glutathione S-transferase pi are required for its peroxidase but not phospholipase A2 activity. Free Radic. Biol. Med. 2016, 94, 145–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolussi, A.; D’Inzeo, S.; Capalbo, C.; Giannini, G.; Coppa, A. The role of peroxiredoxins in cancer. Mol. Clin. Oncol. 2017, 6, 139–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambruso, D.R.; Ellison, M.A.; Thurman, G.W.; Leto, T.L. Peroxiredoxin 6 translocates to the plasma membrane during neutrophil activation and is required for optimal NADPH oxidase activity. Biochim. Biophys. Acta. 2012, 1823, 306–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.W.; Dodia, C.; Feinstein, S.I.; Jain, M.K.; Fisher, A.B. 1-Cys peroxiredoxin, a bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. J. Biol. Chem. 2000, 275, 28421–28427. [Google Scholar] [CrossRef] [PubMed]
- Manevich, Y.; Feinstein, S.I.; Fisher, A.B. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pi GST. Proc. Natl. Acad. Sci. USA 2004, 101, 3780–3785. [Google Scholar] [CrossRef]
- Manevich, Y.; Reddy, K.S.; Shuvaeva, T.; Feinstein, S.I.; Fisher, A.B. Structure and phospholipase function of peroxiredoxin 6: Identification of the catalytic triad and its role in phospholipid substrate binding. J. Lipid Res. 2007, 48, 2306–2318. [Google Scholar] [CrossRef]
- Manevich, Y.; Fisher, A.B. Peroxiredoxin 6, a 1-Cys peroxiredoxin, functions in antioxidant defense and lung phospholipid metabolism. Free Radic. Biol. Med. 2005, 38, 1422–1432. [Google Scholar] [CrossRef]
- Fisher, A.B. The phospholipase A2 activity of peroxiredoxin 6. J. Lipid Res. 2018, 59, 1132–1147. [Google Scholar] [CrossRef]
- Chatterjee, S.; Feinstein, S.I.; Dodia, C.; Sorokina, E.; Lien, Y.C.; Nguyen, S.; Debolt, K.; Speicher, D.; Fisher, A.B. Peroxiredoxin 6 phosphorylation and subsequent phospholipase A2 activity are required for agonist-mediated activation of NADPH oxidase in mouse pulmonary microvascular endothelium and alveolar macrophages. J. Biol. Chem. 2011, 286, 11696–11706. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S. Endothelial Mechanotransduction, Redox Signaling and the Regulation of Vascular Inflammatory Pathways. Front. Physiol. 2018, 9, 524. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Fisher, A.B. Endothelium: A Comprehensive Reference; Cambridge University Press: Cambridge, MA, USA, 2007. [Google Scholar]
- Sarantos, M.R.; Zhang, H.; Schaff, U.Y.; Dixit, N.; Hayenga, H.N.; Lowell, C.A.; Simon, S.I. Transmigration of neutrophils across inflamed endothelium is signaled through LFA-1 and Src family kinase. J. Immunol. 2008, 181, 8660–8669. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Pak, J.H.; Gonzales, L.W.; Feinstein, S.I.; Fisher, A.B. Regulation of 1-cys peroxiredoxin expression in lung epithelial cells. Am. J. Respir. Cell Mol. Biol. 2002, 27, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Browning, E.A.; Chatterjee, S.; Fisher, A.B. Stop the flow: A paradigm for cell signaling mediated by reactive oxygen species in the pulmonary endothelium. Annu. Rev. Physiol. 2012, 74, 403–424. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Fisher, A.B. Mechanotransduction: Forces, sensors, and redox signaling. Antioxid. Redox Signal. 2014, 20, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Fisher, A.B. Mechanotransduction in the endothelium: role of membrane proteins and reactive oxygen species in sensing, transduction, and transmission of the signal with altered blood flow. Antioxid. Redox Signal. 2014, 20, 899–913. [Google Scholar] [CrossRef] [PubMed]
- Chhunchha, B.; Fatma, N.; Bhargavan, B.; Kubo, E.; Kumar, A.; Singh, D.P. Specificity protein, Sp1-mediated increased expression of Prdx6 as a curcumin-induced antioxidant defense in lens epithelial cells against oxidative stress. Cell Death Dis. 2011, 2, e234. [Google Scholar] [CrossRef] [PubMed]
- Burillo, E.; Jorge, I.; Martinez-Lopez, D.; Camafeita, E.; Blanco-Colio, L.M.; Trevisan-Herraz, M.; Ezkurdia, I.; Egido, J.; Michel, J.B.; Meilhac, O.; et al. Quantitative HDL Proteomics Identifies Peroxiredoxin-6 as a Biomarker of Human Abdominal Aortic Aneurysm. Sci. Rep. 2016, 6, 38477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumin, A.; Schafer, M.; Epp, N.; Bugnon, P.; Born-Berclaz, C.; Oxenius, A.; Klippel, A.; Bloch, W.; Werner, S. Peroxiredoxin 6 is required for blood vessel integrity in wounded skin. J. Cell Biol. 2007, 179, 747–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lien, Y.C.; Feinstein, S.I.; Dodia, C.; Fisher, A.B. The roles of peroxidase and phospholipase A2 activities of peroxiredoxin 6 in protecting pulmonary microvascular endothelial cells against peroxidative stress. Antioxid. Redox Signal. 2012, 16, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Levitan, I.; Wei, Z.; Fisher, A.B. KATP channels are an important component of the shear-sensing mechanism in the pulmonary microvasculature. Microcirculation 2006, 13, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Browning, E.A.; Hong, N.; DeBolt, K.; Sorokina, E.M.; Liu, W.; Birnbaum, M.J.; Fisher, A.B. Membrane depolarization is the trigger for PI3K/Akt activation and leads to the generation of ROS. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H105–H114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Matsuzaki, I.; Chatterjee, S.; Fisher, A.B. Activation of endothelial NADPH oxidase during normoxic lung ischemia is KATP channel dependent. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, L954–L961. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chatterjee, S.; Wei, Z.; Liu, W.D.; Fisher, A.B. Rac and PI3 kinase mediate endothelial cell-reactive oxygen species generation during normoxic lung ischemia. Antioxid. Redox Signal. 2008, 10, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Feinstein, S.; Hong, N.K.; Debolt, K. Paradoxical response of ROS production in peroxiredoxin 6 null mice to ischemia. FASEB J. 2007, 21, A1201. [Google Scholar]
- Vazquez-Medina, J.P.; Dodia, C.; Weng, L.; Mesaros, C.; Blair, I.A.; Feinstein, S.I.; Chatterjee, S.; Fisher, A.B. The phospholipase A2 activity of peroxiredoxin 6 modulates NADPH oxidase 2 activation via lysophosphatidic acid receptor signaling in the pulmonary endothelium and alveolar macrophages. FASEB J. 2016, 30, 2885–2898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eismann, T.; Huber, N.; Shin, T.; Kuboki, S.; Galloway, E.; Wyder, M.; Edwards, M.J.; Greis, K.D.; Shertzer, H.G.; Fisher, A.B.; et al. Peroxiredoxin-6 protects against mitochondrial dysfunction and liver injury during ischemia-reperfusion in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G266–G274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordeeva, A.E.; Temnov, A.A.; Charnagalov, A.A.; Sharapov, M.G.; Fesenko, E.E.; Novoselov, V.I. Protective Effect of Peroxiredoxin 6 in Ischemia/Reperfusion-Induced Damage of Small Intestine. Dig. Dis. Sci. 2015, 60, 3610–3619. [Google Scholar] [CrossRef] [PubMed]
- Kubyshkin, A.V.; Novosyolov, S.V.; Fomochkina, I.I.; Kharchenko, V.Z.; Pisarev, A.A.; Gordeeva, A.E.; Beketov, A.A.; Kochkina, A.V.; Fedosov, M.I.; Anisimova, L.V.; et al. Expression of caspase-3 and the cytokine level in experimental reperfusion syndrome upon treatment with peroxiredoxin 6. Biophysics 2017, 62, 848. [Google Scholar] [CrossRef]
- Shanshan, Y.; Beibei, J.; Li, T.; Minna, G.; Shipeng, L.; Li, P.; Yong, Z. Phospholipase A2 of Peroxiredoxin 6 Plays a Critical Role in Cerebral Ischemia/Reperfusion Inflammatory Injury. Front. Cell. Neurosci. 2017, 11, 99. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Chapman, K.E.; Fisher, A.B. Lung ischemia: A model for endothelial mechanotransduction. Cell Biochem. Biophys. 2008, 52, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef]
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef]
- Wang, Y.; Feinstein, S.I.; Manevich, Y.; Ho, Y.S.; Fisher, A.B. Peroxiredoxin 6 gene-targeted mice show increased lung injury with paraquat-induced oxidative stress. Antioxid. Redox Signal. 2006, 8, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Feinstein, S.I.; Wang, Y.; Dodia, C.; Fisher, D.; Yu, K.; Ho, Y.S.; Fisher, A.B. Comparison of glutathione peroxidase 1 and peroxiredoxin 6 in protection against oxidative stress in the mouse lung. Free Radic. Biol. Med. 2010, 49, 1172–1181. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.; Heath, D. The pathology of the lung in paraquat poisoning. J. Clin. Pathol. Suppl. (R. Coll. Pathol.) 1975, 9, 81–93. [Google Scholar] [CrossRef]
- Day, B.J.; Patel, M.; Calavetta, L.; Chang, L.Y.; Stamler, J.S. A mechanism of paraquat toxicity involving nitric oxide synthase. Proc. Natl. Acad. Sci. USA 1999, 96, 12760–12765. [Google Scholar] [CrossRef] [Green Version]
- Tsukamoto, M.; Tampo, Y.; Sawada, M.; Yonaha, M. Paraquat-induced oxidative stress and dysfunction of the glutathione redox cycle in pulmonary microvascular endothelial cells. Toxicol. Appl. Pharmacol. 2002, 178, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Dodia, C.; Chatterjee, S.; Feinstein, S.I.; Fisher, A.B. Protection against LPS-induced acute lung injury by a mechanism based inhibitor of NADPH-oxidase (Type 2). Am. J. Physiol. Lung Cell Mol. Physiol. 2014. [Google Scholar] [CrossRef]
- Parrillo, J.E.; Parker, M.M.; Natanson, C.; Suffredini, A.F.; Danner, R.L.; Cunnion, R.E.; Ognibene, F.P. Septic shock in humans. Advances in the understanding of pathogenesis, cardiovascular dysfunction, and therapy. Ann. Intern. Med. 1990, 113, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.; Rietschel, E.T. Innate immune sensing and its roots: The story of endotoxin. Nat. Rev. Immunol. 2003, 3, 169–176. [Google Scholar] [CrossRef]
- Lagarde, M.; Gualde, N.; Rigaud, M. Metabolic interactions between eicosanoids in blood and vascular cells. Biochem. J. 1989, 257, 313–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hood, E.D.; Greineder, C.F.; Dodia, C.; Han, J.; Mesaros, C.; Shuvaev, V.V.; Blair, I.A.; Fisher, A.B.; Muzykantov, V.R. Antioxidant protection by PECAM-targeted delivery of a novel NADPH-oxidase inhibitor to the endothelium in vitro and in vivo. J. Controll. Release off. J. Controll. Release Soc. 2012, 163, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H.; Park, J.H.; Han, S.B.; Yoon, D.Y.; Jung, Y.Y.; Hong, J.T. Peroxiredoxin 6 overexpression attenuates lipopolysaccharide-induced acute kidney injury. Oncotarget 2017, 8, 51096–51107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, T.; Kajikuri, J.; Tada, T.; Suzuki, Y.; Mabuchi, Y. Angiotensin II-induced modulation of endothelium-dependent relaxation in rabbit mesenteric resistance arteries. J. Physiol. 2003, 548, 893–906. [Google Scholar] [CrossRef]
- Ralat, L.A.; Manevich, Y.; Fisher, A.B.; Colman, R.F. Direct evidence for the formation of a complex between 1-cysteine peroxiredoxin and glutathione S-transferase π with activity changes in both enzymes. Biochemistry 2006, 45, 360–372. [Google Scholar] [CrossRef]
- Shang, W.; Liu, W.H.; Zhao, X.H.; Sun, Q.J.; Bi, J.Z.; Chi, Z.F. Expressions of glutathione S-transferase alpha, mu, and pi in brains of medically intractable epileptic patients. BMC Neurosci. 2008, 9, 67. [Google Scholar] [CrossRef]
- Yang, Y.; Yin, F.; Hang, Q.; Dong, X.; Chen, J.; Li, L.; Cao, P.; Yin, Z.; Luo, L. Regulation of Endothelial Permeability by Glutathione S-Transferase Pi Against Actin Polymerization. Cell. Physiol. Biochem. 2018, 45, 406–418. [Google Scholar] [CrossRef]
- Conklin, D.J.; Haberzettl, P.; Prough, R.A.; Bhatnagar, A. Glutathione-S-transferase P protects against endothelial dysfunction induced by exposure to tobacco smoke. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1586–H1597. [Google Scholar] [CrossRef]
- Walzog, B.; Gaehtgens, P. Adhesion Molecules: The Path to a New Understanding of Acute Inflammation. News Physiol. Sci. 2000, 15, 107–113. [Google Scholar] [CrossRef]
- Browning, E.; Wang, H.; Hong, N.; Yu, K.; Buerk, D.G.; DeBolt, K.; Gonder, D.; Sorokina, E.M.; Patel, P.; De Leon, D.D.; et al. Mechanotransduction drives post ischemic revascularization through K(ATP) channel closure and production of reactive oxygen species. Antioxid. Redox Signal. 2014, 20, 872–886. [Google Scholar] [CrossRef]
- El Eter, E.; Al Masri, A.; Habib, S.; Al Zamil, H.; Al Hersi, A.; Al Hussein, F.; Al Omran, M. Novel links among peroxiredoxins, endothelial dysfunction, and severity of atherosclerosis in type 2 diabetic patients with peripheral atherosclerotic disease. Cell Stress Chaperones 2014, 19, 173–181. [Google Scholar] [CrossRef]
- Wang, X.; Phelan, S.A.; Forsman-Semb, K.; Taylor, E.F.; Petros, C.; Brown, A.; Lerner, C.P.; Paigen, B. Mice with targeted mutation of peroxiredoxin 6 develop normally but are susceptible to oxidative stress. J. Biol. Chem. 2003, 278, 25179–25190. [Google Scholar] [CrossRef] [PubMed]
- Kuroiwa, T. New biomarkers of osteoarthritis. Osteoarthr. Cartil. 2016, 24, S89. [Google Scholar] [CrossRef]
- Kim, S.Y.; Jo, H.Y.; Kim, M.H.; Cha, Y.Y.; Choi, S.W.; Shim, J.H.; Kim, T.J.; Lee, K.Y. H2O2-dependent hyperoxidation of peroxiredoxin 6 (Prdx6) plays a role in cellular toxicity via up-regulation of iPLA2 activity. J. Biol. Chem. 2008, 283, 33563–33568. [Google Scholar] [CrossRef] [PubMed]
- Chhunchha, B.; Fatma, N.; Kubo, E.; Singh, D.P. Aberrant sumoylation signaling evoked by reactive oxygen species impairs protective function of Prdx6 by destabilization and repression of its transcription. FEBS J. 2014, 281, 3357–3381. [Google Scholar] [CrossRef] [PubMed]
- Fatma, N.; Kubo, E.; Takamura, Y.; Ishihara, K.; Garcia, C.; Beebe, D.C.; Singh, D.P. Loss of NF-kappaB control and repression of Prdx6 gene transcription by reactive oxygen species-driven SMAD3-mediated transforming growth factor beta signaling. J. Biol. Chem. 2009, 284, 22758–22772. [Google Scholar] [CrossRef] [PubMed]
- Tu, Q.; Xiong, Y.; Fan, L.; Qiao, B.; Xia, Z.; Hu, L.; Wang, Y.; Peng, G.; Ye, Q. Peroxiredoxin 6 attenuates ischemia and hypoxiainduced liver damage of braindead donors. Mol. Med. Rep. 2016, 13, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Munz, B.; Frank, S.; Hubner, G.; Olsen, E.; Werner, S. A novel type of glutathione peroxidase: Expression and regulation during wound repair. Biochem. J. 1997, 326 Pt 2, 579–585. [Google Scholar] [CrossRef]
- auf dem Keller, U.; Kumin, A.; Braun, S.; Werner, S. Reactive oxygen species and their detoxification in healing skin wounds. J. Investig. Dermatol. Symp. Proc. 2006, 11, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Munz, B.; Werner, S. The human homologue of a bovine non-selenium glutathione peroxidase is a novel keratinocyte growth factor-regulated gene. Oncogene 1997, 14, 915–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumin, A.; Huber, C.; Rulicke, T.; Wolf, E.; Werner, S. Peroxiredoxin 6 is a potent cytoprotective enzyme in the epidermis. Am. J. Pathol. 2006, 169, 1194–1205. [Google Scholar] [CrossRef] [PubMed]
- Ambrozova, N.; Ulrichova, J.; Galandakova, A. Models for the Study of Skin Wound Healing. The Role of Nrf2 and NF-kappaB; Biomedical Papers of the Medical Faculty of Palacky University in Olomouc; Palacky University in Olomouc: Olomouc, Czech Republic, 2017; Volume 161, pp. 1–13. [Google Scholar]
- Hurrle, S.; Hsu, W.H. The etiology of oxidative stress in insulin resistance. Biomed. J. 2017, 40, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Kajimoto, Y.; Kaneto, H. Role of oxidative stress in pancreatic beta-cell dysfunction. Ann. N. Y. Acad. Sci. 2004, 1011, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Wood, Z.A.; Schroder, E.; Robin Harris, J.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef]
- Paula, F.M.; Ferreira, S.M.; Boschero, A.C.; Souza, K.L. Modulation of the peroxiredoxin system by cytokines in insulin-producing RINm5F cells: down-regulation of PRDX6 increases susceptibility of beta cells to oxidative stress. Mol. Cell. Endocrinol. 2013, 374, 56–64. [Google Scholar] [CrossRef]
- Pacifici, F.; Arriga, R.; Sorice, G.P.; Capuani, B.; Scioli, M.G.; Pastore, D.; Donadel, G.; Bellia, A.; Caratelli, S.; Coppola, A.; et al. Peroxiredoxin 6, a novel player in the pathogenesis of diabetes. Diabetes 2014, 63, 3210–3220. [Google Scholar] [CrossRef]
- Avogaro, A.; Albiero, M.; Menegazzo, L.; de Kreutzenberg, S.; Fadini, G.P. Endothelial dysfunction in diabetes: The role of reparatory mechanisms. Diabetes Care 2011, 34 (Suppl. 2), S285–S290. [Google Scholar] [CrossRef]
- Altabas, V. Diabetes, Endothelial Dysfunction, and Vascular Repair: What Should a Diabetologist Keep His Eye on? Int. J. Endocrinol. 2015, 2015, 848272. [Google Scholar] [CrossRef]
- Fadini, G.P.; Sartore, S.; Albiero, M.; Baesso, I.; Murphy, E.; Menegolo, M.; Grego, F.; Vigili de Kreutzenberg, S.; Tiengo, A.; Agostini, C.; et al. Number and function of endothelial progenitor cells as a marker of severity for diabetic vasculopathy. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2140–2146. [Google Scholar] [CrossRef] [PubMed]
- Melhem, H.; Spalinger, M.R.; Cosin-Roger, J.; Atrott, K.; Lang, S.; Wojtal, K.A.; Vavricka, S.R.; Rogler, G.; Frey-Wagner, I. Prdx6 Deficiency Ameliorates DSS Colitis: Relevance of Compensatory Antioxidant Mechanisms. J. Crohns Colitis 2017, 11, 871–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Song, Y.; Wang, X.; Sun, J.; Ben, Y.; An, X.; Tong, L.; Bi, J.; Bai, C. Deletion of peroxiredoxin 6 potentiates lipopolysaccharide-induced acute lung injury in mice. Crit. Care Med. 2011, 39, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Pacifici, F.; Capuani, B.; Piermarini, F.; Pastore, D.; Arriga, R.; Coppola, A.; Rea, S.; Donadel, G.; Bellia, A.; Della-morte, D.; et al. Prdx6 Prevents Diabetic Myopathy by Improving Skeletal Muscle Cell Differentiation; American Diabetes Association: Arlington, VA, USA, 2018; Volume 67. [Google Scholar]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, P.; Chatterjee, S. Peroxiredoxin6 in Endothelial Signaling. Antioxidants 2019, 8, 63. https://doi.org/10.3390/antiox8030063
Patel P, Chatterjee S. Peroxiredoxin6 in Endothelial Signaling. Antioxidants. 2019; 8(3):63. https://doi.org/10.3390/antiox8030063
Chicago/Turabian StylePatel, Priyal, and Shampa Chatterjee. 2019. "Peroxiredoxin6 in Endothelial Signaling" Antioxidants 8, no. 3: 63. https://doi.org/10.3390/antiox8030063
APA StylePatel, P., & Chatterjee, S. (2019). Peroxiredoxin6 in Endothelial Signaling. Antioxidants, 8(3), 63. https://doi.org/10.3390/antiox8030063