Mitochondrial Glutathione: Recent Insights and Role in Disease

 ,
,  ,
,  , , ,
, , ,  and
and 
Abstract
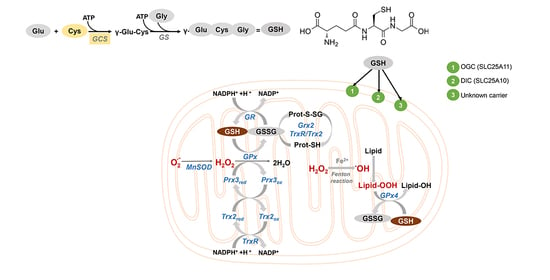
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Mitochondrial Glutathione
Transport of GSH across the Inner Mitochondrial Membrane
3. mGSH and Cell Death
4. mGSH in Pathological Settings
4.1. Alcoholic Liver Disease
4.2. Non Alcoholic Fatty Liver Disease
4.3. Neurodegenerative Disorders
4.4. Diabetic Nephropathy
4.5. Aging and Age-Related Diseases
4.6. Others
- ▪
- Lung diseases—glutathione precursors, particularly N-acetylcysteine, have been prescribed for years to prevent acute pulmonary episodes, bronchitis, or emphysema as an effective method of reducing oxidative stress in clinical settings associated with low GSH levels, such as chronic lung diseases [157]. More recently, glutathione levels have been associated with COVID-19 disease. Since mitochondrial ROS act as signal-transducing molecules that upregulate inflammatory cytokines [158] in conditions with excessive inflammatory response, as happens in severe COVID-19 symptoms, it is expected that mitochondrial antioxidants such as mGSH would play a role during the Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) viral infection.
- ▪
- Chemotherapy—standard chemotherapeutic agents such as doxorubicin and cisplatin are well-known inducers of mitochondrial ROS during their anti-tumoral action [159,160]. However, the mitochondrial effects of other more recently approved anti-cancer drugs, such as sorafenib or regorafenib, are being just revealed [161,162]. More importantly, mitochondrial antioxidants may reduce the effectivity of these drugs, while glutathione depressors potentiate their effect in hepatocellular carcinoma cells (Cucarull et al., unpublished results). Since chemotherapy-induced side effects are frequently also caused by mitochondrial ROS, such as cardiotoxicity or kidney injury after doxorubicin and cisplatin treatments, respectively, these potential redox therapies should be carefully directed to the target cells. Therefore, intake of antioxidants or ROS modulators should be well controlled in order to avoid undesired effects during cancer treatment.
- ▪
- Gender perspective—GSH levels are different in males and females as a consequence of hormonal regulation and aging [163,164]. Although higher mGSH levels in females are expected, this topic has been poorly pursued, with very few studies in animal models and common pathologies. Novel results highlighting the antioxidant differences observed between sexes, frequently reflecting sexual dimorphism in disease incidence will increase the interest in specific mGSH levels and maybe suggest gender-specific biomedical strategies.
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Meister, A.; Anderson, M.E.; Hwang, O. Intracellular cysteine and glutathione delivery systems. J. Am. Coll. Nutr. 1986, 5, 137–151. [Google Scholar] [CrossRef]
- Oestreicher, J.; Morgan, B. Glutathione: Subcellular distribution and membrane transport. Biochem. Cell Biol. 2019, 97, 270–289. [Google Scholar] [CrossRef] [Green Version]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal. 2009, 11. [Google Scholar] [CrossRef] [Green Version]
- Koehler, C.M.; Beverly, K.N.; Leverich, E.P. Redox pathways of the mitochondrion. Antioxid. Redox Signal. 2006, 8, 813–822. [Google Scholar] [CrossRef]
- Han, D.; Hanawa, N.; Saberi, B.; Kaplowitz, N. Mechanisms of liver injury. III. Role of glutathione redox status in liver injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G1–G7. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.P.; Carlson, J.L.; Mody, V.C.; Cai, J.; Lynn, M.J.; Sternberg, P. Redox state of glutathione in human plasma. Free Radic. Biol. Med. 2000, 28, 625–635. [Google Scholar] [CrossRef]
- Hayes, J.D.; McLellan, L.I. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic. Res. 1999, 31, 273–300. [Google Scholar] [CrossRef]
- Pastore, A.; Federici, G.; Bertini, E.; Piemonte, F. Analysis of glutathione: Implication in redox and detoxification. Clin. Chim. Acta 2003, 333, 19–39. [Google Scholar] [CrossRef]
- Griffith, O.W.; Bridges, R.J.; Meister, A. Transport of γ-glutamyl amino acids: Role of glutathione and γ-glutamyl transpeptidase. Proc. Natl. Acad. Sci. USA 1979, 76, 6319–6322. [Google Scholar] [CrossRef] [Green Version]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Sies, H.; Reichert, A.S. Selectively Addressing Mitochondrial Glutathione and Thioredoxin Redox Systems. Cell Chem. Biol. 2019, 26, 316–318. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Gill, R.; Mailloux, R.J. Protein S-glutathionylation: The linchpin for the transmission of regulatory information on redox buffering capacity in mitochondria. Chem. Biol. Interact. 2019, 299, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.G.; Bhatnagar, A. Protein S-glutathiolation: Redox-sensitive regulation of protein function. J. Mol. Cell. Cardiol. 2012, 52, 559–567. [Google Scholar] [CrossRef] [Green Version]
- Jevtović-Todorović, V.; Guenthner, T.M. Depletion of a discrete nuclear glutathione pool by oxidative stress, but not by buthionine sulfoximine. Correlation with enhanced alkylating agent cytotoxicity to human melanoma cells in vitro. Biochem. Pharmacol. 1992, 44, 1383–1393. [Google Scholar] [CrossRef]
- Markovic, J.; Borrás, C.; Ortega, Á.; Sastre, J.; Viña, J.; Pallardó, F.V. Glutathione is recruited into the nucleus in early phases of cell proliferation. J. Biol. Chem. 2007, 282, 20416–20424. [Google Scholar] [CrossRef] [Green Version]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef]
- Chakravarthi, S.; Jessop, C.E.; Bulleid, N.J. The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep. 2006, 7, 271–275. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.J. An update on mitochondrial reactive oxygen species production. Antioxidants 2020, 9, 472. [Google Scholar] [CrossRef]
- Cadenas, E.; Boveris, A.; Ragan, C.I.; Stoppani, A.O.M. Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch. Biochem. Biophys. 1977. [Google Scholar] [CrossRef]
- Boveris, A.; Chance, B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 1973. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J.A. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000. [Google Scholar] [CrossRef]
- Söderdahl, T.; Enoksson, M.; Lundberg, M.; Holmgren, A.; Ottersen, O.P.; Orrenius, S.; Bolcsfoldi, G.; Cotgreave, I.A. Visualization of the compartmentalization of glutathione and protein-glutathione mixed disulfides in cultured cells. FASEB J. 2003, 17, 124–126. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, G.; Morgan, B.; Riemer, J. Mitochondrial Glutathione: Regulation and Functions. Antioxid. Redox Signal. 2017, 27, 1162–1177. [Google Scholar] [CrossRef]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014. [Google Scholar] [CrossRef] [Green Version]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Kaplowitz, N.; Fernández-Checa, J.C. Mitochondrial glutathione: Features, regulation and role in disease. Biochim. Biophys. Acta Gen. Subj. 2013, 1830. [Google Scholar] [CrossRef] [Green Version]
- Lillig, C.H.; Berndt, C.; Holmgren, A. Glutaredoxin systems. Biochim. Biophys. Acta Gen. Subj. 2008. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione Transferases. Annu. Rev. Pharmacol. Toxicol. 2005. [Google Scholar] [CrossRef]
- Conrad, M. Transgenic mouse models for the vital selenoenzymes cytosolic thioredoxin reductase, mitochondrial thioredoxin reductase and glutathione peroxidase 4. Biochim. Biophys. Acta Gen. Subj. 2009. [Google Scholar] [CrossRef]
- Kajarabille, N.; Latunde-Dada, G.O. Programmed cell-death by ferroptosis: Antioxidants as mitigators. Int. J. Mol. Sci. 2019, 20, 4968. [Google Scholar] [CrossRef] [Green Version]
- Collin, F. Chemical basis of reactive oxygen species reactivity and involvement in neurodegenerative diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef] [Green Version]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19. [Google Scholar] [CrossRef]
- Murphy, M.P. Mitochondrial thiols in antioxidant protection and redox signaling: Distinct roles for glutathionylation and other thiol modifications. Antioxid. Redox Signal. 2012. [Google Scholar] [CrossRef]
- Chae, H.Z.; Kang, S.W.; Rhee, S.G. Isoforms of mammalian peroxiredoxin that reduce peroxides in presence of thioredoxin. Methods Enzymol. 1999, 300, 219–226. [Google Scholar] [CrossRef]
- Zhang, H.; Go, Y.M.; Jones, D.P. Mitochondrial thioredoxin-2/peroxiredoxin-3 system functions in parallel with mitochondrial GSH system in protection against oxidative stress. Arch. Biochem. Biophys. 2007, 465, 119–126. [Google Scholar] [CrossRef]
- Mailloux, R.J. Protein S-glutathionylation reactions as a global inhibitor of cell metabolism for the desensitization of hydrogen peroxide signals. Redox Biol. 2020, 32, 101472. [Google Scholar] [CrossRef]
- Chen, Z.; Putt, D.A.; Lash, L.H. Enrichment and functional reconstitution of glutathione transport activity from rabbit kidney mitochondria. Further evidence for the role of the dicarboxylate and 2-oxoglutarate carriers in mitochondrial glutathione transport. Arch. Biochem. Biophys. 2000, 373, 193–202. [Google Scholar] [CrossRef]
- Lash, L.H. Role of glutathione transport processes in kidney function. Toxicol. Appl. Pharmacol. 2005, 204, 329–342. [Google Scholar] [CrossRef]
- Fernandez-Checa, J.C.; Kaplowitz, N. Hepatic mitochondrial glutathione: Transport and role in disease and toxicity. Toxicol. Appl. Pharmacol. 2005, 204, 263–273. [Google Scholar] [CrossRef]
- Fernández-Checa, J.C.; Yi, J.R.; Ruiz, C.G.; Ookhtens, M.; Kaplowitz, N. Plasma membrane and mitochondrial transport of hepatic reduced glutathione. Semin. Liver Dis. 1996, 16, 147–158. [Google Scholar] [CrossRef]
- Lash, L.H. Mitochondrial glutathione transport: Physiological, pathological and toxicological implications. Chem. Biol. Interact. 2006, 163, 54–67. [Google Scholar] [CrossRef] [Green Version]
- Coll, O.; Colell, A.; García-Ruiz, C.; Kaplowitz, N.; Fernández-Checa, J.C. Sensitivity of the 2-oxoglutarate carrier to alcohol intake contributes to mitochondrial glutathione depletion. Hepatology 2003, 38, 692–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marí, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.J.C.; García-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef]
- Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondrial glutathione: Hepatocellular survival-death switch. J. Gastroenterol. Hepatol. 2006, 21. [Google Scholar] [CrossRef]
- Barbero-Camps, E.; Fernández, A.; Martínez, L.; Fernández-Checa, J.C.; Colell, A. APP/PS1 mice overexpressing SREBP-2 exhibit combined Aβ accumulation and tau pathology underlying Alzheimer’s disease. Hum. Mol. Genet. 2013, 22, 3460–3476. [Google Scholar] [CrossRef]
- Fernández, A.; Llacuna, L.; Fernández-Checa, J.C.; Colell, A. Mitochondrial cholesterol loading exacerbates amyloid β peptide-induced inflammation and neurotoxicity. J. Neurosci. 2009, 29, 6394–6405. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Q.; Putt, D.A.; Xu, F.; Lash, L.H. Hepatic mitochondrial transport of glutathione: Studies in isolated rat liver mitochondria and H4IIE rat hepatoma cells. Arch. Biochem. Biophys. 2008, 474, 119–127. [Google Scholar] [CrossRef] [Green Version]
- Booty, L.M.; King, M.S.; Thangaratnarajah, C.; Majd, H.; James, A.M.; Kunji, E.R.S.; Murphy, M.P. The mitochondrial dicarboxylate and 2-oxoglutarate carriers do not transport glutathione. FEBS Lett. 2015, 589, 621–628. [Google Scholar] [CrossRef] [Green Version]
- Monné, M.; Chan, K.W.; Slotboom, D.-J.; Kunji, E.R.S. Functional expression of eukaryotic membrane proteins in Lactococcus lactis. Protein Sci. 2005, 14, 3048–3056. [Google Scholar] [CrossRef] [Green Version]
- Armeni, T.; Cianfruglia, L.; Piva, F.; Urbanelli, L.; Luisa Caniglia, M.; Pugnaloni, A.; Principato, G. S-D-Lactoylglutathione can be an alternative supply of mitochondrial glutathione. Free Radic. Biol. Med. 2014, 67, 451–459. [Google Scholar] [CrossRef]
- de Bari, L.; Atlante, A.; Armeni, T.; Kalapos, M.P. Synthesis and metabolism of methylglyoxal, S-D-lactoylglutathione and D-lactate in cancer and Alzheimer’s disease. Exploring the crossroad of eternal youth and premature aging. Ageing Res. Rev. 2019, 53, 100915. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Treberg, J.R. Protein S-glutathionlyation links energy metabolism to redox signaling in mitochondria. Redox Biol. 2016, 8, 110–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Fariss, M.W.; Chan, C.B.; Patel, M.; Van Houten, B.; Orrenius, S. Role of mitochondria in toxic oxidative stress. Mol. Interv. 2005, 5, 94–111. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Mitochondrial Oxidative Stress: Implications for Cell Death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 143–183. [Google Scholar] [CrossRef]
- Marchetti, P.; Decaudin, D.; Macho, A.; Zamzami, N.; Hirsch, T.; Susin, S.A.; Kroemer, G. Redox regulation of apoptosis: Impact of thiol oxidation status on mitochondrial function. Eur. J. Immunol. 1997, 27, 289–296. [Google Scholar] [CrossRef]
- Yin, F.; Sancheti, H.; Cadenas, E. Mitochondrial thiols in the regulation of cell death pathways. Antioxid. Redox Signal. 2012, 17, 1714–1727. [Google Scholar] [CrossRef] [Green Version]
- Muyderman, H.; Wadey, A.L.; Nilsson, M.; Sims, N.R. Mitochondrial glutathione protects against cell death induced by oxidative and nitrative stress in astrocytes. J. Neurochem. 2007, 102, 1369–1382. [Google Scholar] [CrossRef]
- Hoek, J.B.; Rydstrom, J. Physiological roles of nicotinamide nucleotide transhydrogenase. Biochem. J. 1988, 254, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Arkblad, E.L.; Tuck, S.; Pestov, N.B.; Dmitriev, R.I.; Kostina, M.B.; Stenvall, J.; Tranberg, M.; Rydström, J. A Caenorhabditis elegans mutant lacking functional nicotinamide nucleotide transhydrogenase displays increased sensitivity to oxidative stress. Free Radic. Biol. Med. 2005, 38, 1518–1525. [Google Scholar] [CrossRef]
- Shin, A.H.; Kil, I.S.; Yang, E.S.; Huh, T.L.; Yang, C.H.; Park, J.W. Regulation of high glucose-induced apoptosis by mitochondrial NADP +-dependent isocitrate dehydrogenase. Biochem. Biophys. Res. Commun. 2004, 325, 32–38. [Google Scholar] [CrossRef]
- Albano, E.; Bellomo, G.; Parola, M.; Carini, R.; Dianzani, M.U. Stimulation of lipid peroxidation increases the intracellular calcium content of isolated hepatocytes. BBA Mol. Cell Res. 1991, 1091, 310–316. [Google Scholar] [CrossRef]
- Bacon, B.R.; O’Neill, R.; Britton, R.S. Hepatic mitochondrial energy production in rats with chronic iron overload. Gastroenterology 1993, 105, 1134–1140. [Google Scholar] [CrossRef]
- Bacon, B.R.; O’Neill, R.; Park, C.H. Iron-induced peroxidative injury to isolated rat hepatic mitochondria. J. Free Radic. Biol. Med. 1986, 2, 339–347. [Google Scholar] [CrossRef]
- Masini, A.; Ceccarelli, D.; Trenti, T.; Corongiu, F.P.; Muscatello, U. Perturbation in liver mitochondrial Ca2+ homeostasis in experimental iron overload: A possible factor in cell injury. BBA Mol. Cell Res. 1989, 1014, 133–140. [Google Scholar] [CrossRef]
- Chen, J.J.; Bertrand, H.; Yu, B.P. Inhibition of adenine nucleotide translocator by lipid peroxidation products. Free Radic. Biol. Med. 1995, 19, 583–590. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; Castilho, R.F.; Vercesi, A.E. Opening of the mitochondrial permeability transition pore by uncoupling or inorganic phosphate in the presence of Ca2+ is dependent on mitochondrial-generated reactive oxygen species. FEBS Lett. 1996, 378, 150–152. [Google Scholar] [CrossRef] [Green Version]
- Aniya, Y.; Imaizumi, N. Mitochondrial glutathione transferases involving a new function for membrane permeability transition pore regulation. Drug Metab. Rev. 2011, 43, 292–299. [Google Scholar] [CrossRef]
- Wu, D.; Cederbaum, A.I. Oxidative stress and alcoholic liver disease. Semin. Liver Dis. 2009, 29, 141–154. [Google Scholar] [CrossRef]
- Bruguera, M.; Bertran, A.; Bombi, J.A.; Rodes, J. Giant Mitochondria in Hepatocytes: A Diagnostic Hint for Alcoholic Liver Disease-PubMed. Gastroenterology 1977, 73, 1383–1387. [Google Scholar] [CrossRef]
- Thayer, W.S. Effects of Ethanol on Proteins of Mitochondrial Membranes. Ann. N. Y. Acad. Sci. 1987, 492, 193–206. [Google Scholar] [CrossRef]
- Rottenberg, H.; Robertson, D.E.; Rubin, E. The effect of temperature and chronic ethanol feeding on the proton electrochemical potential and phosphate potential in rat liver mitochondria. BBA Bioenerg. 1985, 809, 1–10. [Google Scholar] [CrossRef]
- Fernández-Checa, J.C.; Colell, A.; García-Ruiz, C. S-Adenosyl-L-methionine and mitochondrial reduced glutathione depletion in alcoholic liver disease. Alcohol 2002, 27, 179–183. [Google Scholar] [CrossRef]
- Fernández-Checa, J.C.; Hirano, T.; Tsukamoto, H.; Kaplowitz, N. Mitochondrial glutathione depletion in alcoholic liver disease. Alcohol 1993, 10, 469–475. [Google Scholar] [CrossRef]
- Marí, M.; Colell, A.; Morales, A.; Caballero, F.; Moles, A.; Fernández, A.; Terrones, O.; Basañez, G.; Antonsson, B.; García-Ruiz, C.; et al. Mechanism of Mitochondrial Glutathione-Dependent Hepatocellular Susceptibility to TNF Despite NF-κB Activation. Gastroenterology 2008, 134. [Google Scholar] [CrossRef] [PubMed]
- Colell, A.; Garcia-Ruiz, C.; Miranda, M.; Ardite, E.; Mari, M.; Morales, A.; Corrales, F.; Kaplowitz, N.; Fernandez-Checa, J.C.C. Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology 1998, 115, 1541–1551. [Google Scholar] [CrossRef]
- Zhao, P.; Slattery, J.T. Effects of ethanol dose and ethanol withdrawal on rat liver mitochondrial glutathione: Implication of potentiated acetaminophen toxicity in alcoholics. Drug Metab. Dispos. 2002, 30, 1413–1417. [Google Scholar] [CrossRef]
- Zhao, P.; Kalhorn, T.F.; Slattery, J.T. Selective mitochondrial glutathione depletion by ethanol enhances acetaminophen toxicity in rat liver. Hepatology 2002, 36, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, M.D.; Nakagami, M.; Bradford, B.U.; Uesugi, T.; Mason, R.P.; Connor, H.D.; Dikalova, A.; Kadiiska, M.; Thurman, R.G. Overexpression of Manganese Superoxide Dismutase Prevents Alcohol-induced Liver Injury in the Rat. J. Biol. Chem. 2001, 276, 36664–36672. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, A.; Matias, N.; Fucho, R.; Ribas, V.; Von Montfort, C.; Nuño, N.; Baulies, A.; Martinez, L.; Tarrats, N.; Mari, M.; et al. ASMase is required for chronic alcohol induced hepatic endoplasmic reticulum stress and mitochondrial cholesterol loading. J. Hepatol. 2013, 59, 805–813. [Google Scholar] [CrossRef] [Green Version]
- Fernández, A.; Colell, A.; Caballero, F.; Matías, N.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial S-adenosyl-I-methionine transport is insensitive to alcohol-mediated changes in membrane dynamics. Alcohol. Clin. Exp. Res. 2009, 33, 1169–1180. [Google Scholar] [CrossRef]
- Marí, M.; Cederbaum, A.I. CYP2E1 overexpression in HepG2 cells induces glutathione synthesis by transcriptional activation of γ-glutamylcysteine synthetase. J. Biol. Chem. 2000, 275. [Google Scholar] [CrossRef] [Green Version]
- Marí, M.; Cederbaum, A.I. Induction of catalase, alpha, and microsomal glutathione S-transferase in CYP2E1 overexpressing HepG2 cells and protection against short-term oxidative stress. Hepatology 2001, 33. [Google Scholar] [CrossRef]
- Marí, M.; Wu, D.; Nieto, N.; Cederbaum, A.I. CYP2E1-dependent toxicity and up-regulation of antioxidant genes. J. Biomed. Sci. 2001, 8. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; Sanyal, A.; Neuschwander-Tetri, B.; Tiribelli, C.; Kleiner, D.E.; Brunt, E.; Bugianesi, E.; Yki-Järvinen, H.; et al. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef]
- Pérez-Carreras, M.; Del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef]
- Cortez-Pinto, H.; Chatham, J.; Chacko, V.P.; Arnold, C.; Rashid, A.; Diehl, A.M. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: A pilot study. J. Am. Med. Assoc. 1999, 282, 1659–1664. [Google Scholar] [CrossRef] [Green Version]
- Hensley, K.; Robinson, K.A.; Gabbita, S.P.; Salsman, S.; Floyd, R.A. Reactive oxygen species, cell signaling, and cell injury. Free Radic. Biol. Med. 2000, 28, 1456–1462. [Google Scholar] [CrossRef]
- Caldwell, S.H.; Swerdlow, R.H.; Khan, E.M.; Iezzoni, J.C.; Hespenheide, E.E.; Parks, J.K.; Parker, W.D. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J. Hepatol. 1999, 31, 430–434. [Google Scholar] [CrossRef]
- Seki, S.; Kitada, T.; Yamada, T.; Sakaguchi, H.; Nakatani, K.; Wakasa, K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J. Hepatol. 2002, 37, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Crespo, J.; Cayón, A.; Fernández-Gil, P.; Hernández-Guerra, M.; Mayorga, M.; Domínguez-Díez, A.; Fernández-Escalante, J.C.; Pons-Romero, F. Gene expression of tumor necrosis factor α and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology 2001, 34, 1158–1163. [Google Scholar] [CrossRef]
- Honda, Y.; Kessoku, T.; Sumida, Y.; Kobayashi, T.; Kato, T.; Ogawa, Y.; Tomeno, W.; Imajo, K.; Fujita, K.; Yoneda, M.; et al. Efficacy of glutathione for the treatment of nonalcoholic fatty liver disease: An open-label, single-arm, multicenter, pilot study. BMC Gastroenterol. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Bjornson, E.; Zhang, C.; Klevstig, M.; Söderlund, S.; Ståhlman, M.; Adiels, M.; Hakkarainen, A.; Lundbom, N.; Kilicarslan, M.; et al. Personal model-assisted identification of NAD + and glutathione metabolism as intervention target in NAFLD. Mol. Syst. Biol. 2017, 13, 916. [Google Scholar] [CrossRef]
- Fernández-Checa, J.C.; Fernández, A.; Morales, A.; Marí, M.; García-Ruiz, C.-R.; Colell, A. Oxidative stress and altered mitochondrial function in neurodegenerative diseases: Lessons from mouse models. CNS Neurol. Disord. Drug Targets 2010, 9. [Google Scholar] [CrossRef]
- Amadoro, G.; Corsetti, V.; Stringaro, A.; Colone, M.; D’Aguanno, S.; Meli, G.; Ciotti, M.; Sancesario, G.; Cattaneo, A.; Bussani, R.; et al. A NH2 tau fragment targets neuronal mitochondria at AD synapses: Possible implications for neurodegeneration. J. Alzheimer’s Dis. 2010, 21, 445–470. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of Aβ accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef]
- Dumont, M.; Ho, D.J.; Calingasan, N.Y.; Xu, H.; Gibson, G.; Beal, M.F. Mitochondrial dihydrolipoyl succinyltransferase deficiency accelerates amyloid pathology and memory deficit in a transgenic mouse model of amyloid deposition. Free Radic. Biol. Med. 2009, 47, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, L.; Raber, J.; Kekonius, L.; Yan, F.; Yu, G.Q.; Bien-Ly, N.; Puoliväli, J.; Scearce-Levie, K.; Masliah, E.; Mucke, L. Reduction in mitochondrial superoxide dismutase modulates Alzheimer’s disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J. Neurosci. 2006, 26, 5167–5179. [Google Scholar] [CrossRef] [Green Version]
- Torres, S.; García-Ruiz, C.M.; Fernandez-Checa, J.C. Mitochondrial Cholesterol in Alzheimer’s Disease and Niemann–Pick Type C Disease. Front. Neurol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- de Dios, C.; Bartolessis, I.; Roca-Agujetas, V.; Barbero-Camps, E.; Mari, M.; Morales, A.; Colell, A. Oxidative inactivation of amyloid beta-degrading proteases by cholesterol-enhanced mitochondrial stress. Redox Biol. 2019, 26. [Google Scholar] [CrossRef] [PubMed]
- Barbero-Camps, E.; Roca-Agujetas, V.; Bartolessis, I.; de Dios, C.; Fernández-Checa, J.C.; Marí, M.; Morales, A.; Hartmann, T.; Colell, A. Cholesterol impairs autophagy-mediated clearance of amyloid beta while promoting its secretion. Autophagy 2018, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddell, J.R.; White, A.R. Nexus between mitochondrial function, iron, copper and glutathione in Parkinson’s disease. Neurochem. Int. 2018, 117, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, I.; Isooka, N.; Imafuku, F.; Sun, J.; Kikuoka, R.; Furukawa, C.; Asanuma, M. Chronic systemic exposure to low-dose rotenone induced central and peripheral neuropathology and motor deficits in mice: Reproducible animal model of parkinson’s disease. Int. J. Mol. Sci. 2020, 21, 3254. [Google Scholar] [CrossRef]
- Esteves, A.R.; Swerdlow, R.H.; Cardoso, S.M. LRRK2, a puzzling protein: Insights into Parkinson’s disease pathogenesis. Exp. Neurol. 2014, 261, 206–216. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K. α-Synuclein and Mitochondria: Partners in Crime? Neurotherapeutics 2013, 10, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Truban, D.; Hou, X.; Caulfield, T.R.; Fiesel, F.C.; Springer, W. PINK1, Parkin, and Mitochondrial Quality Control: What can we Learn about Parkinson’s Disease Pathobiology? J. Parkinson’s Dis. 2017, 7, 13–29. [Google Scholar] [CrossRef] [Green Version]
- Tanner, C.M.; Kame, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef] [Green Version]
- Fitzmaurice, P.S.; Ang, L.; Guttman, M.; Rajput, A.H.; Furukawa, Y.; Kish, S.J. Nigral glutathione deficiency is not specific for idiopathic Parkinson’s disease. Mov. Disord. 2003, 18, 969–976. [Google Scholar] [CrossRef]
- Pearce, R.K.B.; Owen, A.; Daniel, S.; Jenner, P.; Marsden, C.D. Alterations in the distribution of glutathione in the substantia nigra in Parkinson’s disease. J. Neural Transm. 1997, 104, 661–677. [Google Scholar] [CrossRef]
- Perry, T.L.; Yong, V.W. Idiopathic Parkinson’s disease, progressive supranuclear palsy and glutathione metabolism in the substantia nigra of patients. Neurosci. Lett. 1986, 67, 269–274. [Google Scholar] [CrossRef]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Jenner, P.; Marsden, C.D. Glutathione-related enzymes in brain in Parkinson’s disease. Ann. Neurol. 1994, 36, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Sofic, E.; Lange, K.W.; Jellinger, K.; Riederer, P. Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson’s disease. Neurosci. Lett. 1992, 142, 128–130. [Google Scholar] [CrossRef]
- Bharath, S.; Andersen, J.K. Glutathione depletion in a midbrain-derived immortalized dopaminergic cell line results in limited tyrosine nitration of mitochondrial complex I subunits: Implications for Parkinson’s disease. Antioxid. Redox Signal. 2005, 7, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Zeevalk, G.D.; Manzino, L.; Sonsalla, P.K.; Bernard, L.P. Characterization of intracellular elevation of glutathione (GSH) with glutathione monoethyl ester and GSH in brain and neuronal cultures: Relevance to Parkinson’s disease. Exp. Neurol. 2007, 203, 512–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Cozzolino, M.; Carrì, M.T. Mitochondrial dysfunction in ALS. Prog. Neurobiol. 2012, 97, 54–66. [Google Scholar] [CrossRef]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef] [Green Version]
- Ross, E.K.; Winter, A.N.; Wilkins, H.M.; Sumner, W.A.; Duval, N.; Patterson, D.; Linseman, D.A. A cystine-rich whey supplement (Immunocal®) delays disease onset and prevents spinal cord glutathione depletion in the hSOD1G93A mouse model of amyotrophic lateral sclerosis. Antioxidants 2014, 3, 843–865. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.A.; Lin, T.H.; Sheehan, A.E.; Van der Goes van Naters, W.; Neukomm, L.J.; Graves, H.K.; Bis-Brewer, D.M.; Züchner, S.; Freeman, M.R. Glutathione S-Transferase Regulates Mitochondrial Populations in Axons through Increased Glutathione Oxidation. Neuron 2019, 103, 52–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, M.; Zou, F.; Chai, H.S.; Younkin, C.S.; Miles, R.; Nair, A.A.; Crook, J.E.; Pankratz, V.S.; Carrasquillo, M.M.; Rowley, C.N.; et al. Glutathione S-transferase omega genes in Alzheimer and Parkinson disease risk, age-at-diagnosis and brain gene expression: An association study with mechanistic implications. Mol. Neurodegener. 2012, 7, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, P.; Rolo, A.; Sena, C.; Seica, R.; Oliveira, C.; Santos, M. Insulin Attenuates Diabetes-Related Mitochondrial Alterations: A Comparative Study. Med. Chem. (Los Angeles) 2006, 2, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.Y.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P.; et al. Rage-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef] [Green Version]
- Raza, H.; Prabu, S.K.; John, A.; Avadhani, N.G. Impaired mitochondrial respiratory functions and oxidative stress in streptozotocin-induced diabetic rats. Int. J. Mol. Sci. 2011, 12, 3133–3147. [Google Scholar] [CrossRef] [Green Version]
- Sourris, K.C.; Harcourt, B.E.; Tang, P.H.; Morley, A.L.; Huynh, K.; Penfold, S.A.; Coughlan, M.T.; Cooper, M.E.; Nguyen, T.V.; Ritchie, R.H.; et al. Ubiquinone (coenzyme Q10) prevents renal mitochondrial dysfunction in an experimental model of type 2 diabetes. Free Radic. Biol. Med. 2012, 52, 716–723. [Google Scholar] [CrossRef]
- Maddaiah, V.T. Glutathione correlates with lipid peroxidation in liver mitochondria of triiodothyronine-injected hypophysectomized rats. FASEB J. 1990, 4, 1513–1518. [Google Scholar] [CrossRef]
- Madrigal, J.L.M.; Olivenza, R.; Moro, M.A.; Lizasoain, I.; Lorenzo, P.; Rodrigo, J.; Leza, J.C. Glutathione depletion, lipid peroxidation and mitochondrial dysfunction are induced by chronic stress in rat brain. Neuropsychopharmacology 2001, 24, 420–429. [Google Scholar] [CrossRef] [Green Version]
- Lash, L. Mitochondrial Glutathione in Diabetic Nephropathy. J. Clin. Med. 2015, 4, 1428–1447. [Google Scholar] [CrossRef] [Green Version]
- Alderson, N.L.; Chachich, M.E.; Frizzell, N.; Canning, P.; Metz, T.O.; Januszewski, A.S.; Youssef, N.N.; Stitt, A.W.; Baynes, J.W.; Thorpe, S.R. Effect of antioxidants and ACE inhibition on chemical modification of proteins and progression of nephropathy in the streptozotocin diabetic rat. Diabetologia 2004, 47, 1385–1395. [Google Scholar] [CrossRef] [Green Version]
- Beisswenger, P.J.; Drummond, K.S.; Nelson, R.G.; Howell, S.K.; Szwergold, B.S.; Mauer, M. Susceptibility to diabetic nephropathy is related to dicarbonyl and oxidative stress. Diabetes 2005, 54, 3274–3281. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, S.; Starnes, J.; Shi, S.; Lonis, B.; Tran, R. Diabetic nephropathy is associated with oxidative stress and decreased renal nitric oxide production. J. Am. Soc. Nephrol. 2007, 18, 2945–2952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Winiarska, K.; Drozak, J.; Wegrzynowicz, M.; Fraczyk, T.; Bryla, J. Diabetes-induced changes in glucose synthesis, intracellular glutathione status and hydroxyl free radical generation in rabbit kidney-cortex tubules. Mol. Cell. Biochem. 2004, 261, 91–98. [Google Scholar] [CrossRef]
- Ueno, Y.; Kizaki, M.; Nakagiri, R.; Kamiya, T.; Sumi, H.; Osawa, T. Dietary glutathione protects rats from diabetic nephropathy and neuropathy. J. Nutr. 2002, 132, 897–900. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Wang, W. Effect of glutathione liposomes on diabetic nephropathy based on oxidative stress and polyol pathway mechanism. J. Liposome Res. 2020, 1–9. [Google Scholar] [CrossRef]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.P. Redox theory of aging. Redox Biol. 2015, 5, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Go, Y.M.; Jones, D.P. Redox theory of aging: Implications for health and disease. Clin. Sci. 2017, 131, 1669–1688. [Google Scholar] [CrossRef] [Green Version]
- Beal, M.F. Mitochondria take center stage in aging and neurodegeneration. Ann. Neurol. 2005, 58, 495–505. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.X.; Correia, S.C.; Zhu, X.; Smith, M.A.; Moreira, P.I.; Castellani, R.J.; Nunomura, A.; Perry, G. Mitochondrial DNA oxidative damage and repair in aging and Alzheimer’s disease. Antioxid. Redox Signal. 2013, 18, 2444–2457. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.; Han, D.; Sancheti, H.; Yap, L.P.; Kaplowitz, N.; Cadenas, E. Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J. Biol. Chem. 2010, 285, 39646–39654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradshaw, P.C. Cytoplasmic and mitochondrial NADPH-coupled Redox systems in the regulation of aging. Nutrients 2019, 11, 504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastre, J.; Pallardó, F.V.; Viña, J. The role of mitochondrial oxidative stress in aging. Free Radic. Biol. Med. 2003, 35, 1–8. [Google Scholar] [CrossRef]
- Suh, J.H.; Heath, S.H.; Hagen, T.M. Two subpopulations of mitochondria in the aging rat heart display heterogenous levels of oxidative stress. Free Radic. Biol. Med. 2003, 35, 1064–1072. [Google Scholar] [CrossRef] [Green Version]
- Suh, J.H.; Shenvi, S.V.; Dixon, B.M.; Liu, H.; Jaiswal, A.K.; Liu, R.M.; Hagen, T.M. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc. Natl. Acad. Sci. USA 2004, 101, 3381–3386. [Google Scholar] [CrossRef] [Green Version]
- McCarty, M.F.; DiNicolantonio, J.J. An increased need for dietary cysteine in support of glutathione synthesis may underlie the increased risk for mortality associated with low protein intake in the elderly. Age (Omaha) 2015, 37. [Google Scholar] [CrossRef] [Green Version]
- Sekhar, R.V.; Patel, S.G.; Guthikonda, A.P.; Reid, M.; Balasubramanyam, A.; Taffet, G.E.; Jahoor, F. Deficient synthesis of glutathione underlies oxidative stress in aging and can be corrected by dietary cysteine and glycine supplementation. Am. J. Clin. Nutr. 2011, 94, 847–853. [Google Scholar] [CrossRef] [Green Version]
- Bayne, A.C.V.; Mockett, R.J.; Orr, W.C.; Sohal, R.S. Enhanced catabolism of mitochondrial superoxide/hydrogen peroxide and aging in transgenic Drosophila. Biochem. J. 2005, 391, 277–284. [Google Scholar] [CrossRef]
- Herbener, G.H. A morphometric study of age-dependent changes in mitochondrial population of mouse liver and heart. J. Gerontol. 1976, 31. [Google Scholar] [CrossRef] [PubMed]
- Stocco, D.M.; Hutson, J.C. Quantitation of mitochondrial DNA and protein in the liver of Fischer 344 rats during aging. J. Gerontol. 1978, 33. [Google Scholar] [CrossRef]
- Stio, M.; Iantomasi, T.; Favilli, F.; Marraccini, P.; Lunghi, B.; Vincenzini, M.T.; Treves, C. Glutathione metabolism in heart and liver of the aging rat. Biochem. Cell Biol. 1994, 72, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Yen, T.C.; Chen, Y.S.; King, K.L.; Yeh, S.H.; Wei, Y.H. Liver mitochondrial respiratory functions decline with age. Biochem. Biophys. Res. Commun. 1989, 165, 994–1003. [Google Scholar] [CrossRef]
- Tauchi, H.; Sato, T. Age changes in size and number of mitochondria of human hepatic cells. J. Gerontol. 1968, 23. [Google Scholar] [CrossRef]
- Stocco, D.M.; Cascarano, J.; Wilson, M.A. Quantitation of mitochondrial DNA, RNA, and protein in starved and starved-refed rat liver. J. Cell. Physiol. 1977, 90, 295–306. [Google Scholar] [CrossRef]
- Silvagno, F.; Vernone, A.; Pescarmona, G.P. The Role of Glutathione in Protecting against the Severe Inflammatory Response Triggered by COVID-19. Antioxidants (Basel Switz.) 2020, 9, 624. [Google Scholar] [CrossRef]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef]
- Tadokoro, T.; Ikeda, M.; Ide, T.; Deguchi, H.; Ikeda, S.; Okabe, K.; Ishikita, A.; Matsushima, S.; Koumura, T.; Yamada, K.I.; et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Yang, S.-K.; Han, Y.-C.; He, J.-R.; Yang, M.; Zhang, W.; Zhan, M.; Li, A.-M.; Li, L.; Na-Song; Liu, Y.-T.; et al. Mitochondria targeted peptide SS-31 prevent on cisplatin-induced acute kidney injury via regulating mitochondrial ROS-NLRP3 pathway. Biomed. Pharmacother. 2020, 130, 110521. [Google Scholar] [CrossRef]
- Tutusaus, A.; Stefanovic, M.; Boix, L.; Cucarull, B.; Zamora, A.; Blasco, L.; de Frutos, P.G.; Reig, M.; Fernandez-Checa, J.C.; Marí, M.; et al. Antiapoptotic BCL-2 proteins determine sorafenib/regorafenib resistance and BH3-mimetic efficacy in hepatocellular carcinoma. Oncotarget 2018, 9, 16701–16717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cucarull, B.; Tutusaus, A.; Subías, M.; Stefanovic, M.; Hernáez-Alsina, T.; Boix, L.; Reig, M.; de Frutos, P.G.; Marí, M.; Colell, A.; et al. Regorafenib alteration of the BCL-xL/MCL-1 ratio provides a therapeutic opportunity for BH3-mimetics in hepatocellular carcinoma models. Cancers (Basel) 2020, 12, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Ahn, Y.J.; Asmis, R. Sexual dimorphism in glutathione metabolism and glutathione-dependent responses. Redox Biol. 2020, 31. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, H.; Liu, R.M. Gender difference in glutathione metabolism during aging in mice. Exp. Gerontol. 2003, 38, 507–517. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marí, M.; de Gregorio, E.; de Dios, C.; Roca-Agujetas, V.; Cucarull, B.; Tutusaus, A.; Morales, A.; Colell, A. Mitochondrial Glutathione: Recent Insights and Role in Disease. Antioxidants 2020, 9, 909. https://doi.org/10.3390/antiox9100909
Marí M, de Gregorio E, de Dios C, Roca-Agujetas V, Cucarull B, Tutusaus A, Morales A, Colell A. Mitochondrial Glutathione: Recent Insights and Role in Disease. Antioxidants. 2020; 9(10):909. https://doi.org/10.3390/antiox9100909
Chicago/Turabian StyleMarí, Montserrat, Estefanía de Gregorio, Cristina de Dios, Vicente Roca-Agujetas, Blanca Cucarull, Anna Tutusaus, Albert Morales, and Anna Colell. 2020. "Mitochondrial Glutathione: Recent Insights and Role in Disease" Antioxidants 9, no. 10: 909. https://doi.org/10.3390/antiox9100909
APA StyleMarí, M., de Gregorio, E., de Dios, C., Roca-Agujetas, V., Cucarull, B., Tutusaus, A., Morales, A., & Colell, A. (2020). Mitochondrial Glutathione: Recent Insights and Role in Disease. Antioxidants, 9(10), 909. https://doi.org/10.3390/antiox9100909