Regulation of Vascular Calcification by Reactive Oxygen Species

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
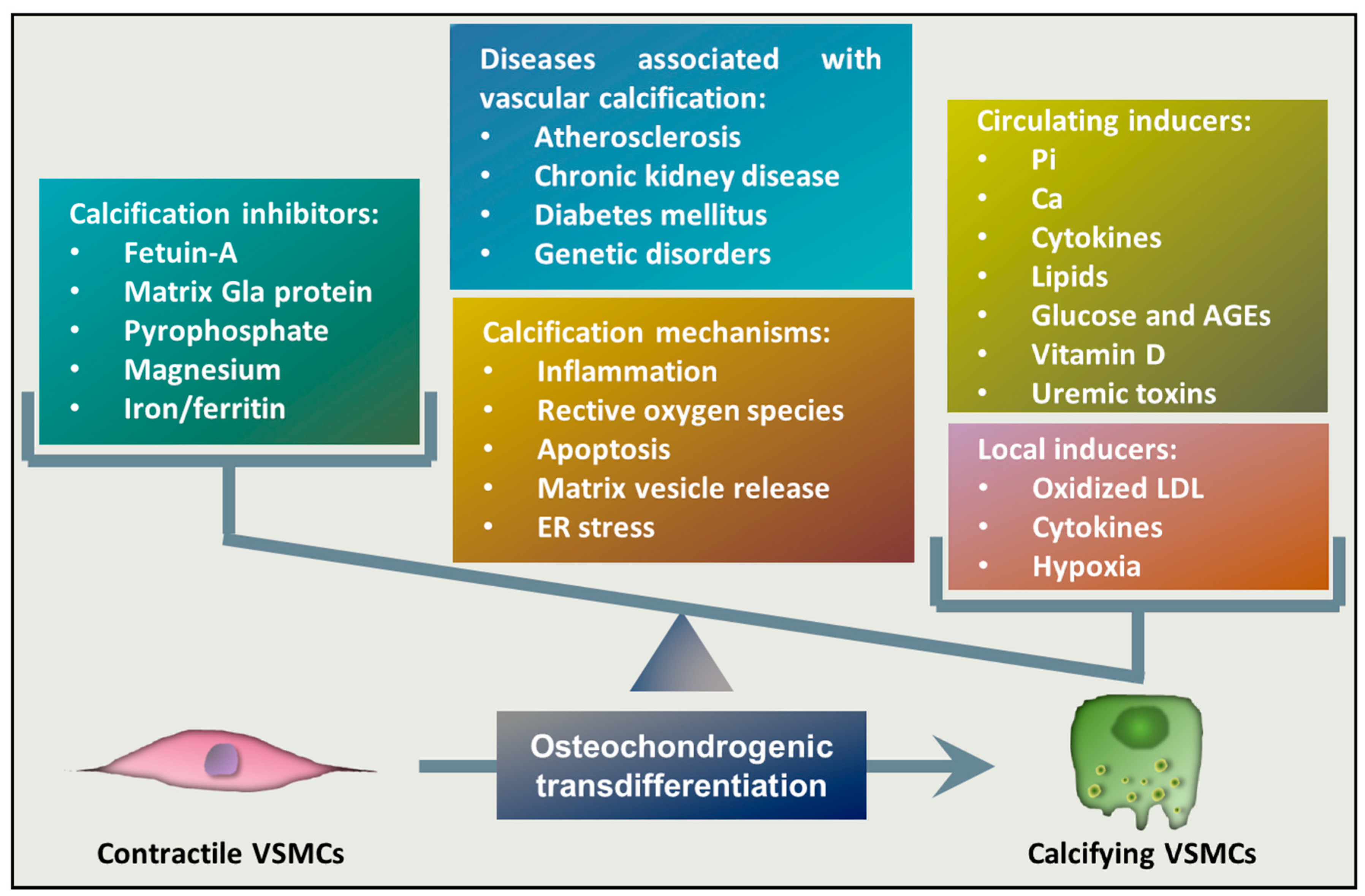
2. Vascular Calcification
3. Osteochondrogenic Transdifferentiation of VSMCs
3.1. Inducers and Inhibitors of Osteochondrogenic Transdifferentiation of VSMCs
3.2. Transcriptional Regulation of VSMCs Osteochondrogenic Transdifferentiation
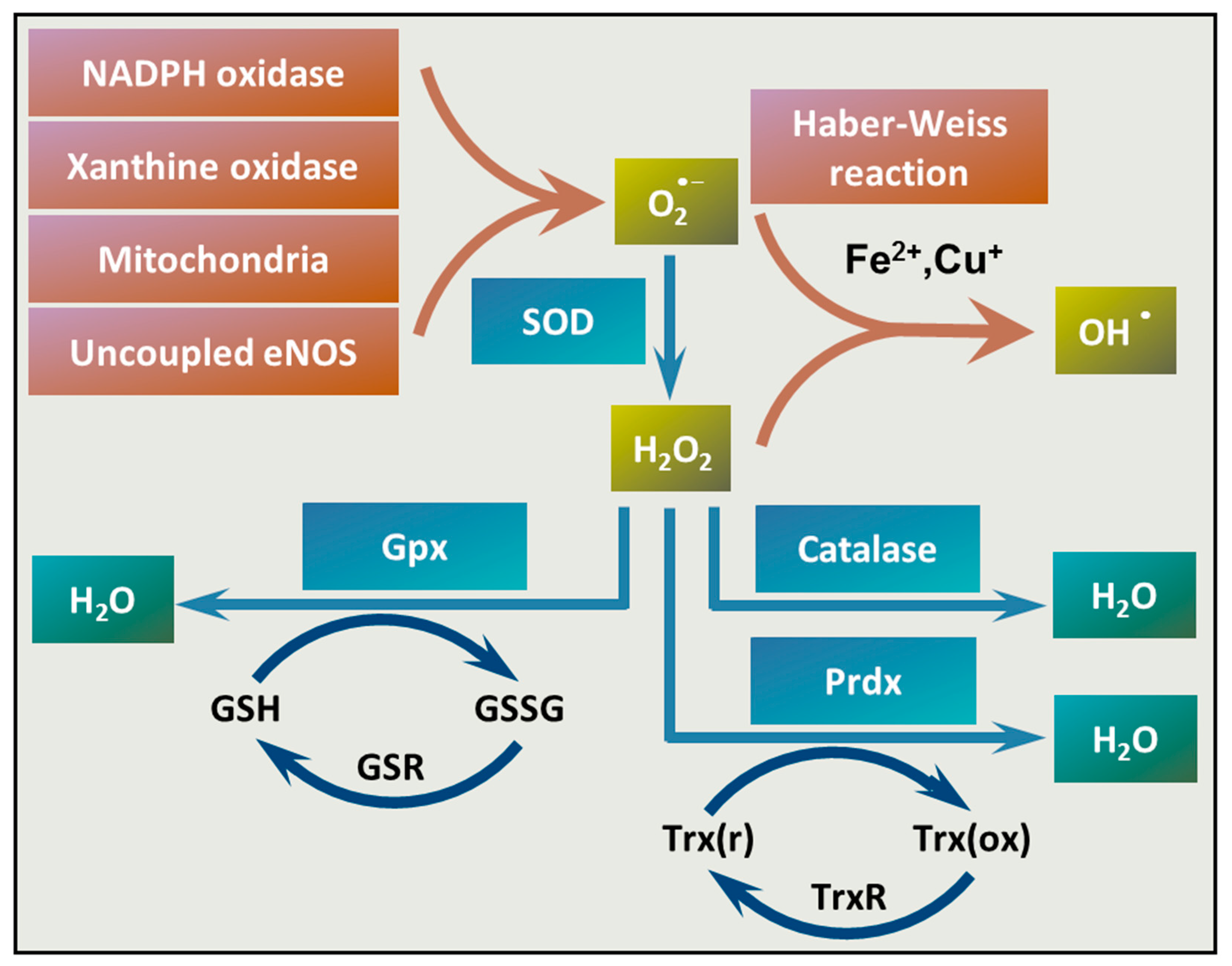
4. The Involvement of ROS in Vascular Calcification
4.1. Induction of Vascular Degeneration, Calcification and Osteochondrogenic Transdifferentiation by Excess Levels of ROS
4.2. ROS Production and Elimination in the Vasculature
4.3. Unfettered ROS Production in Vascular Calcification
5. Redox Regulation of Osteochondrogenic Signal Transduction Pathways
5.1. BMP-2/Msx2/Wnt Signaling and Oxidative Stress
5.2. Hypoxia/HIF-1 Signaling and Oxidative Stress
5.3. PERK/eIF2α/ATF4/CHOP Pathway
5.4. Nuclear Factor Kappa B (NF-κB) Pathway
5.5. Mitogen-Activated Protein Kinase (MAPK) and the PI3K/Akt Pathways
6. Controlling ROS Production as a Therapeutic Approach to Prevent Vascular Calcification
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AGE | advanced glycation end product |
| ALP | alkaline phosphatase |
| ARE | antioxidant response elements |
| α-SMA | smooth muscle α-actin |
| ATF | activating transcription factor |
| BMPs | bone morphogenetic proteins |
| CHOP | CCAAT-enhancer-binding protein homologous protein |
| CKD | chronic kidney disease |
| COPD | chronic obstructive pulmonary disease |
| eIF2α | eukaryotic initiation factor 2α |
| eNOS | endothelial NO synthase |
| ER | endoplasmic reticulum |
| Ero1 | ER oxidoreductin 1 |
| Fzd | Frizzled |
| Gpx | glutathione peroxidase |
| HIF | hypoxia inducible factor |
| HIF-1α | hypoxia inducible factor alpha subunit |
| HREs | hypoxia response elements |
| IκB | inhibitor of κB |
| IKK | IκB kinase |
| IL-1β | interleukin-1 beta |
| Keap1 | Kelch-like ECH-associated protein 1 |
| LEF | lymphoid enhancer factor |
| LPS | lipopolysaccharide |
| MAPK | mitogen-activated protein kinase |
| Msx | muscle segment homeobox homolog |
| Nox | NADPH oxidase |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| NF-κB | nuclear factor kappa B |
| OCN | osteocalcin |
| 8-OHdG | 8-Oxo-7,8-dihydro-2’-deoxyguanosine |
| oxLDL | oxidized low-density lipoprotein |
| P | phosphate |
| PERK | pancreatic ER kinase (PKR)-like ER kinase |
| PHDs | prolyl hydroxylase enzymes |
| Pi | inorganic phosphate |
| Pit1 | type III sodium-dependent phosphate cotransporter 1 |
| PPi | pyrophosphate |
| Prdx | peroxiredoxin |
| RAGE | receptor for AGEs |
| ROS | reactive oxygen species |
| SOD | superoxide dismutase |
| Sox9 | Sry-related HMG box-9 |
| TCF | T cell transcription factor |
| TGF-β | transforming growth factor beta |
| TNF-α | tumor necrosis factor alpha |
| UPR | unfolded protein response |
| VSMCs | vascular smooth muscle cells |
| Wnt | wingless/mouse mammary tumor virus integration site |
| XBP1 | X box-binding protein 1 |
References
- Paloian, N.J.; Giachelli, C.M. A current understanding of vascular calcification in CKD. Am. J. Physiol. Physiol. 2014, 307, F891–F900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial Calcification in Chronic Kidney Disease: Key Roles for Calcium and Phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanzer, P.; Boehm, M.; Sorribas, V.; Thiriet, M.; Janzen, J.; Zeller, T.; Hilaire, C.S.; Shanahan, C. Medial vascular calcification revisited: Review and perspectives. Eur. Hear. J. 2014, 35, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Demer, L.L.; Tintut, Y. Vascular Calcification. Circulation 2008, 117, 2938–2948. [Google Scholar] [CrossRef]
- Abedin, M.; Tintut, Y.; Demer, L.L. Vascular Calcification. Arter. Thromb. Vasc. Biol. 2004, 24, 1161–1170. [Google Scholar] [CrossRef]
- Yoshida, T.; Owens, G.K. Molecular Determinants of Vascular Smooth Muscle Cell Diversity. Circ. Res. 2005, 96, 280–291. [Google Scholar] [CrossRef]
- Iyemere, V.P.; Proudfoot, D.; Weissberg, P.L.; Shanahan, C.M. Vascular smooth muscle cell phenotypic plasticity and the regulation of vascular calcification. J. Intern. Med. 2006, 260, 192–210. [Google Scholar] [CrossRef]
- Alexander, M.R.; Owens, G.K. Epigenetic Control of Smooth Muscle Cell Differentiation and Phenotypic Switching in Vascular Development and Disease. Annu. Rev. Physiol. 2012, 74, 13–40. [Google Scholar] [CrossRef]
- Shanahan, C. Mechanisms of vascular calcification in renal disease. Clin. Nephrol. 2005, 63, 146–157. [Google Scholar] [CrossRef]
- Giachelli, C.M.; Jono, S.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H. Vascular calcification and inorganic phosphate. Am. J. Kidney Dis. 2001, 38, S34–S37. [Google Scholar] [CrossRef]
- Giachelli, C.M. Vascular Calcification: In Vitro Evidence for the Role of Inorganic Phosphate. J. Am. Soc. Nephrol. 2003, 14, 300–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giachelli, C. Inducers and inhibitors of biomineralization: Lessons from pathological calcification. Orthod. Craniofacial Res. 2005, 8, 229–231. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Virmani, R.; Joner, M.; Sakakura, K. Recent Highlights of ATVB: Calcification. Arter. Thromb. Vasc. Biol. 2014, 34, 1329–1332. [Google Scholar] [CrossRef] [Green Version]
- Block, G.A.; Port, F.K. Re-evaluation of risks associated with hyperphosphatemia and hyperparathyroidism in dialysis patients: Recommendations for a change in management. Am. J. Kidney Dis. 2000, 35, 1226–1237. [Google Scholar] [CrossRef]
- Rumberger, J.A.; Simons, D.B.; Fitzpatrick, L.A.; Sheedy, P.F.; Schwartz, R.S. Coronary Artery Calcium Area by Electron-Beam Computed Tomography and Coronary Atherosclerotic Plaque Area. Circulation 1995, 92, 2157–2162. [Google Scholar] [CrossRef]
- Sangiorgi, G.; A Rumberger, J.; Severson, A.; Edwards, W.D.; Gregoire, J.; A Fitzpatrick, L.; Schwartz, R.S. Arterial Calcification and Not Lumen Stenosis Is Highly Correlated With Atherosclerotic Plaque Burden in Humans: A Histologic Study of 723 Coronary Artery Segments Using Nondecalcifying Methodology. J. Am. Coll. Cardiol. 1998, 31, 126–133. [Google Scholar] [CrossRef] [Green Version]
- Beadenkopf, W.G.; Daoud, A.S.; Love, B.M. Calcification in the Coronary Arteries and Its Relationship to Arteriosclerosis and Myocardial Infarction. Am. J. Roentgenol. Radium Ther. Nucl. Med. 1964, 92, 865–871. [Google Scholar]
- Loecker, T.H.; Schwartz, R.S.; Cotta, C.W.; Hickman, J.R. Fluoroscopic coronary artery calcification and associated coronary disease in asymptomatic young men. J. Am. Coll. Cardiol. 1992, 19, 1167–1172. [Google Scholar] [CrossRef] [Green Version]
- Budoff, M.J.; Young, R.; Burke, G.L.; Carr, J.J.; Detrano, R.; Folsom, A.R.; Kronmal, R.; Lima, J.A.C.; Liu, K.; McClelland, R.L.; et al. Ten-year association of coronary artery calcium with atherosclerotic cardiovascular disease (ASCVD) events: The multi-ethnic study of atherosclerosis (MESA). Eur. Hear. J. 2018, 39, 2401–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehto, S.; Niskanen, L.; Suhonen, M.; Ronnemaa, T.; Laasko, M. Medial artery calcification. A neglected harbinger of cardiovascular complications in non-insulin-dependent diabetes mellitus. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 978–983. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.C.; Edmundowicz, D.; Becker, D.J.; Kuller, L.H.; Orchard, T.J. Coronary calcium in adults with type 1 diabetes: A stronger correlate of clinical coronary artery disease in men than in women. Diabetes 2000, 49, 1571–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Yang, L.; Gan, L.; Fan, Z.; Zhou, B.; Deng, Z.; Dey, D.; Berman, D.S.; Li, D.; Xie, Y. Spotty Calcium on Cervicocerebral Computed Tomography Angiography Associates With Increased Risk of Ischemic Stroke. Stroke 2019, 50, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, P.J.; Ports, T.A.; Yock, P.G. Contribution of localized calcium deposits to dissection after angioplasty. An observational study using intravascular ultrasound. Circulation 1992, 86, 64–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niskanen, L.; Suhonen, M.; Siitonen, O.; Lehtinen, J.; Uusitupa, M. Aortic and lower limb artery calcification in type 2 (non-insulin-dependent) diabetic patients and non-diabetic control subjects A five year follow-up study. Atherosclerosis 1990, 84, 61–71. [Google Scholar] [CrossRef]
- Burke, A.; Taylor, A.; Farb, A.; Malcolm, G.; Virmani, R. Coronary calcification: Insights from sudden coronary death victims. Z. Kardiol. 2000, 89, S049–S053. [Google Scholar] [CrossRef]
- Taylor, A.J.; Burke, A.P.; O’Malley, P.G.; Farb, A.; Malcom, G.T.; Smialek, J.; Virmani, R. A Comparison of the Framingham Risk Index, Coronary Artery Calcification, and Culprit Plaque Morphology in Sudden Cardiac Death. Circulation 2000, 101, 1243–1248. [Google Scholar] [CrossRef]
- Wu, M.; Rementer, C.; Giachelli, C.M. Vascular calcification: An update on mechanisms and challenges in treatment. Calcif. Tissue Int. 2013, 93, 365–373. [Google Scholar] [CrossRef]
- Mori, H.; Torii, S.; Kutyna, M.; Sakamoto, A.; Finn, A.V.; Virmani, R. Coronary Artery Calcification and its Progression. JACC: Cardiovasc. Imaging 2018, 11, 127–142. [Google Scholar] [CrossRef]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Hruska, K.A.; Mathew, S.; Lund, R.; Qiu, P.; Pratt, R. Hyperphosphatemia of chronic kidney disease. Kidney Int. 2008, 74, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, G.A.; Hulbert-Shearon, E.T.; Levin, N.W.; Port, F.K. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: A national study. Am. J. Kidney Dis. 1998, 31, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Kestenbaum, B.; Sampson, J.N.; Rudser, K.D.; Patterson, D.J.; Seliger, S.L.; Young, B.; Sherrard, N.J.; Andress, D.L. Serum Phosphate Levels and Mortality Risk among People with Chronic Kidney Disease. J. Am. Soc. Nephrol. 2004, 16, 520–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- London, G.M.; Guérin, A.P.; Marchais, S.J.; Métivier, F.; Pannier, B.; Adda, H. Arterial media calcification in end-stage renal disease: Impact on all-cause and cardiovascular mortality. Nephrol. Dial. Transplant. 2003, 18, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Marchais, S.J.; Metivier, F.; Guerin, A.P.; London, G.M. Association of hyperphosphataemia with haemodynamic disturbances in end-stage renal disease. Nephrol. Dial. Transplant. 1999, 14, 2178–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parhami, F.; Boström, K.; Watson, K.; Demer, L.L. Role of Molecular Regulation in Vascular Calcification. J. Atheroscler. Thromb. 1996, 3, 90–94. [Google Scholar] [CrossRef] [Green Version]
- Shroff, R.C.; Shanahan, C.M. Vascular Biology of Calcification. Semin. Dial. 2007, 20, 103–109. [Google Scholar] [CrossRef]
- Leszczynska, A.; Murphy, J.M. Vascular Calcification: Is it rather a Stem/Progenitor Cells Driven Phenomenon? Front. Bioeng. Biotechnol. 2018, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Shigematsu, T.; Kono, T.; Satoh, K.; Yokoyama, K.; Yoshida, T.; Hosoya, T.; Shirai, K. Phosphate overload accelerates vascular calcium deposition in end-stage renal disease patients. Nephrol. Dial. Transplant. 2003, 18, 86iii–89. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, J.L.; Joannides, A.J.; Skepper, J.N.; McNair, R.; Schurgers, L.J.; Proudfoot, D.; Jahnen-Dechent, W.; Weissberg, P.L.; Shanahan, C.M. Human Vascular Smooth Muscle Cells Undergo Vesicle-Mediated Calcification in Response to Changes in Extracellular Calcium and Phosphate Concentrations: A Potential Mechanism for Accelerated Vascular Calcification in ESRD. J. Am. Soc. Nephrol. 2004, 15, 2857–2867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jono, S.; McKee, M.D.; E Murry, C.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H.; Giachelli, C.M. Phosphate regulation of vascular smooth muscle cell calcification. Circ. Res. 2000, 87, e10–e17. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Byon, C.H.; Yang, Y.; Bradley, W.E.; Dell’Italia, L.J.; Sanders, P.W.; Agarwal, A.; Wu, H.; Chen, Y. Dietary potassium regulates vascular calcification and arterial stiffness. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazono, K.; Kamiya, Y.; Morikawa, M. Bone morphogenetic protein receptors and signal transduction. J. Biochem. 2009, 147, 35–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, J.; Pardali, E.; Sanchez-Duffhues, G.; Dijke, P.T. BMP signaling in vascular diseases. FEBS Lett. 2012, 586, 1993–2002. [Google Scholar] [CrossRef]
- Li, X.; Yang, H.-Y.; Giachelli, C.M. BMP-2 promotes phosphate uptake, phenotypic modulation, and calcification of human vascular smooth muscle cells. Atherosclerosis 2008, 199, 271–277. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.X.; Moe, S.M. Arterial calcification in diabetes. Curr. Diabetes Rep. 2003, 3, 28–32. [Google Scholar] [CrossRef]
- Chen, N.X.; Duan, D.; O’Neill, K.D.; Moe, S.M. High glucose increases the expression of Cbfa1 and BMP-2 and enhances the calcification of vascular smooth muscle cells. Nephrol. Dial. Transplant. 2006, 21, 3435–3442. [Google Scholar] [CrossRef] [Green Version]
- Tanikawa, T.; Okada, Y.; Tanikawa, R.; Tanaka, Y. Advanced Glycation End Products Induce Calcification of Vascular Smooth Muscle Cells through RAGE/p38 MAPK. J. Vasc. Res. 2009, 46, 572–580. [Google Scholar] [CrossRef]
- Mori, K.; Shioi, A.; Jono, S.; Nishizawa, Y.; Morii, H. Dexamethasone enhances In vitro vascular calcification by promoting osteoblastic differentiation of vascular smooth muscle cells. Arter. Thromb. Vasc. Biol. 1999, 19, 2112–2118. [Google Scholar] [CrossRef] [Green Version]
- Jono, S.; Nishizawa, Y.; Shioi, A.; Morii, H. 1,25-Dihydroxyvitamin D3 increases in vitro vascular calcification by modulating secretion of endogenous parathyroid hormone-related peptide. Circulation 1998, 98, 1302–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balica, M.; Bostrom, K.; Shin, V.; Tillisch, K.; Demer, L.L. Calcifying subpopulation of bovine aortic smooth muscle cells is responsive to (17)beta-estradiol. Circulation 1997, 95, 1954–1960. [Google Scholar] [CrossRef] [PubMed]
- Shioi, A.; Katagi, M.; Okuno, Y.; Mori, K.; Jono, S.; Koyama, H.; Nishizawa, Y. Induction of bone-type alkaline phosphatase in human vascular smooth muscle cells: Roles of tumor necrosis factor-alpha and oncostatin M derived from macrophages. Circ. Res. 2002, 91, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aikawa, E.; Nahrendorf, M.; Figueiredo, J.-L.; Swirski, F.K.; Shtatland, T.; Kohler, R.H.; Jaffer, F.A.; Aikawa, M.; Weissleder, R. Osteogenesis Associates With Inflammation in Early-Stage Atherosclerosis Evaluated by Molecular Imaging In Vivo. Circulation 2007, 116, 2841–2850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceneri, N.; Zhao, L.; Young, B.D.; Healy, A.; Coskun, S.; Vasavada, H.; Yarovinsky, T.O.; Ike, K.; Pardi, R.; Qin, L.; et al. Rac2 Modulates Atherosclerotic Calcification by Regulating Macrophage Interleukin-1 beta Production. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awan, A.Z.; Denis, M.; Roubtsova, A.; Essalmani, R.; Marcinkiewicz, J.; Awan, A.; Gram, H.; Seidah, N.G.; Genest, J. Reducing Vascular Calcification by Anti-IL-1? Monoclonal Antibody in a Mouse Model of Familial Hypercholesterolemia. Angiology 2015, 67, 157–167. [Google Scholar] [CrossRef]
- Tintut, Y.; Patel, J.; Territo, M.; Saini, T.; Parhami, F.; Demer, L.L. Monocyte/Macrophage Regulation of Vascular Calcification In Vitro. Circulation 2002, 105, 650–655. [Google Scholar] [CrossRef]
- Zickler, D.; Luecht, C.; Willy, K.; Chen, L.; Witowski, W.; Girndt, M.; Fiedler, R.; Storr, M.; Kamhieh-Milz, J.; Schoon, J.; et al. Tumour necrosis factor-alpha in uraemic serum promotes osteoblastic transition and calcification of vascular smooth muscle cells via extracellular signal-regulated kinases and activator protein 1/c-FOS-mediated induction of interleukin 6 expression. Nephrol Dial Transpl. 2017, 33, 574–585. [Google Scholar] [CrossRef]
- Parhami, F.; Morrow, A.D.; Balucan, J.; Leitinger, N.; Watson, A.D.; Tintut, Y.; Berliner, J.A.; Demer, L.L. Lipid oxidation products have opposite effects on calcifying vascular cell and bone cell differentiation—A possible explanation for the paradox of arterial calcification in osteoporotic patients. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 680–687. [Google Scholar] [CrossRef]
- Bear, M.; Butcher, M.; Shaughnessy, S.G. Oxidized low-density lipoprotein acts synergistically with beta-glycerophosphate to induce osteoblast differentiation in primary cultures of vascular smooth muscle cells. J. Cell. Biochem. 2008, 105, 185–193. [Google Scholar] [CrossRef]
- Mokas, S.; Larivière, R.; Lamalice, L.; Gobeil, S.; Cornfield, D.N.; Agharazii, M.; Richard, D.E. Hypoxia-inducible factor-1 plays a role in phosphate-induced vascular smooth muscle cell calcification. Kidney Int. 2016, 90, 598–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balogh, E.; Tóth, A.; Méhes, G.; Trencsényi, G.; Paragh, G.; Jeney, V. Hypoxia Triggers Osteochondrogenic Differentiation of Vascular Smooth Muscle Cells in an HIF-1 (Hypoxia-Inducible Factor 1)-Dependent and Reactive Oxygen Species-Dependent Manner. Arter. Thromb. Vasc. Biol. 2019, 39, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Ducy, P.; McKee, M.D.; Pinero, G.J.; Loyer, E.; Behringer, R.R.; Karsenty, G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nat. Cell Biol. 1997, 386, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Schinke, T.; Amendt, C.; Trindl, A.; Poeschke, O.; Muller, W.; Jahnen, W. The serum protein alpha(2)-HS glycoprotein/fetuin inhibits apatite formation in vitro and in mineralizing calvaria cells—A possible role in mineralization and calcium homeostasis. J. Biol. Chem. 1996, 271, 20789–20796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, J.J.; Zhu, D.; Opdebeeck, B.; D’Haese, P.; Millán, J.L.; Bourne, L.E.; Wheeler-Jones, C.P.D.; Arnett, T.R.; Macrae, V.E.; Orriss, I.R. Inhibition of arterial medial calcification and bone mineralization by extracellular nucleotides: The same functional effect mediated by different cellular mechanisms. J. Cell. Physiol. 2017, 233, 3230–3243. [Google Scholar] [CrossRef] [PubMed]
- Orriss, I.R.; Arnett, T.R.; Russell, R.G. Pyrophosphate: A key inhibitor of mineralisation. Curr. Opin. Pharmacol. 2016, 28, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ter Braake, A.D.; Shanahan, C.M.; De Baaij, J.H.F. Magnesium Counteracts Vascular Calcification Passive Interference or Active Modulation? Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1431–1445. [Google Scholar] [CrossRef] [Green Version]
- Zarjou, A.; Jeney, V.; Arosio, P.; Poli, M.; Antal-Szalmás, P.; Agarwal, A.; Balla, G.; Balla, J. Ferritin prevents calcification and osteoblastic differentiation of vascular smooth muscle cells. J. Am. Soc. Nephrol. 2009, 20, 1254–1263. [Google Scholar] [CrossRef] [Green Version]
- Matsubara, T.; Kida, K.; Yamaguchi, A.; Hata, K.; Ichida, F.; Meguro, H.; Aburatani, H.; Nishimura, R.; Yoneda, T. BMP2 Regulates Osterix through Msx2 and Runx2 during Osteoblast Differentiation. J. Biol. Chem. 2008, 283, 29119–29125. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, V.; Bryant, H.U.; MacDougald, O.A. Regulation of bone mass by Wnt signaling. J. Clin. Investig. 2006, 116, 1202–1209. [Google Scholar] [CrossRef]
- Noda, M.; Rodan, G.A. Type beta transforming growth factor (TGF beta) regulation of alkaline phosphatase expression and other phenotype-related mRNAs in osteoblastic rat osteosarcoma cells. J. Cell. Physiol. 1987, 133, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Byon, C.H.; Yuan, K.; Chen, J.; Mao, X.; Heath, J.M.; Javed, A.; Zhang, K.; Anderson, P.G.; Chen, Y. Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circ. Res. 2012, 111, 543–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steitz, S.A.; Speer, M.Y.; Curinga, G.; Yang, H.-Y.; Haynes, P.A.; Aebersold, R.; Schinke, T.; Karsenty, G.; Giachelli, C.M. Smooth Muscle Cell Phenotypic Transition Associated With Calcification. Circ. Res. 2001, 89, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Tyson, K.L.; Reynolds, J.L.; McNair, R.; Zhang, Q.; Weissberg, P.L.; Shanahan, C.M. Osteo/Chondrocytic Transcription Factors and Their Target Genes Exhibit Distinct Patterns of Expression in Human Arterial Calcification. Arter. Thromb. Vasc. Biol. 2003, 23, 489–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neven, E.; Dauwe, S.; De Broe, M.; Haese, P.C.D.; Persy, V.P. Endochondral bone formation is involved in media calcification in rats and in men. Kidney Int. 2007, 72, 574–581. [Google Scholar] [CrossRef] [Green Version]
- Moe, S.M.; Duan, D.; Doehle, B.P.; O’Neill, K.D.; Chen, N.X. Uremia induces the osteoblast differentiation factor Cbfa1 in human blood vessels. Kidney Int. 2003, 63, 1003–1011. [Google Scholar] [CrossRef] [Green Version]
- Minol, J.-P.; Reinsch, I.; Luik, M.; Leferink, A.; Barth, M.; Assmann, A.; Lichtenberg, A.; Akhyari, P. Focal induction of ROS-release to trigger local vascular degeneration. PLoS ONE 2017, 12, e0179342. [Google Scholar] [CrossRef] [Green Version]
- Münzel, T.; Camici, G.G.; Maack, C.; Bonetti, N.R.; Fuster, V.; Kovacic, J.C. Impact of Oxidative Stress on the Heart and Vasculature. J. Am. Coll. Cardiol. 2017, 70, 212–229. [Google Scholar] [CrossRef]
- Lassègue, B.; Griendling, K.K. NADPH oxidases: Functions and pathologies in the vasculature. Arter. Thromb. Vasc. Biol. 2009, 30, 653–661. [Google Scholar] [CrossRef]
- Byon, C.H.; Heath, J.M.; Chen, Y. Redox signaling in cardiovascular pathophysiology: A focus on hydrogen peroxide and vascular smooth muscle cells. Redox Biol. 2016, 9, 244–253. [Google Scholar] [CrossRef] [Green Version]
- Agharazii, M.; St-Louis, R.; Gautier-Bastien, A.; Ung, R.-V.; Mokas, S.; Larivière, R.; Richard, D.E. Inflammatory Cytokines and Reactive Oxygen Species as Mediators of Chronic Kidney Disease-Related Vascular Calcification. Am. J. Hypertens. 2014, 28, 746–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberman, M.; Bassi, E.; Martinatti, M.K.; Lario, F.C.; Wosniak, J.; Pomerantzeff, P.M.; Laurindo, F.R.; Lario, F.C. Oxidant Generation Predominates Around Calcifying Foci and Enhances Progression of Aortic Valve Calcification. Arter. Thromb. Vasc. Biol. 2008, 28, 463–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mody, N. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free. Radic. Biol. Med. 2001, 31, 509–519. [Google Scholar] [CrossRef]
- Byon, C.H.; Javed, A.; Dai, Q.; Kappes, J.C.; Clemens, T.L.; Darley-Usmar, V.M.; McDonald, J.M.; Chen, Y. Oxidative Stress Induces Vascular Calcification through Modulation of the Osteogenic Transcription Factor Runx2 by AKT Signaling. J. Biol. Chem. 2008, 283, 15319–15327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alesutan, I.; Feger, M.; Tuffaha, R.; Castor, T.; Musculus, K.; Buehling, H.H.; Heine, C.L.; Kuro-O, M.; Pieske, B.; Schmidt, K.; et al. Augmentation of phosphate-induced osteo-/chondrogenic transformation of vascular smooth muscle cells by homoarginine. Cardiovasc. Res. 2016, 110, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Tada, Y.; Yano, S.; Yamaguchi, T.; Okazaki, K.; Ogawa, N.; Morita, M.; Sugimoto, T. Advanced Glycation End Products-induced Vascular Calcification is Mediated by Oxidative Stress: Functional Roles of NAD(P)H-oxidase. Horm. Metab. Res. 2012, 45, 267–272. [Google Scholar] [CrossRef]
- Wei, Q.; Ren, X.; Jiang, Y.; Jin, H.; Liu, N.-F.; Li, J. Advanced glycation end products accelerate rat vascular calcification through RAGE/oxidative stress. BMC Cardiovasc. Disord. 2013, 13, 13. [Google Scholar] [CrossRef] [Green Version]
- Gawdzik, J.; Mathew, L.; Kim, G.H.; Puri, T.S.; Bowman, M.A.H. Vascular remodeling and arterial calcification are directly mediated by S100A12 (EN-RAGE) in chronic kidney disease. Am. J. Nephrol. 2011, 33, 250–259. [Google Scholar] [CrossRef] [Green Version]
- Liberman, M.; Johnson, R.C.; Handy, D.E.; Loscalzo, J.; Leopold, J.A. Bone morphogenetic protein-2 activates NADPH oxidase to increase endoplasmic reticulum stress and human coronary artery smooth muscle cell calcification. Biochem. Biophys. Res. Commun. 2011, 413, 436–441. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.X.; O’Neill, K.D.; Moe, S.M. Matrix vesicles induce calcification of recipient vascular smooth muscle cells through multiple signaling pathways. Kidney Int. 2018, 93, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Wuthier, R.E.; Lipscomb, G.F. Matrix vesicles: Structure, composition, formation and function in calcification. Front. Biosci. 2011, 16, 2812–2902. [Google Scholar] [CrossRef] [PubMed]
- Furmanik, M.; Chatrou, M.; Van Gorp, R.H.; Akbulut, A.; Willems, B.; Schmidt, H.H.; Van Eys, G.; Bochaton-Piallat, M.-L.; Proudfoot, D.; Al Biessen, E.; et al. Reactive Oxygen-Forming Nox5 Links Vascular Smooth Muscle Cell Phenotypic Switching and Extracellular Vesicle-Mediated Vascular Calcification. Circ. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.-M.; Xu, M.-J.; Cai, Y.; Zhao, G.; Guan, Y.; Kong, W.; Tang, C.; Wang, X. Mitochondrial reactive oxygen species promote p65 nuclear translocation mediating high-phosphate-induced vascular calcification in vitro and in vivo. Kidney Int. 2011, 79, 1071–1079. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Li, Z.; Chang, X.; Cong, G.; Hao, L. Quercetin attenuates vascular calcification by inhibiting oxidative stress and mitochondrial fission. Vasc. Pharmacol. 2017, 88, 21–29. [Google Scholar] [CrossRef]
- Green, F.H.Y.; Butt, J.C.; James, A.L.; Carroll, N.G. Abnormalities of the Bronchial Arteries in Asthma. Chest 2006, 130, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Murchison, J.T.; Edwards, L.; Agustí, A.; Bakke, P.; A Calverley, P.M.; Celli, B.; O Coxson, H.; Crim, C.; Lomas, D.A.; et al. Coronary artery calcification is increased in patients with COPD and associated with increased morbidity and mortality. Thorax 2014, 69, 718–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tachikawa, R.; Koyasu, S.; Matsumoto, T.; Hamada, S.; Azuma, M.; Murase, K.; Tanizawa, K.; Inouchi, M.; Oga, T.; Mishima, M.; et al. Obstructive sleep apnea and abdominal aortic calcification: Is there an association independent of comorbid risk factors? Atherosclerosis 2015, 241, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Paddenberg, R.; Ishaq, B.; Goldenberg, A.; Faulhammer, P.; Rose, F.; Weissmann, N.; Braun-Dullaeus, R.C.; Kummer, W. Essential role of complex II of the respiratory chain in hypoxia-induced ROS generation in the pulmonary vasculature. Am. J. Physiol. Cell. Mol. Physiol. 2003, 284, L710–L719. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1 alpha during hypoxia—A mechanism of O-2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [Green Version]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr. Opin. Cell Biol. 2009, 21, 894–899. [Google Scholar] [CrossRef] [Green Version]
- Waypa, G.B.; Smith, K.A.; Schumacker, P.T. O2 sensing, mitochondria and ROS signaling: The fog is lifting. Mol. Asp. Med. 2016, 47–48, 76–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, H.; Kanai, H.; Uchiyama, T.; Iso, T.; Ohyama, Y.; Sakamoto, H.; Tamura, J.; Nagai, R.; Kurabayashi, M. Mitochondrial reactive oxygen species and c-Src play a critical role in hypoxic response in vascular smooth muscle cells. Cardiovasc. Res. 2005, 67, 714–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Chen, G.; Li, Y.-P. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016, 4, 16009. [Google Scholar] [CrossRef] [PubMed]
- Hruska, K.A.; Mathew, S.; Saab, G. Bone Morphogenetic Proteins in Vascular Calcification. Circ. Res. 2005, 97, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Boström, K.; E Watson, K.; Horn, S.; Wortham, C.; Herman, I.M.; Demer, L.L. Bone morphogenetic protein expression in human atherosclerotic lesions. J. Clin. Investig. 1993, 91, 1800–1809. [Google Scholar] [CrossRef]
- Dhore, C.R.; Cleutjens, J.P.; Lutgens, E.; Cleutjens, K.B.; Geusens, P.P.; Kitslaar, P.J.; Tordoir, J.H.; Spronk, H.M.; Vermeer, C.; Daemen, M.J. Differential Expression of Bone Matrix Regulatory Proteins in Human Atherosclerotic Plaques. Arter. Thromb. Vasc. Biol. 2001, 21, 1998–2003. [Google Scholar] [CrossRef] [Green Version]
- Sirard, C.; Kim, S.; Mirtsos, C.; Tadich, P.; Hoddless, P.A.; Itie, A.; Maxson, R.; Wrana, J.L.; Mak, T.W. Targeted disruption in murine cells reveals variable requirement for Smad4 in transforming growth factor beta-related signaling. J Biol Chem 2000, 275, 2063–2070. [Google Scholar] [CrossRef] [Green Version]
- Dalfino, G.; Simone, S.; Porreca, S.; Cosola, C.; Balestra, C.; Manno, C.; Schena, F.P.; Grandaliano, G.; Pertosa, G. Bone morphogenetic protein-2 may represent the molecular link between oxidative stress and vascular stiffness in chronic kidney disease. Atherosclerosis 2010, 211, 418–423. [Google Scholar] [CrossRef]
- Csiszar, A.; Smith, K.E.; Koller, A.; Kaley, G.; Edwards, J.G.; Ungvari, Z. Regulation of Bone Morphogenetic Protein-2 Expression in Endothelial Cells. Circulation 2005, 111, 2364–2372. [Google Scholar] [CrossRef] [Green Version]
- Mandal, C.C.; Ganapathy, S.; Gorin, Y.; Mahadev, K.; Block, K.; Abboud, H.E.; Harris, S.E.; Ghosh-Choudhury, G.; Ghosh-Choudhury, N. Reactive oxygen species derived from Nox4 mediate BMP2 gene transcription and osteoblast differentiation. Biochem. J. 2010, 433, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.-L.; Shao, J.-S.; Charlton-Kachigian, N.; Loewy, A.P.; Towler, D.A. Msx2 Promotes Osteogenesis and Suppresses Adipogenic Differentiation of Multipotent Mesenchymal Progenitors. J. Biol. Chem. 2003, 278, 45969–45977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, J.-S.; Cheng, S.-L.; Pingsterhaus, J.M.; Charlton-Kachigian, N.; Loewy, A.P.; Towler, D.A. Msx2 promotes cardiovascular calcification by activating paracrine Wnt signals. J. Clin. Investig. 2005, 115, 1210–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackall, C.L.; Nusse, R. Towards an integrated view of Wnt signaling in development. Dev. 2009, 136, 3205–3214. [Google Scholar] [CrossRef] [Green Version]
- De, A. Wnt/Ca2+ signaling pathway: A brief overview. Acta Biochim. Biophys. Sin. 2011, 43, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Sun, Y.; Sun, W.; Xu, T.; Ren, C.; Fan, X.; Sun, L.; Liu, L.; Feng, J.; Ma, J.; et al. High phosphorus level leads to aortic calcification via beta-catenin in chronic kidney disease. Am. J. Nephrol. 2015, 41, 28–36. [Google Scholar] [CrossRef]
- Cai, T.; Sun, D.; Duan, Y.; Wen, P.; Dai, C.; Yang, J.; He, W. WNT/beta-catenin signaling promotes VSMCs to osteogenic transdifferentiation and calcification through directly modulating Runx2 gene expression. Exp. Cell Res. 2016, 345, 206–217. [Google Scholar] [CrossRef] [Green Version]
- Deng, D.; Diao, Z.; Han, X.; Liu, W. Secreted Frizzled-Related Protein 5 Attenuates High Phosphate-Induced Calcification in Vascular Smooth Muscle Cells by Inhibiting the Wnt/ß-Catenin Pathway. Calcif. Tissue Int. 2016, 99, 66–75. [Google Scholar] [CrossRef]
- Caliceti, C.; Nigro, P.; Rizzo, P.; Ferrari, R. ROS, Notch, and Wnt Signaling Pathways: Crosstalk between Three Major Regulators of Cardiovascular Biology. BioMed Res. Int. 2014, 2014, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen Sensing, Homeostasis, and Disease. N. Engl. J. Med. 2011, 365, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. HIF-1 and mechanisms of hypoxia sensing. Curr. Opin. Cell Biol. 2001, 13, 167–171. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Hypoxia-Inducible Factor 1 (HIF-1) Pathway. Sci. STKE 2007, 2007, cm8. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, D.C.; Brüne, B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. 2017, 12, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cash, T.P.; Pan, Y.; Simon, M.C. Reactive oxygen species and cellular oxygen sensing. Free. Radic. Biol. Med. 2007, 43, 1219–1225. [Google Scholar] [CrossRef] [Green Version]
- Hielscher, A.; Gerecht, S. Hypoxia and free radicals: Role in tumor progression and the use of engineering-based platforms to address these relationships. Free. Radic. Biol. Med. 2015, 79, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q.; Colgan, S.P.; Shelley, C.S. Hypoxia: The Force that Drives Chronic Kidney Disease. Clin. Med. Res. 2016, 14, 15–39. [Google Scholar] [CrossRef] [Green Version]
- Movafagh, S.; Raj, D.; Sanaei-Ardekani, M.; Bhatia, D.; Vo, K.; Mahmoudieh, M.; Rahman, R.; Kim, E.; Harralson, A. Hypoxia Inducible Factor 1: A Urinary Biomarker of Kidney Disease. Clin. Transl. Sci. 2017, 10, 201–207. [Google Scholar] [CrossRef]
- Liu, J.; Wei, Q.; Guo, C.; Dong, G.; Liu, Y.; Tang, C.; Dong, Z. Hypoxia, HIF, and Associated Signaling Networks in Chronic Kidney Disease. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Zemplényi, T.; Crawford, D.W.; Cole, M.A. Adaptation to arterial wall hypoxia demonstrated in vivo with oxygen microcathodes. Atherosclerosis 1989, 76, 173–179. [Google Scholar] [CrossRef]
- Jurrus, E.; Weiss, H. In vitro tissue oxygen tensions in the rabbit aortic arch. Atherosclerosis 1977, 28, 223–232. [Google Scholar] [CrossRef]
- Crawford, D.; Blankenhorn, D. Arterial wall oxygenation, oxyradicals, and atherosclerosis. Atherosclerosis 1991, 89, 97–108. [Google Scholar] [CrossRef]
- Björnheden, T.; Levin, M.; Evaldsson, M.; Wiklund, O. Evidence of Hypoxic Areas Within the Arterial Wall In Vivo. Arter. Thromb. Vasc. Biol. 1999, 19, 870–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sluimer, J.C.; Gasc, J.-M.; Van Wanroij, J.L.; Kisters, N.; Groeneweg, M.; Gelpke, M.D.S.; Cleutjens, J.P.; Akker, L.H.V.D.; Corvol, P.; Wouters, B.G.; et al. Hypoxia, Hypoxia-Inducible Transcription Factor, and Macrophages in Human Atherosclerotic Plaques Are Correlated With Intraplaque Angiogenesis. J. Am. Coll. Cardiol. 2008, 51, 1258–1265. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, E.R.; Garvin, M.R.; Dev, R.; Stewart, D.K.; Hinohara, T.; Simpson, J.B.; Schwartz, S.M. Angiogenesis in human coronary atherosclerotic plaques. Am. J. Pathol. 1994, 145, 883–894. [Google Scholar]
- Jeziorska, M.; Woolley, D.E. Neovascularization in early atherosclerotic lesions of human carotid arteries: Its potential contribution to plaque development. Hum. Pathol. 1999, 30, 919–925. [Google Scholar] [CrossRef]
- Geiringer, E. Intimal vascularization and atherosclerosis. J. Pathol. Bacteriol. 1951, 63, 201–211. [Google Scholar] [CrossRef]
- Moulton, K.S.; Heller, E.; Konerding, M.A.; Flynn, E.; Palinski, W.; Folkman, J. Angiogenesis Inhibitors Endostatin or TNP-470 Reduce Intimal Neovascularization and Plaque Growth in Apolipoprotein E–Deficient Mice. Circulation 1999, 99, 1726–1732. [Google Scholar] [CrossRef]
- Filho, I.P.T.; Leunig, M.; Yuan, F.; Intaglietta, M.; Jain, R.K. Noninvasive measurement of microvascular and interstitial oxygen profiles in a human tumor in SCID mice. Proc. Natl. Acad. Sci. USA 1994, 91, 2081–2085. [Google Scholar] [CrossRef] [Green Version]
- Nakano, D.; Hayashi, T.; Tazawa, N.; Yamashita, C.; Inamoto, S.; Okuda, N.; Mori, T.; Sohmiya, K.; Kitaura, Y.; Okada, Y.; et al. Chronic Hypoxia Accelerates the Progression of Atherosclerosis in Apolipoprotein E-Knockout Mice. Hypertens. Res. 2005, 28, 837–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, J.C.; Reinke, C.; Bedja, D.; Berkowitz, D.; Bevans-Fonti, S.; Li, J.; Barouch, L.A.; Gabrielson, K.; Polotsky, V.Y. Effect of intermittent hypoxia on atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis 2010, 209, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Folco, E. Tension in the Plaque: Hypoxia Modulates Metabolism in Atheroma. Circ. Res. 2011, 109, 1100–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Lu, W.-H.; Ai, R.; Yang, J.-H.; Chen, F.; Zhong-Zhi, T. The relationship between serum hypoxia-inducible factor 1α and coronary artery calcification in asymptomatic type 2 diabetic patients. Cardiovasc. Diabetol. 2014, 13, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrotta, I.; Moraca, F.M.; Sciangula, A.; Aquila, S.; Mazzulla, S. HIF-1α and VEGF: Immunohistochemical Profile and Possible Function in Human Aortic Valve Stenosis. Ultrastruct. Pathol. 2015, 39, 198–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffenach, G.; Chabot, S.; Tanguay, V.F.; Courboulin, A.; Boucherat, O.; Potus, F.; Meloche, J.; Pflieger, A.; Breuils-Bonnet, S.; Nadeau, V.; et al. Role for Runt-related Transcription Factor 2 in Proliferative and Calcified Vascular Lesions in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2016, 194, 1273–1285. [Google Scholar] [CrossRef] [PubMed]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A Review in the Theme: Cellular Mechanisms of Endoplasmic Reticulum Stress Signaling in Health and Disease. Am. J. Physiol. Physiol. 2015, 308, C415–C425. [Google Scholar] [CrossRef] [Green Version]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [Green Version]
- Oakes, S.A.; Papa, F.R. The Role of Endoplasmic Reticulum Stress in Human Pathology. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 173–194. [Google Scholar] [CrossRef] [Green Version]
- Shanahan, C.M. Endoplasmic Reticulum Stress in Arterial Smooth Muscle Cells: A Novel Regulator of Vascular Disease. Curr. Cardiol. Rev. 2017, 13, 94–105. [Google Scholar] [CrossRef]
- Duan, X.; Zhou, Y.; Teng, X.; Tang, C.; Qi, Y.-F. Endoplasmic reticulum stress-mediated apoptosis is activated in vascular calcification. Biochem. Biophys. Res. Commun. 2009, 387, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.-H.; Chang, J.-R.; Zhang, J.; Zhang, B.-H.; Li, Y.; Teng, X.; Zhu, Y.; Du, J.; Tang, C.-S.; Qi, Y.-F. Activating transcription factor 4 is involved in endoplasmic reticulum stress-mediated apoptosis contributing to vascular calcification. Apoptosis 2013, 18, 1132–1144. [Google Scholar] [CrossRef] [PubMed]
- Masuda, M.; Ting, T.; Levi, M.; Saunders, S.J.; Miyazaki-Anzai, S.; Miyazaki, M. Activating transcription factor 4 regulates stearate-induced vascular calcification. J. Lipid Res. 2012, 53, 1543–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, M.; Miyazaki-Anzai, S.; Levi, M.; Ting, T.C.; Miyazaki, M. PERK-eIF2 alpha-ATF4-CHOP Signaling Contributes to TNF alpha-Induced Vascular Calcification. J. Am. Heart Association 2013, 2, e000238. [Google Scholar] [CrossRef] [Green Version]
- Zeeshan, H.M.A.; Lee, G.-H.; Kim, H.-R.; Chae, H.-J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [Green Version]
- Dandekar, A.; Mendez, R.; Zhang, K. Cross Talk Between ER Stress, Oxidative Stress, and Inflammation in Health and Disease. Immunosurveill. Immunodefic. Lymphoproliferations 2015, 1292, 205–214. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Miao, H.; Zhang, K.; Wolfson, A.; Pennathur, S.; Pipe, S.W.; Kaufman, R.J. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. USA 2008, 105, 18525–18530. [Google Scholar] [CrossRef] [Green Version]
- Santos, C.X.C.; Tanaka, L.Y.; Wosniak, J.; Laurindo, F.R. Mechanisms and Implications of Reactive Oxygen Species Generation During the Unfolded Protein Response: Roles of Endoplasmic Reticulum Oxidoreductases, Mitochondrial Electron Transport, and NADPH Oxidase. Antioxidants Redox Signal. 2009, 11, 2409–2427. [Google Scholar] [CrossRef]
- Gross, E.; Sevier, C.S.; Heldman, N.; Vitu, E.; Bentzur, M.; Kaiser, C.A.; Thorpe, C.; Fass, D. Generating disulfides enzymatically: Reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc. Natl. Acad. Sci. USA 2006, 103, 299–304. [Google Scholar] [CrossRef] [Green Version]
- Laurindo, F.R.; Araujo, T.L.; Abrahão, T.B. Nox NADPH Oxidases and the Endoplasmic Reticulum. Antioxidants Redox Signal. 2014, 20, 2755–2775. [Google Scholar] [CrossRef] [Green Version]
- Sen, R.; Baltimore, D. Inducibility of kappa immunoglobulin enhancer-binding protein Nf-kappa B by a posttranslational mechanism. Cell 1986, 47, 921–928. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Signaling to NF-kappaB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, M.; Lin, A. NF-kappa B at the crossroads of life and death. Nat. Immunol. 2002, 3, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Karin, M. Missing pieces in the NF-kappaB puzzle. Cell 2002, 109, S81–S96. [Google Scholar] [CrossRef] [Green Version]
- Napetschnig, J.; Wu, H. Molecular basis of NF-kappaB signaling. Annu. Rev. Biophys. 2013, 42, 443–468. [Google Scholar] [CrossRef] [Green Version]
- Xanthoulea, S.; Curfs, D.M.J.; Hofker, M.H.; De Winther, M.P.J. Nuclear factor kappaB signaling in macrophage function and atherogenesis. Curr. Opin. Lipidol. 2005, 16, 536–542. [Google Scholar] [CrossRef]
- Raines, E.W.; Garton, K.J.; Ferri, N. Beyond the Endothelium. Circ. Res. 2004, 94, 706–708. [Google Scholar] [CrossRef] [Green Version]
- Voelkl, J.; Lang, F.; Eckardt, K.-U.; Amann, K.; Kuro-O, M.; Pasch, A.; Pieske, B.; Alesutan, I. Signaling pathways involved in vascular smooth muscle cell calcification during hyperphosphatemia. Cell. Mol. Life Sci. 2019, 76, 2077–2091. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Yamashita, M.; Hormai, C.; Hayashi, M. Smooth Muscle-Selective Nuclear Factor-kappaB Inhibition Reduces Phosphate-Induced Arterial Medial Calcification in Mice With Chronic Kidney Disease. J. Am. Heart Assoc. 2017, 6, e007248. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-L.; Woo, K.M.; Ryoo, H.-M.; Baek, J.-H. Tumor necrosis factor-α increases alkaline phosphatase expression in vascular smooth muscle cells via MSX2 induction. Biochem. Biophys. Res. Commun. 2010, 391, 1087–1092. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Z.-Y.; Chen, X.-Q.; Wang, X.; Cao, H.; Liu, S.-W. Advanced glycation end products promote human aortic smooth muscle cell calcification in vitro via activating NF-κB and down-regulating IGF1R expression. Acta Pharmacol. Sin. 2013, 34, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Hou, M.; Li, Z.; Luo, C.; Ou, J.-S.; Yu, H.; Yan, J.; Lu, L. TLR4/NF-kappaB/Ceramide signaling contributes to Ox-LDL-induced calcification of human vascular smooth muscle cells. Eur. J. Pharmacol. 2017, 794, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Voelkl, J.; Luong, T.T.; Tuffaha, R.; Musculus, K.; Auer, T.; Lian, X.; Daniel, C.; Zickler, D.; Boehme, B.; Sacherer, M.; et al. SGK1 induces vascular smooth muscle cell calcification through NF-κB signaling. J. Clin. Investig. 2018, 128, 3024–3040. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.R.; Cotter, T.G. Hydrogen peroxide: A Jekyll and Hyde signalling molecule. Cell Death Dis. 2011, 2, e213. [Google Scholar] [CrossRef] [Green Version]
- Radcliff, K.; Tang, T.-B.; Lim, J.; Zhang, Z.; Abedin, M.; Demer, L.L.; Tintut, Y. Insulin-Like Growth Factor-I Regulates Proliferation and Osteoblastic Differentiation of Calcifying Vascular Cells via Extracellular Signal-Regulated Protein Kinase And Phosphatidylinositol 3-Kinase Pathways. Circ. Res. 2005, 96, 398–400. [Google Scholar] [CrossRef] [Green Version]
- Camalier, C.E.; Yi, M.; Yu, L.-R.; Hood, B.L.; Conrads, K.A.; Lee, Y.J.; Lin, Y.; Garneys, L.M.; Bouloux, G.F.; Young, M.R.; et al. An integrated understanding of the physiological response to elevated extracellular phosphate. J. Cell. Physiol. 2013, 228, 1536–1550. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Xu, Q.; Peng, H.; Liu, Z.; Yang, T.; Yu, Z.; Cheng, G.; Li, X.; Zhang, G.; Shi, R. The role of vascular peroxidase 1 in ox-LDL-induced vascular smooth muscle cell calcification. Atherosclerosis 2015, 243, 357–363. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, A.-P.; Yang, H.-P.; Ai, L.; Zhang, H.-J.; Wang, Y.-M.; Bi, Y.-L.; Fan, H.-H.; Gao, J.; Zhang, H.-Y.; et al. Apelin-13 attenuates high glucose-induced calcification of MOVAS cells by regulating MAPKs and PI3K/AKT pathways and ROS-mediated signals. Biomed. Pharmacother. 2020, 128, 110271. [Google Scholar] [CrossRef]
- Blanc, A.; Pandey, N.R.; Srivastava, A.K. Synchronous activation of ERK 1/2, p38(mapk) and PKB/Akt signaling by H2O2 in vascular smooth muscle cells: Potential involvement in vascular disease (Review). Int. J. Mol. Med. 2003, 11, 229–234. [Google Scholar]
- Tang, F.; Chan, E.; Lu, M.; Zhang, X.; Dai, C.; Mei, M.; Zhang, S.; Wang, H.; Song, Q. Calpain-1 Mediated Disorder of Pyrophosphate Metabolism Contributes to Vascular Calcification Induced by oxLDL. PLoS ONE 2015, 10, e0129128. [Google Scholar] [CrossRef]
- Koike, S.; Yano, S.; Tanaka, S.; Sheikh, A.M.; Nagai, A.; Sugimoto, T. Advanced Glycation End-Products Induce Apoptosis of Vascular Smooth Muscle Cells: A Mechanism for Vascular Calcification. Int. J. Mol. Sci. 2016, 17, 1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.P.; Huang, P.H.; Lai, C.F.; Chen, J.W.; Lin, S.J.; Chen, J.S. Simvastatin Attenuates Oxidative Stress, NF-kappaB Activation, and Artery Calcification in LDLR-/- Mice Fed with High Fat Diet via Down-regulation of Tumor Necrosis Factor-alpha and TNF Receptor 1. PLoS ONE 2015, 10, e0143686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, C.-T.; Yeh, H.-Y.; Tsai, Y.-T.; Chuang, P.-H.; Yuan, T.-H.; Huang, J.-W.; Chen, H.-W. Natural and non-natural antioxidative compounds: Potential candidates for treatment of vascular calcification. Cell Death Discov. 2019, 5, 145. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, X.; Zhong, X.; Li, Z.; Cai, S.; Yang, P.; Ou, C.; Chen, M. Puerarin inhibits vascular calcification of uremic rats. Eur. J. Pharmacol. 2019, 855, 235–243. [Google Scholar] [CrossRef]
- Ji, R.; Sun, H.; Peng, J.; Ma, X.; Bao, L.; Fu, Y.; Zhang, X.; Luo, C.; Gao, C.; Jin, Y.; et al. Rosmarinic acid exerts an antagonistic effect on vascular calcification by regulating the Nrf2 signalling pathway. Free. Radic. Res. 2019, 53, 187–197. [Google Scholar] [CrossRef]
- Chang, X.-Y.; Cui, L.; Wang, X.-Z.; Zhang, L.; Zhu, D.; Zhou, X.-R.; Hao, L.-R. Quercetin Attenuates Vascular Calcification through Suppressed Oxidative Stress in Adenine-Induced Chronic Renal Failure Rats. BioMed Res. Int. 2017, 2017, 1–7. [Google Scholar] [CrossRef]
- Feng, W.; Zhang, F.; Liu, Y.; Chen, J.; Cai, Q.; Zhang, Y.; Wang, M.; Wang, J.; Huang, H. Apocynin attenuates angiotensin II-induced vascular smooth muscle cells osteogenic switching via suppressing extracellular signal-regulated kinase 1/2. Oncotarget 2016, 7, 83588–83600. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Li, H.; Tao, H.; Wu, N.; Yu, L.; Zhang, D.; Lu, X.; Zhu, J.; Lu, Z.; Zhu, Q. Metformin Inhibits Vascular Calcification in Female Rat Aortic Smooth Muscle Cells via the AMPK-eNOS-NO Pathway. Endocrinology 2013, 154, 3680–3689. [Google Scholar] [CrossRef] [Green Version]
- Ivanovski, O.; Szumilak, D.; Nguyen-Khoa, T.; Nikolov, I.G.; Joki, N.; Mothu, N.; Maizel, J.; Westenfeld, R.; Ketteler, M.; Lacour, B.; et al. Effect of Simvastatin in Apolipoprotein E Deficient Mice With Surgically Induced Chronic Renal Failure. J. Urol. 2008, 179, 1631–1636. [Google Scholar] [CrossRef]
- Yamada, S.; Taniguchi, M.; Tokumoto, M.; Toyonaga, J.; Fujisaki, K.; Suehiro, T.; Noguchi, H.; Iida, M.; Tsuruya, K.; Kitazono, T. The antioxidant tempol ameliorates arterial medial calcification in uremic rats: Important role of oxidative stress in the pathogenesis of vascular calcification in chronic kidney disease. J. Bone Miner. Res. 2012, 27, 474–485. [Google Scholar] [CrossRef]
- Arefin, S.; Buchanan, S.; Hobson, S.; Steinmetz, J.; AlSalhi, S.; Shiels, P.G.; Kublickiene, K.; Stenvinkel, P. Nrf2 in early vascular ageing: Calcification, senescence and therapy. Clin. Chim. Acta 2020, 505, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Sykiotis, G.P.; Bohmann, D. Stress-Activated Cap’n’collar Transcription Factors in Aging and Human Disease. Sci. Signal. 2010, 3, re3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-Antioxidant Response Element Signaling Pathway and Its Activation by Oxidative Stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, C.; Park, S.; Choi, Y.-K.; Jeong, J.-Y.; Oh, C.J.; Bae, K.-H.; Lee, S.J.; Kim, J.-H.; Park, K.; Jun, D.Y.; et al. Activation of Nrf2 by dimethyl fumarate improves vascular calcification. Vasc. Pharmacol. 2014, 63, 29–36. [Google Scholar] [CrossRef]
- Zhang, P.; Li, Y.; Du, Y.; Li, G.; Wang, L.; Zhou, F. Resveratrol Ameliorated Vascular Calcification by Regulating Sirt-1 and Nrf2. Transplant. Proc. 2016, 48, 3378–3386. [Google Scholar] [CrossRef]
- Aghagolzadeh, P.; Radpur, R.; Bachtler, M.; Van Goor, H.; Smith, E.R.; Lister, A.; Odermatt, A.; Feelisch, M.; Pasch, A. Hydrogen sulfide attenuates calcification of vascular smooth muscle cells via KEAP1/NRF2/NQO1 activation. Atherosclerosis 2017, 265, 78–86. [Google Scholar] [CrossRef] [Green Version]
- Yao, L.; Wang, J.; Tian, B.-Y.; Xu, T.-H.; Sheng, Z.-T. Activation of the Nrf2-ARE Signaling Pathway Prevents Hyperphosphatemia-Induced Vascular Calcification by Inducing Autophagy in Renal Vascular Smooth Muscle Cells. J. Cell. Biochem. 2017, 118, 4708–4715. [Google Scholar] [CrossRef]
- Wei, R.; Enaka, M.; Muragaki, Y. Activation of KEAP1/NRF2/P62 signaling alleviates high phosphate-induced calcification of vascular smooth muscle cells by suppressing reactive oxygen species production. Sci. Rep. 2019, 9, 10366. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tóth, A.; Balogh, E.; Jeney, V. Regulation of Vascular Calcification by Reactive Oxygen Species. Antioxidants 2020, 9, 963. https://doi.org/10.3390/antiox9100963
Tóth A, Balogh E, Jeney V. Regulation of Vascular Calcification by Reactive Oxygen Species. Antioxidants. 2020; 9(10):963. https://doi.org/10.3390/antiox9100963
Chicago/Turabian StyleTóth, Andrea, Enikő Balogh, and Viktória Jeney. 2020. "Regulation of Vascular Calcification by Reactive Oxygen Species" Antioxidants 9, no. 10: 963. https://doi.org/10.3390/antiox9100963
APA StyleTóth, A., Balogh, E., & Jeney, V. (2020). Regulation of Vascular Calcification by Reactive Oxygen Species. Antioxidants, 9(10), 963. https://doi.org/10.3390/antiox9100963