The Impact of Mitochondrial Deficiencies in Neuromuscular Diseases


Abstract
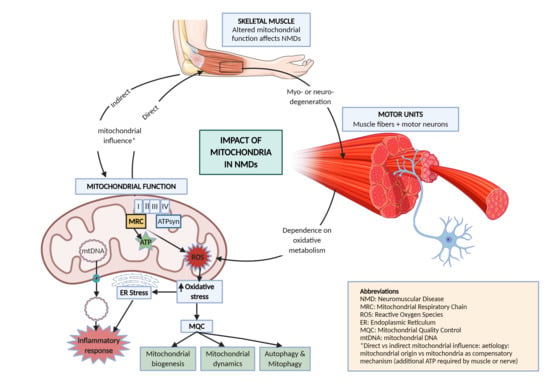
:
1. Introduction
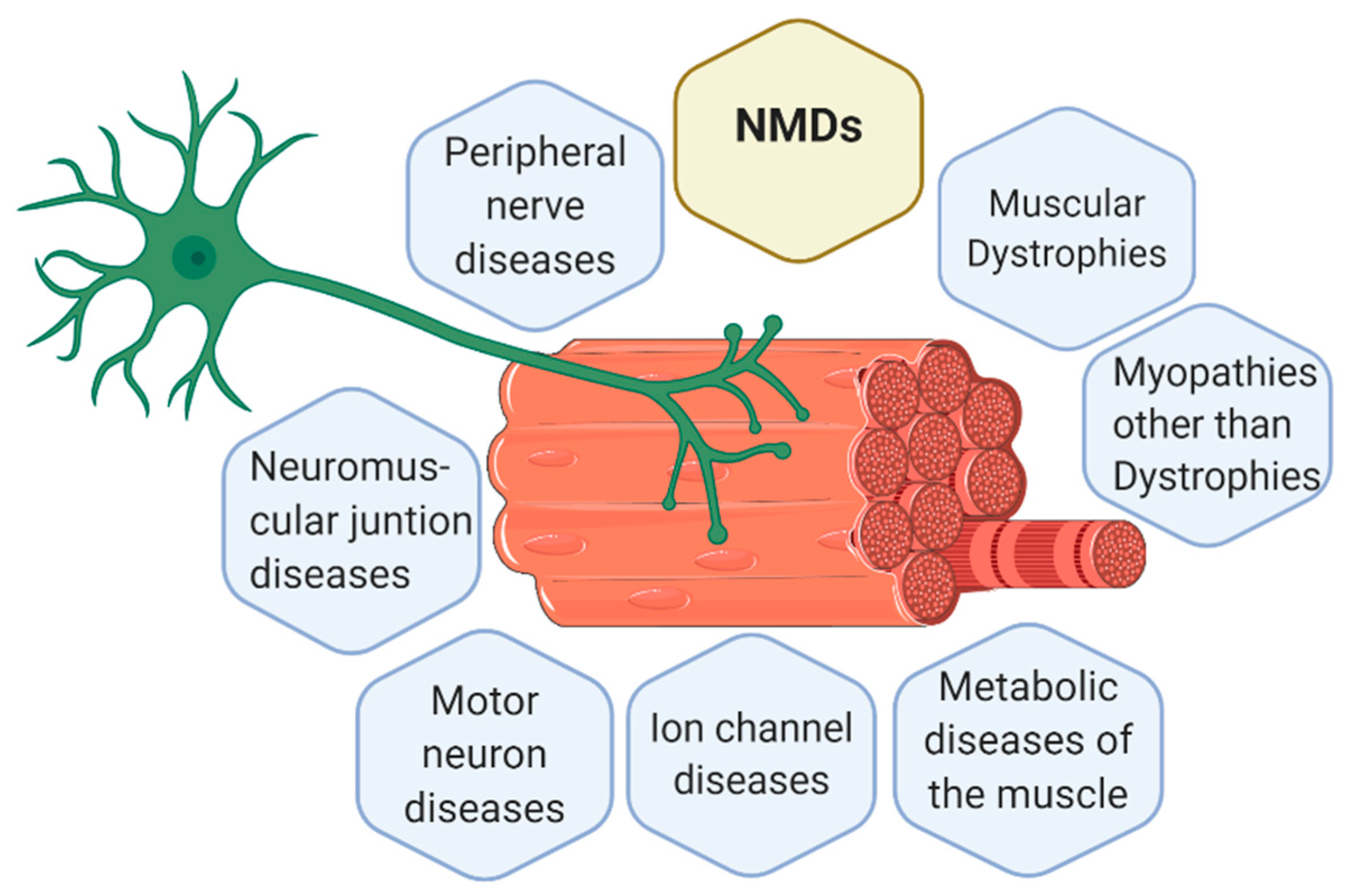
1.1. Neuromuscular Diseases (NMDs)
Classification of NMDs
- Muscular dystrophies (MD): affect the structure of the muscle cells, causing weakness and degeneration of the skeletal muscles. MD subtypes are described in Table 1.
- Myopathies other than dystrophies: affect tone and contraction of muscles controlling voluntary movements; may include inflammation of muscles or related tissues, resulting in muscular weakness. Numerous myopathies have been described and classified in different groups (Table 2).
- Neuromuscular junction (NMJ) diseases: result from the destruction, dysfunction, or absence of one or more key proteins involved in the transmission of signals between nerves and muscles (Table 3).
- Motor neuron diseases: involve nerve cells in the spinal cord (lower motor neurons). Lower motor neurons progressively lose their function, causing the muscles they control to become weak and eventually non-functional (Table 3).
- Peripheral nerve diseases: involve motor and sensory nerves that connect the brain and spinal cord to the rest of the body causing impaired sensations, movement or other functions (Table 4).
- Mitochondrial diseases: involve errors in metabolism that affect energy production in muscle cells (Table 4).
- Ion channel diseases: diseases associated with defects in proteins forming ion channels, leading to muscular weakness, absent muscle tone, or episodic muscle paralysis (Table 5).
1.2. Muscle and Nerve System
1.3. Mitochondrial Function
2. Mitochondrial Pathways Altered in NMD
2.1. Mitochondrial Genome and mtDNA Mutations
2.2. Mitochondrial Respiratory Chain (MRC)
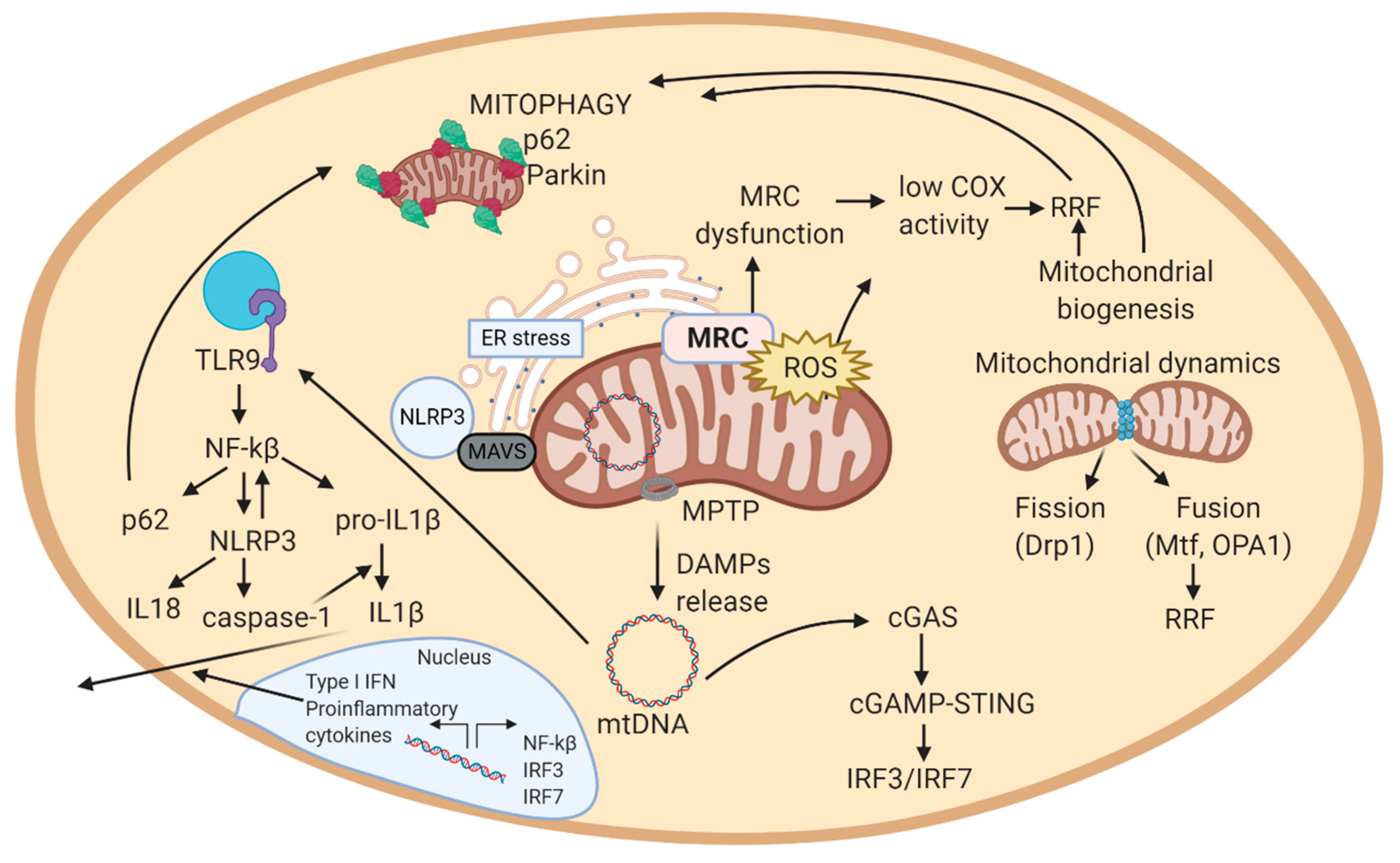
2.3. Mitochondrial Quality Control
2.3.1. Mitochondrial Biogenesis
2.3.2. Mitochondrial Dynamics
2.3.3. Autophagy and Mitophagy
2.4. Mitochondrial ROS and ER Stress
2.5. Mitochondrially Induced Inflammatory Response
2.5.1. Implication of mtDNA in Inflammation
2.5.2. Implication of Mitochondrial ROS in Inflammation
2.5.3. Feedback Regulation of the Mitochondrial Inflammatory Response
3. Treatment Strategies in NMDs
4. Discussion
5. Conclusions
- NMDs have a direct or indirect influence of mitochondria in their etiology. Mitochondrial genetic defects directly affect nerves and muscles in NMDs; but when there is an alternative cause of disease, unproper mitochondrial function can limit energetic supply of compensatory mechanisms, thus indirectly conditioning the progression of disease.
- Defects in oxidative metabolism can lead to accumulation of ROS, potentially affecting the ER and triggering inflammation, which eventually may lead to cell death and tissue damage.
- To avoid cell damage and excessive oxidative stress, mitochondrial quality control processes closely monitor changes in mitochondrial metabolism. In case of trouble, different responses arise by the increase in the number of mitochondria (mitochondrial biogenesis), by changing their morphology and size (mitochondrial dynamics) or recycling them (mitophagy). Most of these processes are altered in NMDs, highlighting the relevance of mitochondria in the development and progression of these disorders.
- Understanding nerves, muscles, and mitochondrial defects in NMDs is essential to improve diagnosis and treatments for these major incapacitating diseases.
- Despite growing efforts to clarify the etiology of NMD, the complexity of reality is still far beyond current knowledge and information provided in the present review, challenging new researchers to develop novel approaches.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Diagnosis—European Reference Network—EURO-NMD. Available online: https://ern-euro-nmd.eu/ (accessed on 22 April 2020).
- Scotton, C.; Passarelli, C.; Neri, M.; Ferlini, A. Biomarkers in rare neuromuscular diseases. Exp. Cell Res. 2013, 325, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Bonne, G.; Rivier, F.; Hamroun, D. The 2018 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromuscul. Disord. 2017, 27, 1152–1183. [Google Scholar] [CrossRef] [PubMed]
- Deenen, C.W.J.; Horlings, G.C.C.; Verschuuren, J.G.M.J.; Verbeek, L.M.A.; van Engelen, G.M.B. The Epidemiology of Neuromuscular Disorders: A Comprehensive Overview of the Literature. J. Neuromuscul. Dis. 2015, 2, 73–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Putten, M.; Hmeljak, J.; Aartsma-Rus, A.; Dowling, J.J. Moving neuromuscular disorders research forward: From novel models to clinical studies. Dis. Model. Mech. 2020, 13, 44370. [Google Scholar] [CrossRef] [Green Version]
- Neurology Neuromuscular Disorders Causes & Symptoms Beaumont Health. Available online: https://www.beaumont.org/conditions/neuromuscular-disorders (accessed on 19 May 2020).
- Nardin, R.A.; Johns, D.R. Mitochondrial dysfunction and neuromuscular disease. Muscle Nerve 2001, 24, 170–191. [Google Scholar] [CrossRef]
- Cowling, B.S.; Thielemans, L. Translational medicine in neuromuscular disorders: From academia to industry. Dis. Model. Mech. 2019, 13, 41434. [Google Scholar] [CrossRef] [Green Version]
- Find A Neuromuscular Disease Muscular Dystrophy Association. Available online: https://www.mda.org/disease/list (accessed on 19 May 2020).
- GeneTable. Available online: http://www.musclegenetable.fr/index.html (accessed on 7 July 2020).
- Hewitt, J.E.; Pollard, A.K.; Lesanpezeshki, L.; Deane, C.S.; Gaffney, C.J.; Etheridge, T.; Szewczyk, N.J.; Vanapalli, S.A. Muscle strength deficiency and mitochondrial dysfunction in a muscular dystrophy model of Caenorhabditis elegans and its functional response to drugs. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Heydemann, A. Skeletal muscle metabolism in duchenne and becker muscular dystrophy—Implications for therapies. Nutrients 2018, 10, 796. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.R.; Sahenk, Z.; Malik, V.; Gomez, A.M.; Flanigan, K.M.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Meadows, E.; Lewis, S.; et al. A phase 1/2a follistatin gene therapy trial for becker muscular dystrophy. Mol. Ther. 2015, 23, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Katsetos, C.D.; Koutzaki, S.; Melvin, J.J. Mitochondrial dysfunction in neuromuscular disorders. Semin. Pediatric Neurol. 2013, 20, 202–215. [Google Scholar] [CrossRef]
- Grumati, P.; Grumati, P.; Coletto, L.; Sabatelli, P.; Cescon, M.; Angelin, A.; Bertaggia, E.; Blaauw, B.; Urciuolo, A.; Tiepolo, T.; et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat. Med. 2010, 16, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Jongpiputvanich, S.; Sueblinvong, T.; Norapucsunton, T. Mitochondrial respiratory chain dysfunction in various neuromuscular diseases. J. Clin. Neurosci. 2005, 12, 426–428. [Google Scholar] [CrossRef] [PubMed]
- Pleasure, D. Advances in Translational Research in Neuromuscular Diseases. Arch. Neurol. 2011, 68, 429. [Google Scholar] [CrossRef] [Green Version]
- Lamar, K.-M.; Mcnally, E.M. Genetic Modifiers for Neuromuscular Diseases. J. Neuromuscul. Dis. 2014, 1, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Rybalka, E.; Timpani, C.A.; Cooke, M.B.; Williams, A.D.; Hayes, A. Defects in mitochondrial ATP synthesis in dystrophin-deficient Mdx skeletal muscles may be caused by complex I insufficiency. PLoS ONE 2014, 9, 115763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godin, R.; Daussin, F.; Matecki, S.; Li, T.; Petrof, B.J.; Burelle, Y. Peroxisome proliferator-activated receptor γ coactivator 1-α gene transfer restores mitochondrial biomass and improves mitochondrial calcium handling in post-necrotic mdx mouse skeletal muscle. J. Physiol. 2012, 590, 5487–5502. [Google Scholar] [CrossRef]
- Pauly, M.; Daussin, F.; Burelle, Y.; Li, T.; Godin, R.; Fauconnier, J.; Koechlin-Ramonatxo, C.; Hugon, G.; Lacampagne, A.; Coisy-Quivy, M.; et al. AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am. J. Pathol. 2012, 181, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Zuela, N.; Dorfman, J.; Gruenbaum, Y. Global transcriptional changes caused by an EDMD mutation correlate to tissue specific disease phenotypes in C. elegans. Nucleus 2017, 8, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Puckelwartz, M.; McNally, E.M. Emery-Dreifuss muscular dystrophy. Handb. Clin. Neurol. 2011, 101, 155–166. [Google Scholar] [CrossRef]
- Orphanet. Available online: https://www.orpha.net/consor4.01/www/cgi-bin/?lng=ES (accessed on 22 May 2020).
- Lemmers, R.J.L.F.; Van Der Vliet, P.J.; Klooster, R.; Sacconi, S.; Camaño, P.; Dauwerse, J.G.; Snider, L.; Straasheijm, K.R.; Van Ommen, G.J.; Padberg, G.W.; et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010, 329, 1650–1653. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.-X.; Klein, C.J.; Yan, J.; Shi, Y.; Wu, Y.; Fecto, F.; Yau, H.-J.; Yang, Y.; Zhai, H.; Siddique, N.; et al. Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4. Nat. Genet. 2010, 42, 165–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demonbreun, A.R.; Wyatt, E.J.; Fallon, K.S.; Oosterbaan, C.C.; Page, P.G.; Hadhazy, M.; Quattrocelli, M.; Barefield, D.Y.; McNally, E.M. A gene-edited mouse model of limb-girdle muscular dystrophy 2C for testing exon skipping. DMM Dis. Model. Mech. 2020, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lilleker, J.B.; Keh, Y.S.; Roncaroli, F.; Sharma, R.; Roberts, M. Metabolic myopathies: A practical approach. Pract. Neurol. 2018, 18, 14–26. [Google Scholar] [CrossRef] [PubMed]
- CNBP Gene—Genetics Home Reference—NIH. Available online: https://ghr.nlm.nih.gov/gene/CNBP#conditions (accessed on 25 May 2020).
- Malerba, A.; Klein, P.; Bachtarzi, H.; Jarmin, S.A.; Cordova, G.; Ferry, A.; Strings, V.; Espinoza, M.P.; Mamchaoui, K.; Blumen, S.C.; et al. PABPN1 gene therapy for oculopharyngeal muscular dystrophy. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malerba, A.; Roth, F.; Harish, P.; Dhiab, J.; Lu-Nguyen, N.; Cappellari, O.; Jarmin, S.; Mahoudeau, A.; Ythier, V.; Lainé, J.; et al. Pharmacological modulation of the ER stress response ameliorates oculopharyngeal muscular dystrophy. Hum. Mol. Genet. 2019, 28, 1694–1708. [Google Scholar] [CrossRef]
- Chartier, A.; Klein, P.; Pierson, S.; Barbezier, N.; Gidaro, T.; Casas, F.; Carberry, S.; Dowling, P.; Maynadier, L.; Bellec, M.; et al. Mitochondrial Dysfunction Reveals the Role of mRNA Poly(A) Tail Regulation in Oculopharyngeal Muscular Dystrophy Pathogenesis. PLoS Genet. 2015, 11, 1005092. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D.; Palmio, J.; Evilä, A.; Galli, L.; Barone, V.; Caldwell, T.A.; Policke, R.A.; Aldkheil, E.; Berndsen, C.E.; Wright, N.T.; et al. A novel FLNC frameshift and an OBSCN variant in a family with distal muscular dystrophy. PLoS ONE 2017, 12, 186642. [Google Scholar] [CrossRef]
- Vincent, A.E.; Rosa, H.S.; Alston, C.L.; Grady, J.P.; Rygiel, K.A.; Rocha, M.C.; Barresi, R.; Taylor, R.W.; Turnbull, D.M. Dysferlin mutations and mitochondrial dysfunction. Neuromuscul. Disord. 2016, 26, 782–788. [Google Scholar] [CrossRef] [Green Version]
- GeneTable. Available online: http://www.musclegenetable.fr/4DACTION/Blob_groupe11/ (accessed on 6 July 2020).
- Fusto, A.; Moyle, L.A.; Gilbert, P.M.; Pegoraro, E. Cored in the act: The use of models to understand core myopathies. DMM Dis. Model. Mech. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Gilbreath, H.R.; Castro, D.; Iannaccone, S.T. Congenital myopathies and muscular dystrophies. Neurol. Clin. 2014, 32, 689–703. [Google Scholar] [CrossRef]
- Siciliano, G.; Monzani, F.; Manca, M.L.; Tessa, A.; Caraccio, N.; Tozzi, G.; Piemonte, F.; Mancuso, M.; Santorelli, F.M.; Ferrannini, E.; et al. Human Mitochondrial Transcription Factor A Reduction and Mitochondrial Dysfunction in Hashimoto’s Hypothyroid Myopathy. Mol. Med. 2002, 8, 326–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloreta, J.; Roquer, J.; Corominas, J.M.; Serrano, S. Hyperthyroid myopathy with mitochondrial paracrystalline rectangular inclusions. Ultrastruct. Pathol. 1996, 20, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Gong, Y.; Dong, M.; Pei, Z.; Ren, J. Role of autophagy in inherited metabolic and endocrine myopathies. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Selva-O’callaghan, A.; Pinal-Fernandez, I.; Trallero-Araguás, E.; Milisenda, J.C.; Grau-Junyent, M.; Mammen, A.L. Classification and management of adult inflammatory myopathies. Lancet Neurol. 2018, 17, 816–828. [Google Scholar] [CrossRef]
- Catalan-Garcia, M.; Garrabou, G.; Moren, C.; Guitart-Mampel, M.; Hernando, A.; Diaz-Ramos, A.; Gonzalez-Casacuberta, I.; Juarez, D.-L.; Bano, M.; Enrich-Bengoa, J.; et al. Mitochondrial DNA disturbances and deregulated expression of oxidative phosphorylation and mitochondrial fusion proteins in sporadic inclusion body myositis. Clin. Sci. 2016, 130, 1741–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, L.J.; Haller, R.G. Metabolic and mitochondrial myopathies. Neurol. Clin. 2014, 32, 777–799. [Google Scholar] [CrossRef]
- Selcen, D.; Ohno, K.; Engel, A.G. Myofibrillar myopathy: Clinical, morphological and genetic studies in 63 patients. Brain 2004, 127, 439–451. [Google Scholar] [CrossRef]
- Perrone, L.; Squillaro, T.; Napolitano, F.; Terracciano, C.; Sampaolo, S.; Anna, M.; Melone, B. The Autophagy Signaling Pathway: A Potential Multifunctional Therapeutic Target of Curcumin in Neurological and Neuromuscular Diseases. Nutrients 2019, 11, 1881. [Google Scholar] [CrossRef] [Green Version]
- Vincent, A.E.; Grady, J.P.; Rocha, M.C.; Alston, C.L.; Rygiel, K.A.; Barresi, R.; Taylor, R.W.; Turnbull, D.M. Mitochondrial dysfunction in myofibrillar myopathy. Neuromuscul. Disord. 2016, 26, 691–701. [Google Scholar] [CrossRef] [Green Version]
- Pá, E.; Bedekovics Istvá, T.; Ti, G. Familial Scapuloperoneal Myopathy and Mitochondrial DNA Defect. Eur Neurol. 1999, 42, 211–216. [Google Scholar]
- Zukosky, K.; Meilleur, K.; Traynor, B.J.; Dastgir, J.; Medne, L.; Devoto, M.; Collins, J.; Rooney, J.; Zou, Y.; Yang, M.L.; et al. Association of a novel ACTA1 mutation with a dominant progressive scapuloperoneal myopathy in an extended family. JAMA Neurol. 2015, 72, 689–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.H.; Raskind, W.H.; Parson, W.W.; Sonnen, J.A.; Vu, T.; Zheng, Y.; Matsushita, M.; Wolff, J.; Lipe, H.; Bird, T.D. A novel mutation in FHL1 in a family with X-linked scapuloperoneal myopathy: Phenotypic spectrum and structural study of FHL1 mutations. J. Neurol. Sci. 2010, 296, 22–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, A.G.; Shen, X.M.; Selcen, D.; Sine, S.M. Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet Neurol. 2015, 14, 420–434. [Google Scholar] [CrossRef]
- Chaouch, A.; Porcelli, V.; Cox, D.; Edvardson, S.; Scarcia, P.; De Grassi, A.; Pierri, C.L.; Cossins, J.; Laval, S.H.; Griffin, H.; et al. Chaouch et al. Mutations in the Mitochondrial Citrate Carrier SLC25A1. J. Neuromuscul. Dis. 2014, 1, 75–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finsterer, J. Congenital myasthenic syndromes. Orphanet J. Rare Dis. 2019, 14, 57. [Google Scholar] [CrossRef]
- Bogdanik, L.P.; Burgess, R.W. A valid mouse model of AGRIN-associated congenital myasthenic syndrome. Hum. Mol. Genet. 2011, 20, 4617–4633. [Google Scholar] [CrossRef] [Green Version]
- Kesner, V.G.; Oh, S.J.; Dimachkie, M.M.; Barohn, R.J. Lambert-Eaton Myasthenic Syndrome. Neurol. Clin. 2018, 36, 379–394. [Google Scholar] [CrossRef]
- Hülsbrink, R.; Hashemolhosseini, S. Lambert-Eaton myasthenic syndrome-Diagnosis, pathogenesis and therapy. Clin. Neurophysiol. 2014, 125, 2328–2336. [Google Scholar] [CrossRef]
- Herrmann, D.N.; Horvath, R.; Sowden, J.E.; Gonzales, M.; Sanchez-Mejias, A.; Guan, Z.; Whittaker, R.G.; Almodovar, J.L.; Lane, M.; Bansagi, B.; et al. Synaptotagmin 2 mutations cause an autosomal-dominant form of lambert-eaton myasthenic syndrome and nonprogressive motor neuropathy. Am. J. Hum. Genet. 2014, 95, 332–339. [Google Scholar] [CrossRef] [Green Version]
- Marchiori, P.E.; Levy, J.A.; Carvalho-Alegro, M.S.; Lusvarghi, E.S.; Tsanaclis, A.M.; De Assis, J.L.; Scaff, M. Mitochondrial dysfunction in myasthenia gravis. Report of a case. Arq. Neuropsiquiatr. 1989, 47, 355–358. [Google Scholar] [CrossRef] [Green Version]
- Gilhus, N.E. Myasthenia Gravis. N. Engl. J. Med. 2016, 375, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nat. Lett. 2010, 465. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.; Paul, P.; Chen, H.-J.; Morris, A.; Payling, M.; Falchi, M.; Habgood, J.; Panoutsou, S.; Winkler, S.; Tisato, V.; et al. Familial amyotrophic lateral sclerosis is associated with a mutation in D-amino acid oxidase. Proc. Natl. Acad. Sci. USA 2010, 107, 7556–7561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardiman, O.; Van Den Berg, L.H.; Kiernan, M.C. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 639–649. [Google Scholar] [CrossRef]
- Chiò, A.; Mora, G.; Lauria, G. Pain in amyotrophic lateral sclerosis. Lancet Neurol. 2017, 16, 144–157. [Google Scholar] [CrossRef]
- Comi, G.P.; Bordoni, A.; Salani, S.; Franceschina, L.; Sciacco, M.; Prelle, A.; Fortunato, F.; Zeviani, M.; Napoli, L.; Bresolin, N.; et al. Cytochrome c oxidase subunit I microdeletion in a patient with motor neuron disease. Ann. Neurol. 1998, 43, 110–116. [Google Scholar] [CrossRef]
- Figueroa-Romero, C.; Guo, K.; Murdock, B.J.; Paez-Colasante, X.; Bassis, C.M.; Mikhail, K.A.; Raue, K.D.; Evans, M.C.; Taubman, G.F.; McDermott, A.J.; et al. Temporal evolution of the microbiome, immune system and epigenome with disease progression in ALS mice. DMM Dis. Model. Mech. 2020, 13. [Google Scholar] [CrossRef] [Green Version]
- Lynch, E.; Semrad, T.; Belsito, V.S.; FitzGibbons, C.; Reilly, M.; Hayakawa, K.; Suzuki, M. C9ORF72-related cellular pathology in skeletal myocytes derived from ALS-patient induced pluripotent stem cells. DMM Dis. Model. Mech. 2019, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ranganathan, S.; Harmison, G.G.; Meyertholen, K.; Pennuto, M.; Burnett, B.G.; Fischbeck, K.H. Mitochondrial abnormalities in spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2008, 18, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Ravi, B.; Antonellis, A.; Sumner, C.J.; Lieberman, A.P. Genetic approaches to the treatment of inherited neuromuscular diseases. Hum. Mol. Genet. 2019, 28, 1–55. [Google Scholar] [CrossRef] [Green Version]
- Giorgetti, E.; Lieberman, A.P. Polyglutamine androgen receptor-mediated neuromuscular disease. Cell Mol. Life Sci. 2016, 73, 3991–3999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunseich, C.; Miller, R.; Swan, T.; Glass, D.J.; El Mouelhi, M.; Fornaro, M.; Petricoul, O.; Vostiar, I.; Roubenoff, R.; Meriggioli, M.N.; et al. Safety, tolerability, and preliminary efficacy of an IGF-1 mimetic in patients with spinal and bulbar muscular atrophy: A randomised, placebo-controlled trial. Lancet Neurol. 2018, 17, 1043–1052. [Google Scholar] [CrossRef]
- Hashizume, A.; Katsuno, M.; Suzuki, K.; Banno, H.; Takeuchi, Y.; Kawashima, M.; Suga, N.; Mano, T.; Araki, A.; Hijikata, Y.; et al. Efficacy and safety of leuprorelin acetate for subjects with spinal and bulbar muscular atrophy: Pooled analyses of two randomized-controlled trials. J. Neurol. 2019, 266, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Passini, M.A.; Bu, J.; Roskelley, E.M.; Richards, A.M.; Sardi, S.P.; O’Riordan, C.R.; Klinger, K.W.; Shihabuddin, L.S.; Cheng, S.H. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J. Clin. Investig. 2010, 120, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Tinelli, E.; Pereira, J.A.; Suter, U. Muscle-specific function of the centronuclear myopathy and Charcot-Marie-Tooth neuropathy-associated dynamin 2 is required for proper lipid metabolism, mitochondria, muscle fibers, neuromuscular junctions and peripheral nerves. Hum. Mol. Genet. 2013, 22, 4417–4429. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.I.; Roa, B.B.; Welcher, A.A.; Schoener-Scott, R.; Trask, B.J.; Pentao, L.; Snipes, G.J.; Garcia, C.A.; Francke, U.; Shooter, E.M.; et al. The gene for the peripheral myelin protein PMP–22 is a candidate for Charcot–Marie–Tooth disease type 1A. Nat. Genet. 1992, 1, 159–165. [Google Scholar] [CrossRef]
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef]
- Pezeshkpour, G.; Krarup, C.; Buchthal, F.; DiMauro, S.; Bresolin, N.; McBurney, J. Peripheral neuropathy in mitochondrial disease. J. Neurol. Sci. 1987, 77, 285–304. [Google Scholar] [CrossRef]
- Bomont, P.; Cavalier, L.; Blondeau, F.; Hamida, C.B.; Belal, S.; Tazir, M.; Demir, E.; Topaloglu, H.; Korinthenberg, R.; Tüysüz, B.; et al. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat. Lett. 2000, 26, 370–374. [Google Scholar] [CrossRef]
- Milone, M.; Wong, L.-J. Diagnosis of mitochondrial myopathies. Mol. Genet. Metab. 2013, 110, 35–41. [Google Scholar] [CrossRef]
- Khan, N.A.; Auranen, M.; Paetau, I.; Pirinen, E.; Euro, L.; Forsström, S.; Pasila, L.; Velagapudi, V.; Carroll, C.J.; Auwerx, J.; et al. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol. Med. 2014, 6, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.E.; Ng, Y.S.; White, K.; Davey, T.; Mannella, C.; Falkous, G.; Feeney, C.; Schaefer, A.M.; Mcfarland, R.; Gorman, G.S.; et al. The Spectrum of Mitochondrial Ultrastructural Defects in Mitochondrial Myopathy. Sci. Rep. 2016, 6, 30610. [Google Scholar] [CrossRef] [Green Version]
- Cook, A.; Giunti, P. Friedreich’s ataxia: Clinical features, pathogenesis and management. Br. Med. Bull. 2017, 124, 19–30. [Google Scholar] [CrossRef]
- Alfedi, G.; Luffarelli, R.; Condò, I.; Pedini, G.; Mannucci, L.; Massaro, D.S.; Benini, M.; Toschi, N.; Alaimo, G.; Panarello, L.; et al. Drug repositioning screening identifies etravirine as a potential therapeutic for friedreich’s ataxia. Mov. Disord. 2019, 34, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Strickland, M.; Yacoubi-Loueslati, B.; Bouhaouala-Zahar, B.; Pender, S.L.F.; Larbi, A. Relationships between ion channels, mitochondrial functions and inflammation in human aging. Front. Physiol. 2019, 10, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurkat-Rott, K.; Lehmann-Horn, F. State of the art in hereditary muscle channelopathies. Acta Myol. 2010, 29, 343–350. [Google Scholar]
- Pi, Y.; Goldenthal, M.J.; Marín-García, J.; Pi, Y.; Goldenthal, M.J.; Marín-García, J. Mitochondrial channelopathies in aging. J. Mol. Med. 2007, 85, 937–951. [Google Scholar] [CrossRef]
- Vicart, S.; Sternberg, D.; Fontaine, B.; Meola, G. Human skeletal muscle sodium channelopathies. Neurol. Sci. 2005, 26, 194–202. [Google Scholar] [CrossRef]
- Rudolf, R.; Khan, M.M.; Witzemann, V. Motor Endplate—Anatomical, Functional, and Molecular Concepts in the Historical Perspective. Cells 2019, 8, 387. [Google Scholar] [CrossRef] [Green Version]
- Calderón, J.C.; Bolaños, P.; Caputo, C. The excitation-contraction coupling mechanism in skeletal muscle. Biophys. Rev. 2014, 6, 133–160. [Google Scholar] [CrossRef] [Green Version]
- Bach-y-Rita, P.; Ito, F. In vivo studies on fast and slow muscle fibers in cat extraocular muscles. J. Gen. Physiol. 1966, 49, 1177–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-García, C.; Valcárcel-Ares, M.N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion 2013. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.F.; Feener, C.; Kunkel’tt, L.M. Complete Cloning of the Duchenne Muscular Dystrophy (DMD) cDNA and Preliminary Genomic Organization of the DMD Gene in Normal and Affected Individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- Siekevitz, P. Powerhouse of the Cell. Nature 1957, 197, 131–144. [Google Scholar] [CrossRef]
- Kauppila, T.E.S.; Kauppila, J.H.K.; Ran Larsson, N.-G. Cell Metabolism Review Mammalian Mitochondria and Aging: An Update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef]
- Palikaras, K.; Tavernarakis, N. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 2014, 56, 182–188. [Google Scholar] [CrossRef]
- Piantadosi, C.A.; Suliman, H.B. Transcriptional control of mitochondrial biogenesis and its interface with inflammatory processes. Biochim. Biophys. Acta 2012, 1820, 532–541. [Google Scholar] [CrossRef] [Green Version]
- Kepp, O.; Galluzzi, L.; Kroemer, G. Mitochondrial control of the NLRP3 inflammasome. Nat. Immunol. 2011, 12. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Vance, J.E. MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 595–609. [Google Scholar] [CrossRef]
- Manevski, M.; Muthumalage, T.; Devadoss, D.; Sundar, I.K.; Wang, Q.; Singh, K.P.; Unwalla, H.J.; Chand, H.S.; Rahman, I. Cellular stress responses and dysfunctional Mitochondrial–cellular senescence, and therapeutics in chronic respiratory diseases. Redox Biol. 2020, 33, 101443. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Jang, H.; Uematsu, S.; Yang, B.-G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1b production. Nat. Lett. 2008, 456. [Google Scholar] [CrossRef]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Nuevo, A.; Díaz-Ramos, A.; Noguera, E.; Díaz-Sáez, F.; Duran, X.; Muñoz, J.P.; Romero, M.; Plana, N.; Sebastián, D.; Tezze, C.; et al. Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Couvillion, M.T.; Soto, I.C.; Shipkovenska, G.; Stirling, L. Synchronized mitochondrial and cytosolic translation programs. Nature 2016, 533, 499–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokuyama, T.; Hirai, A.; Shiiba, I.; Ito, N.; Matsuno, K.; Takeda, K.; Saito, K.; Mii, K.; Matsushita, N.; Fukuda, T.; et al. Mitochondrial Dynamics Regulation in Skin Fibroblasts from Mitochondrial Disease Patients. Biomolecules 2020, 10, 450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F.; Bernabei, R.; Marzetti, E. Fueling inflamm-aging through mitochondrial dysfunction: Mechanisms and molecular targets. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Frank, M.; Duvezin-Caubet, S.; Koob, S.; Occhipinti, A.; Jagasia, R.; Petcherski, A.; Ruonala, M.O.; Priault, M.; Salin, B.; Reichert, A.S. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 2297–2310. [Google Scholar] [CrossRef]
- Babbar, M.; Basu, S.; Yang, B.; Croteau, D.L.; Bohr, V.A. Mitophagy and DNA damage signaling in human aging. Mech. Ageing Dev. 2020, 186. [Google Scholar] [CrossRef]
- Bhatt, P.S.; Tzoulis, C.; Balafkan, N.; Miletic, H.; Tran, G.T.T.; Sanaker, P.S.; Bindoff, L.A. Mitochondrial DNA depletion in sporadic inclusion body myositis. Neuromuscul. Disord. 2019, 29, 242–246. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C.; Pressman, B.C.; Gianetto, R.; Wattiaux, R.; Appelmans, F. Tissue fractionation studies. Biochem. J. 1955, 60, 604–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koopman, W.J.H.; Willems, P.H.G.M.; Smeitink, J.A.M. Monogenic Mitochondrial Diseases. N. Engl. J. Med. 2012, 366, 1132–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keogh, M.J.; Chinnery, P.F. Mitochondrial DNA mutations in neurodegeneration. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 1401–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lightowlers, R.N.; Taylor, R.W.; Turnbull, D.M. Mutations causing mitochondrial disease: What is new and what challenges remain? Science 2015, 349, 1494–1499. [Google Scholar] [CrossRef]
- Argov, Z.; Renshaw, P.F.; Boden, B.; Winokur, A.; Bank, W.J. Effects of thyroid hormones on skeletal muscle bioenergetics. In vivo phosphorus-31 magnetic resonance spectroscopy study of humans and rats. J. Clin. Investig. 1988, 81, 1695–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monzani, F.; Caraccio, N.; Siciliano, G.; Manca, L.; Murri, L.; Ferrannini, E. Clinical and Biochemical Features of Muscle Dysfunction in Subclinical Hypothyroidism. J. Clin. Endocrinol. Metab. 1997, 82, 3315–3318. [Google Scholar] [CrossRef]
- Deng, J.; Wang, P.; Chen, X.; Cheng, H.; Liu, J.; Fushimi, K.; Zhu, L.; Wu, J.Y. FUS interacts with ATP synthase beta subunit and induces mitochondrial unfolded protein response in cellular and animal models. Proc. Natl. Acad. Sci. USA 2018, 115, E9678–E9686. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, R.; Saada, A.; Flannery, P.J.; Burté, F.; Soiferman, D.; Khayat, M.; Eisner, V.; Vladovski, E.; Taylor, R.W.; Bindoff, L.A.; et al. Fatal infantile mitochondrial encephalomyopathy, hypertrophic cardiomyopathy and optic atrophy associated with a homozygous OPA1 mutation. J. Med. Genet. 2016, 53, 127–131. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef] [Green Version]
- Waterham, H.R.; Koster, J.; van Roermund, C.W.T.; Mooyer, P.A.W.; Wanders, R.J.A.; Leonard, J.V. A Lethal Defect of Mitochondrial and Peroxisomal Fission. N. Engl. J. Med. 2007, 356, 1736–1741. [Google Scholar] [CrossRef] [PubMed]
- Kodavati, M.; Wang, H.; Hegde, M.L. Altered Mitochondrial Dynamics in Motor Neuron Disease: An Emerging Perspective. Cells 2020, 9, 1065. [Google Scholar] [CrossRef] [PubMed]
- Clark, S. Newborn mice studied with the electron mircroscope. Biophys. Biochem. Cytol. 1957, 3, 349–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Seabright, A.P.; Fine, N.H.F.; Barlow, J.P.; Lord, S.O.; Musa, I.; Gray, A.; Bryant, J.A.; Banzhaf, M.; Lavery, G.G.; Hardie, D.G.; et al. AMPK activation induces mitophagy and promotes mitochondrial fission while activating TBK1 in a PINK1-Parkin independent manner. FASEB J. 2020, 34, 6284–6301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nat. Lett. 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Hu, X.; Shen, Q.; Xing, D. Mitochondria-specific drug release and reactive oxygen species burst induced by polyprodrug nanoreactors can enhance chemotherapy. Nat. Commun. 2019, 10, 1–14. [Google Scholar]
- Lerner, C.A.; Sundar, I.K.; Rahman, I. Mitochondrial redox system, dynamics, and dysfunction in lung inflammaging and COPD. Int. J. Biochem. Cell Biol. 2016, 81, 294–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domènech, B.E.; Marfany, G. The Relevance of Oxidative Stress in the Pathogenesis and Therapy of Retinal Dystrophies. Antioxidants 2020, 9, 347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterman, E.M.; Sullivan, C.; Goody, M.F.; Rodriguez-Nunez, I.; Yoder, J.A.; Kim, C.H. Neutralization of mitochondrial superoxide by superoxide dismutase 2 promotes bacterial clearance and regulates phagocyte numbers in zebrafish. Infect. Immun. 2015, 83, 430–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, P.M.; Nilsson, P.; Forsgren, L.; Marklund, S.L. CuZn-Superoxide Dismutase, Extracellular Superoxide Dismutase, and Glutathione Peroxidase in Blood from Individuals Homozygous for Asp90Ala CuZn-Superoxide Dismutase Mutation. J. Neurochem. 2002, 70, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef] [PubMed]
- Naon, D.; Scorrano, L. At the right distance: ER-mitochondria juxtaposition in cell life and death. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2184–2194. [Google Scholar] [CrossRef]
- Nadalutti, C.A.; Stefanick, D.F.; Zhao, M.-L.; Horton, J.K.; Prasad, R.; Brooks, A.M.; Griffith, J.D.; Wilson, S.H. Mitochondrial dysfunction and DNA damage accompany enhanced levels of formaldehyde in cultured primary human fibroblasts. Sci. Rep. 2020, 10, 5575. [Google Scholar] [CrossRef] [Green Version]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Pyo Kim, H.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 8, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Treviño, P.; Velásquez, M.; García, N. Mechanisms of mitochondrial DNA escape and its relationship with different metabolic diseases. BBA Mol. Basis Dis. 2020, 1866, 165761. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Nuevo, A.; Zorzano, A. The sensing of mitochondrial DAMPs by non-immune cells. Cell Stress 2019, 3, 195–207. [Google Scholar] [CrossRef] [PubMed]
- West, A.P. Mitochondrial dysfunction as a trigger of innate immune responses and inflammation. Toxicology 2017, 391, 54–63. [Google Scholar] [CrossRef]
- Baroja-Mazo, A.; Martín-Sánchez, F.; Gomez, A.I.; Martínez, C.M.; Amores-Iniesta, J.; Compan, V.; Barberà-Cremades, M.; Yagüe, J.; Ruiz-Ortiz, E.; Antón, J. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat. Immunol. 2014, 15, 738–748. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef]
- Cappelletti, C.; Salerno, F.; Canioni, E.; Mora, M.; Mantegazza, R.; Bernasconi, P.; Maggi, L. Up-regulation of toll-like receptors 7 and 9 and its potential implications in the pathogenic mechanisms of lmna-related myopathies. Nucleus 2018, 9, 398–409. [Google Scholar] [CrossRef] [Green Version]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Juliana, C.; Hong, S.; Datta, P.; Hwang, I.; Fernandes-Alnemri, T.; Yu, J.-W.; Alnemri, E.S. The mitochondrial anti-viral protein MAVS associates with NLRP3 and regulates its inflammasome activity 1. J. Immunol. 2013, 191, 4358–4366. [Google Scholar] [CrossRef] [Green Version]
- Deswaerte, V.; Ruwanpura, S.M.; Jenkins, B.J. Transcriptional regulation of inflammasome-associated pattern recognition receptors, and the relevance to disease pathogenesis. Mol. Immunol. 2017, 86, 3–9. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Kornegay, J.N.; Spurney, C.F.; Nghiem, P.P.; Brinkmeyer-Langford, C.L.; Hoffman, E.P.; Nagaraju, K. Pharmacologic Management of Duchenne Muscular Dystrophy: Target Identification and Preclinical Trials. ILAR J. 2014, 55, 119–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antosh, M.; Whitaker, R.; Kroll, A.; Hosier, S.; Chang, C.; Bauer, J.; Cooper, L.; Neretti, N.; Helfand, S.L. Comparative transcriptional pathway bioinformatic analysis of dietary restriction, Sir2, p53 and resveratrol life span extension in Drosophila. Cell Cycle 2011, 10, 904–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Interventional Studies Neuromuscular Diseases. Available online: https://clinicaltrials.gov/ct2/results?cond=Neuromuscular+Diseases&term=mitochondria&type=Intr&rslt=&age_v=&gndr=&intr=&titles=&outc=&spons=&lead=&id=&cntry=&state=&city=&dist=&locn=&rsub=&strd_s=&strd_e=&prcd_s=&prcd_e=&sfpd_s=&sfpd_e=&rfpd_s=&rfpd_e=&lu (accessed on 29 May 2020).
- Liang, J.; Zeng, Z.; Zhang, Y.; Chen, N. Regulatory role of exercise-induced autophagy for sarcopenia. Exp. Gerontol. 2020, 130, 110789. [Google Scholar] [CrossRef]
- Schoch, K.M.; Miller, T.M. Antisense Oligonucleotides: Translation from Mouse Models to Human Neurodegenerative Diseases. Neuron 2017, 94, 1056–1070. [Google Scholar] [CrossRef] [Green Version]
- Levin, A.A. Treating disease at the RNA level with oligonucleotides. N. Engl. J. Med. 2019, 380, 57–70. [Google Scholar] [CrossRef]
- Saraiva, J.; Nobre, R.J.; Pereira De Almeida, L. Gene therapy for the CNS using AAVs: The impact of systemic delivery by AAV9. J. Control. Release 2016, 241, 94–109. [Google Scholar] [CrossRef]
- Mendell, J.R. Single-Dose Gene-Replacement Therapy for SMA. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Young, C.S.; Pyle, A.D.; Spencer, M.J. CRISPR for Neuromuscular Disorders: Gene Editing and Beyond. Physiology 2019, 34, 341–353. [Google Scholar] [CrossRef]
- Understanding Neuromuscular Disease Care—IQVIA. Available online: https://www.iqvia.com/insights/the-iqvia-institute/reports/understanding-neuromuscular-disease-care (accessed on 28 May 2020).
- Patridge, E.V.; Gareiss, P.C.; Kinch, M.S.; Hoyer, D.W. An analysis of original research contributions toward FDA-approved drugs. Drug Discov. Today 2015, 20, 1182–1187. [Google Scholar] [CrossRef]
- S4R Registration Share4Rare. Available online: https://www.share4rare.org/registration/understanding-neuromuscular-diseases (accessed on 19 May 2020).



{kind=link}
{kind=link}
{kind=link}
| NMD Group | Name of Main Diseases | Description of Principal Disease Features | Description of Main Mitochondrial Alteration | Most Affected Mitochondrial Pathways | Main Evidence Level/Disease Model | Relevant References | Examples of Mutations in Genes |
|---|---|---|---|---|---|---|---|
| Muscular Dystro-phies (MD) | Becker MD (BMD) | Atrophy of the skeletal, cardiac, and pulmonary muscles | Reduced mitochondrial mass, complex I activity and ATP levels, and increased Ca2+ levels | Increased MPTP opening, ROS levels, and inflammation | in vitro, cells, animal (C. elegans, mdx mice), patient (CT) | [11,12,13] | DMD |
| Congenital MD (CMD) | Muscle weakness and possible joint deformities with slow progression and shortened life span | Most common CMD are collagen VI myopathies, with visible mitochondrial dysfunction | Increased MPTP opening and defective autophagy | in vitro, cells, animal (Col6a1 -/- mice), patient (CT) | [2,14,15] | LMNA, DPM3, DAG1, TRAPPC11 | |
| Duchenne MD (DMD) | General muscle weakness and wasting due to lack of dystrophin protein. Shortened lifespan. Rarely affects women (milder symptoms and better prognosis) | Massive aggregates of mitochondria, lower activities of MRC complexes III and IV and increased Ca2+. | Increased MPTP opening, ROS levels and inflammation | in vitro, cells, animal (C. elegans, mdx mice), patient (CT) | [8,11,12,14,16,17,18,19,20,21] | DMD | |
| Emery-Dreifuss MD (EDMD) | Muscular weakness and atrophy of shoulder, upper arm, and shin muscles, with early joint contractures and cardiomyopathy | Reduced expression of MRC complex genes and upregulation of mitochondrial disassembly genes, altered mitochondrial location and morphology | Decreased MRC and altered mitochondrial biogenesis | in vitro and animal models (C. elegans, mice) | [2,22,23,24] | LMNA, EMD, FHL1 | |
| Faciosca-pulohume-ral MD (FSHD) | Muscle weakness that affects mainly facial, shoulder, and arm muscles | Reduced antioxidative response molecules (low levels of zinc, selenium, and vitamin C) | Higher ROS and mitochondrial dysfunction | cells and patient (CT) | [14,17,25,26] | TRPV4, DUX4, SMCHD1 | |
| Limb-girdle MD (LGMD) | Weakness and wasting of the muscles in hips and shoulders | Morphologic mitochondrial abnormalities, including RRF and decreased COX | MPTP dysregulation and mitochondrial dysfunction | in vitro, cells, animal (521ΔT mice), patient (CT) | [2,5,14,27] | SGCG, LMNA, DYSF, TCAP, TRIM32, TNPO3 * | |
| Myotonic dystrophy (MD) | Muscle loss and weakness due to inability to relax them. It affects facial muscles first, but also feet, hands, and neck | Altered mitochondrial proteins (decreased EF-Tu, hsp60, GRP75, dienoyl CoA isomerase) and disruption of ubiquitin-proteasome systems in MD2 (affects proximal muscles) | ER stress and mitochondrial dysfunction | cells and patient (CT) | [14,28,29] | CNBP (ZNF9), DMPK | |
| Oculopha-ryngeal MD (OPMD) | Weakness of eye, face, and throat muscles leading to drooping eyelids and problems with swallowing | PABPN1 protein aggregates, reduced complex I and V mitochondrial proteins, altered UPR and apoptosis | ER stress, mitochondrial dysfunction and apoptosis | in vitro, cells, animal, patient (CT) | [24,30,31,32] | PABPN1 | |
| Distal MD (DD) (or Distal myopathy) | Weakness and wasting of muscles of the hands, forearms, and lower legs with slow progression. Many DD diseases | Decreased mitochondrial membrane potential, increased mitochondrial oxygen consumption and Ca2+ and deficiencies in MRC complexes I and IV | MPTP opening and ATP depletion | in vitro, cells, patient (CT) | [14,33,34] | FLNC, TTN, DYSF |
| NMD Group | Name of Main Diseases | Description of Principal Disease Features | Description of Main Mitochondrial Alteration | Most Affected Mitochondrial Pathways | Main Evidence Level/Disease Model | Relevant References | Examples of Mutations in Genes |
|---|---|---|---|---|---|---|---|
| Myopa-thies | Congenital myopathies | Inherited diseases that affect the tone and contraction of skeletal muscles causing general muscle weakness | The most frequent congenital myopathies are core myopathies, with reduced MRC activity and near-total depletion of mitochondria | reduced MRC and mitochondrial depletion | in vitro, cells, animal, patient (CT) | [14,29,36,37] | RYR1, DNM2, MTM1, TNPO3, COL6A1/2/3, COL12A1, PYROXD1, MSTO1 |
| Endocrine myopathies | Weakness and atrophy (shrinking) of the muscles around the shoulders and hips, muscle stiffness, cramps, and slowed reflexes caused by abnormal activity of the thyroid gland. Two types: hypothyroid (reduced hormone levels) and hyperthyroid myopathies (excess in hormones) | Hypothyroid myopathy: decreased TFAM, reduced mitochondrial DNA copy number and mitochondrial alterations (COX- fibers). Hyperthyroid: moderate increase in mitochondrial size and protein aggregates | mitochondrial structure, ER stress (proteotoxicity), autophagy failure | cells | [38,39,40] | ||
| Inflammatory myopathies | Chronic muscle inflammation accompanied by prolonged muscle fatigue and weakness. sIBM is the inflammatory myopathy with more mitochondrial alteration | Abnormal mitochondria in sIBM (RRFs and COX- fibers). In sIBM and PM, mtDNA deletions in muscle and altered autophagy | Altered mitochondrial structure, mtDNA deletions and autophagy | in vitro, cells, animal, patient (CT) | [7,14,41,42] | VCP, HNRNPA1, GNE | |
| Metabolic myopathies | Group of disorders caused each by a different genetic defect that impairs the body’s metabolism causing muscle weakness, exercise intolerance, muscle pain or cramps | Altered MRC: substrates are not properly processed or cannot enter mitochondria affecting energy production of the cell. mtDNA or nDNA mutations | reduced MRC and ATP depletion | in vitro, cells, patient (CT) | [7,28,43] | GAA, AGL, GBE1, PYGM, PFKM, PHKA1 | |
| Myofibrillar myopathies (MFM) | Characterized by muscle weakness, cardiomyopathy, myalgia, loss of sensation and weakness in the limbs (peripheral neuropathy), and respiratory failure | Mitochondrial abnormalities with RRF and enlarged mitochondria. Protein aggregates affecting distribution and function of mitochondria. Deficiencies in MRC complex I and IV | MRC dysfunction, autophagy and ER stress, reduced mitochondrial biogenesis | in vitro, cells, patient | [14,44,45,46] | MYOT, DES, PYROXD1, CRYAB, LDB3, FLNC, BAG3, TTN | |
| Scapulopero-neal myopathy | Rare genetic disorder characterized by weakness and wasting of specific muscles: shoulder blade area (scapula) and the smaller of the two leg muscle groups below the knee (peroneal) | mtDNA mutations reported in a few cases | mtDNA | cells | [47,48,49] | VCP, FHL1 |
| NMD Group | Name of Main Diseases | Description of Principal Disease Features | Description of Main Mitochondrial Alteration | Most Affected Mitochondrial Pathways | Main Evidence Level/Disease Model | Relevant References | Examples of Mutations in Genes |
|---|---|---|---|---|---|---|---|
| Congenital myasthenic syndromes (CMS) | Weakness and fatigue resulting from problems at NMJ. Different types of CMS, according to the part of the NMJ affected: presynaptic (the nerve cell), postsynaptic (the muscle cell) or synaptic (the space in between) | Gene defect in mitochondrial citrate carrier SLC25A1 underlie deficits in NMJ transmission. SLC25A1 is involved in many biological processes (e.g., glycolysis, autophagy) | NMJ signaling | in vitro, cells, animal (zebrafish, mice), patient (CT) | [50,51,52,53] | CHRNA1, PLEC, CHRNB1, CHRND, CHRNE, COLQ, CHAT, SYT2, AGRN, SLC5A7, SYT2 | |
| Neuro-muscular junction diseases (NMJ) | Lambert-Eaton myasthenic syndrome (LEMS) | Autoimmune disease that attacks the calcium channels in the NMJ and interferes with the ability of nerve cells to send acetylcholine to muscle cells, affecting muscle contraction and causing muscle weakness | Altered calcium channels | NMJ signaling | cells, patient (CT) | [50,54,55,56] | SYT2 |
| Myasthenia gravis (MG) | Chronic autoimmune disorder in which antibodies destroy neuromuscular connections. It affects voluntary muscles of the body, especially the eyes, mouth, throat, and limbs | Mitochondrial morphological alterations and RRF | mitochondrial morphology, neuromuscular connections | cells, animal (rat, mice), patient (CT) | [55,57,58] | CHAT | |
| Amyotrophic lateral sclerosis (ALS) | Fatal disease with degeneration of nerve cells in the spinal cord and brain. It affects voluntary control of arms and legs and eventually leads to trouble breathing | Accumulation of mitochondrial in proximal axons, mitochondrial injury by ROS excess, COX I mtDNA mutation and RRF | Increased ROS and altered mitochondrial structure | in vitro, cells, animal (SOD1G93A mice), patient (CT) | [7,14,17,18,45,59,60,61,62,63,64,65] | SOD1, ALS2, SPG111, HNRNPA1, SQSTM1* | |
| Motor neuron diseases | Spinal-bulbar muscular atrophy (SBMA) | Genetic disorder in which loss of lower motor neurons affect voluntary muscle movement, specifically facial, swallowing muscles and limbs. Only affects men | Reduced MMP. Increased expression of apoptotic proteins that activate mitochondrial caspase pathway. AR unfolding and oligomerization induces toxicity | MPTP opening, increased ROS and apoptosis. Lower mitochondrial mass and ER stress | in vitro, cells, animal, patient (CT) | [66,67,68,69,70] | AR |
| Spinal muscular atrophy (SMA) | Genetic disease affecting the central and peripheral nervous system, and voluntary muscle movement, mainly shoulders, hips, thighs, and upper back | Decreased enzyme activities involving MRC complexes I-IV causing mitochondrial dysfunction | mitochondrial dysfunction and altered MRC | in vitro, cells, animal, patient (CT) | [8,14,16,17,18,67,69,70,71] | SMN1, IGHMBP2, SIGMAR1, PLEKHG5, DNAJB2, VRK1, TRPV4 |
| NMD Group | Name of Main Diseases | Description of Principal Disease Features | Description of Main Mitochondrial Alteration | Most Affected Mitochondrial Pathways | Main Evidence Level/Disease Model | Relevant References | Examples of Mutations in Genes |
|---|---|---|---|---|---|---|---|
| Peripheral nerve diseases | Charcot-Marie-Tooth disease (CMT) | Inherited disorder that affects nerves outside of your brain and spinal cord, nerves that supply feet, legs, hands, and arms. Two subtypes: CMT1 (demyelinating), CMT2 (axonal) | Altered mitochondrial dynamics and axonal transport of mitochondria, causing axonal degeneration. Mutations in mitochondrial fusion regulatory genes | mitochondrial dynamics and ER stress | in vitro, cells, animal, patient (CT) | [14,17,26,45,67,72,73,74] | CMT1: PMP22, MPZ. CMT2: MFN2, LMNA, VCP, DNM2, IGHMBP2, DNAJB2, AARS, DYNC1H1, SOD2 |
| Giant axonal neuropathy (GAN) | Inherited condition characterized by abnormally large and dysfunctional axons. First, limbs have problems with walking, followed by difficulties coordinating movements (ataxia), and require wheelchair assistance | Abnormal and enlarged mitochondria in Schwann cells, deficiencies of complexes I and IV, several mtDNA point mutations and multiple mtDNA deletions. | altered mitochondrial size and mitochondrial dynamics, reduced MRC function | in vitro, cells, animal, patient (CT) | [7,75,76] | GAN | |
| Mitochon-drial diseases | Mitochon-drial myopathies | Genetic defects that affect mitochondria can cause muscular and neurological problems (e.g., muscle weakness, exercise intolerance, trouble with balance and coordination). Many mitochondrial myopathies are known | Mutations in mtDNA or nDNA affecting proteins involved in MRC, mitochondrial morphology (RRF) and protein aggregation (UPR) | Altered energy production and redox signaling, mitochondrial morphology and ER stress | in vitro, cells, animal (ANT- mice), patient (CT) | [9,77,78,79] | MRPS25, TIMM22, NDUFAF1, COX6A2, SLC25A42 |
| Friedreich’s ataxia (FA) | Muscle weakness and ataxia, loss of balance and coordination due to reduced synthesis of the mitochondrial protein frataxin. It mostly affects the spinal cord, peripheral nerves and cerebellum | Altered synthesis of frataxin affects mitochondrial iron metabolism and homeostasis and antioxidant protection | oxidative stress, mitochondrial dysfunction | in vitro, cells, patient (CT) | [7,80,81] | FXN |
| NMD Group | Name of Main Diseases | Description of Principal Disease Features | Description of Main Mitochondrial Alteration | Most Affected Mitochondrial Pathways | Main Evidence Level/Disease Model | Relevant References | Examples of Mutations in Genes |
|---|---|---|---|---|---|---|---|
| Ion channel diseases (or Channe-lopathies) | Andersen-Tawil syndrome | Altered potassium channel gene affect the heartbeat and the ability of muscles to stay ready to contract. Paralysis may occur | Mitochondrial channelopathies affect the K+, Ca2+, VDAC and MPTP channels. Reduced channel activity rate results in reduced MMP and delayed repolarization, causing mitochondrial dysfunction. Intracellular Ca2+ homeostasis, mitochondrial bioenergetic metabolism, and modulation of cell survival and death are also affected | reduced MMP, mitochondrial dysfunction, apoptosis | in vitro, cells, animal (mice, rat, C. elegans, patient (CT) | [82,83,84,85] | KCNJ2 |
| Hyperkalemic periodic paralysis | Genetic alterations in sodium channels results in temporary muscle weakness, and eventually, temporary paralysis | SCN4A, T704M, M1592V | |||||
| Hypokalemic periodic paralysis | Genetic defects in calcium or sodium channel cause a loss of muscle excitability when serum potassium is low | SCN4A, CACNA1S, ATP1A2, KCNE3 | |||||
| Myotonia congenita | Disease caused by mutations in the gene encoding a chloride channel necessary for stopping muscle contraction. Delayed muscle relaxation triggers muscle stiffness | CLCN1 (CLC-1), SCN4A | |||||
| Paramyotonia congenita | Mutations in the muscle sodium channel gene prolong the channel’s opening, higher muscle excitation triggering episodes of muscle stiffness and weakness, mostly in the face, neck and upper extremities | SCN4A | |||||
| Potassium-aggravated myotonia (or Sodium Channel myotonias) | Sustained muscle tensing causes muscle stiffness that worsens after exercise and may be aggravated by eating potassium-rich foods | SCN4A |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cantó-Santos, J.; Grau-Junyent, J.M.; Garrabou, G. The Impact of Mitochondrial Deficiencies in Neuromuscular Diseases. Antioxidants 2020, 9, 964. https://doi.org/10.3390/antiox9100964
Cantó-Santos J, Grau-Junyent JM, Garrabou G. The Impact of Mitochondrial Deficiencies in Neuromuscular Diseases. Antioxidants. 2020; 9(10):964. https://doi.org/10.3390/antiox9100964
Chicago/Turabian StyleCantó-Santos, Judith, Josep M. Grau-Junyent, and Glòria Garrabou. 2020. "The Impact of Mitochondrial Deficiencies in Neuromuscular Diseases" Antioxidants 9, no. 10: 964. https://doi.org/10.3390/antiox9100964
APA StyleCantó-Santos, J., Grau-Junyent, J. M., & Garrabou, G. (2020). The Impact of Mitochondrial Deficiencies in Neuromuscular Diseases. Antioxidants, 9(10), 964. https://doi.org/10.3390/antiox9100964