The Late-Stage Protective Effect of Mito-TEMPO against Acetaminophen-Induced Hepatotoxicity in Mouse and Three-Dimensional Cell Culture Models

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Materials
2.2. Animals and Protocols
2.3. Drug Administration
2.4. Measurement of Alanine Aminotransaminase (ALT) Activity
2.5. Hepatic Histopathology
2.6. Immunostaining
2.7. Hepatic Total Glutathione Content
2.8. Western Blotting
2.9. Hepatic mRNA Isolation and Real-Time RT-PCR
2.10. Cell Cultures and Induction of the APAP Hepatotoxicity Model in 3-Dimensional HepG2 Cells
2.11. Cell Viability Assays in the 3D-HepG2 Cell Model
2.12. Measurement of Mitochondrial Oxidative Stress in the 3D-HepG2 Cell Model
2.13. Statistical Analysis
3. Results
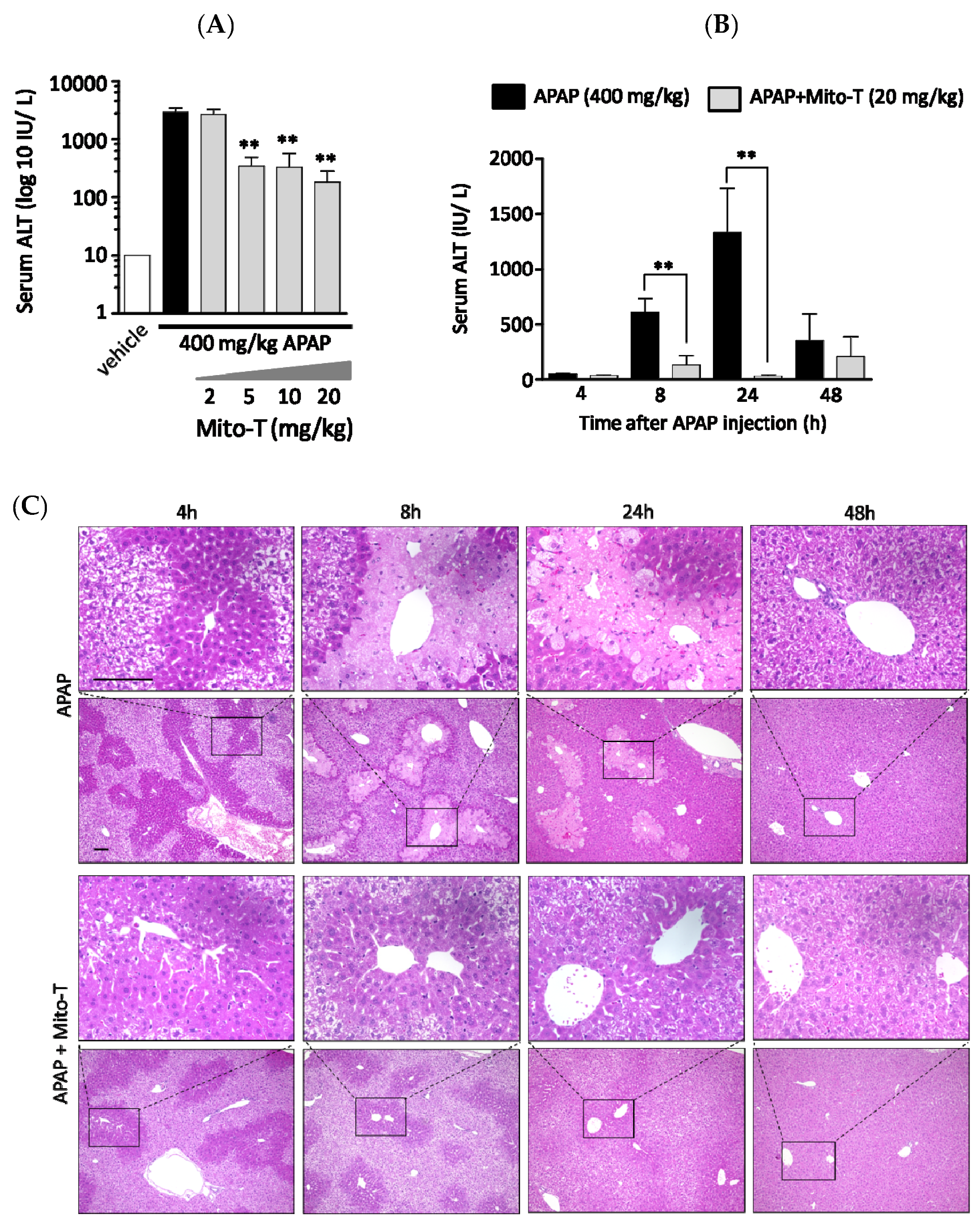
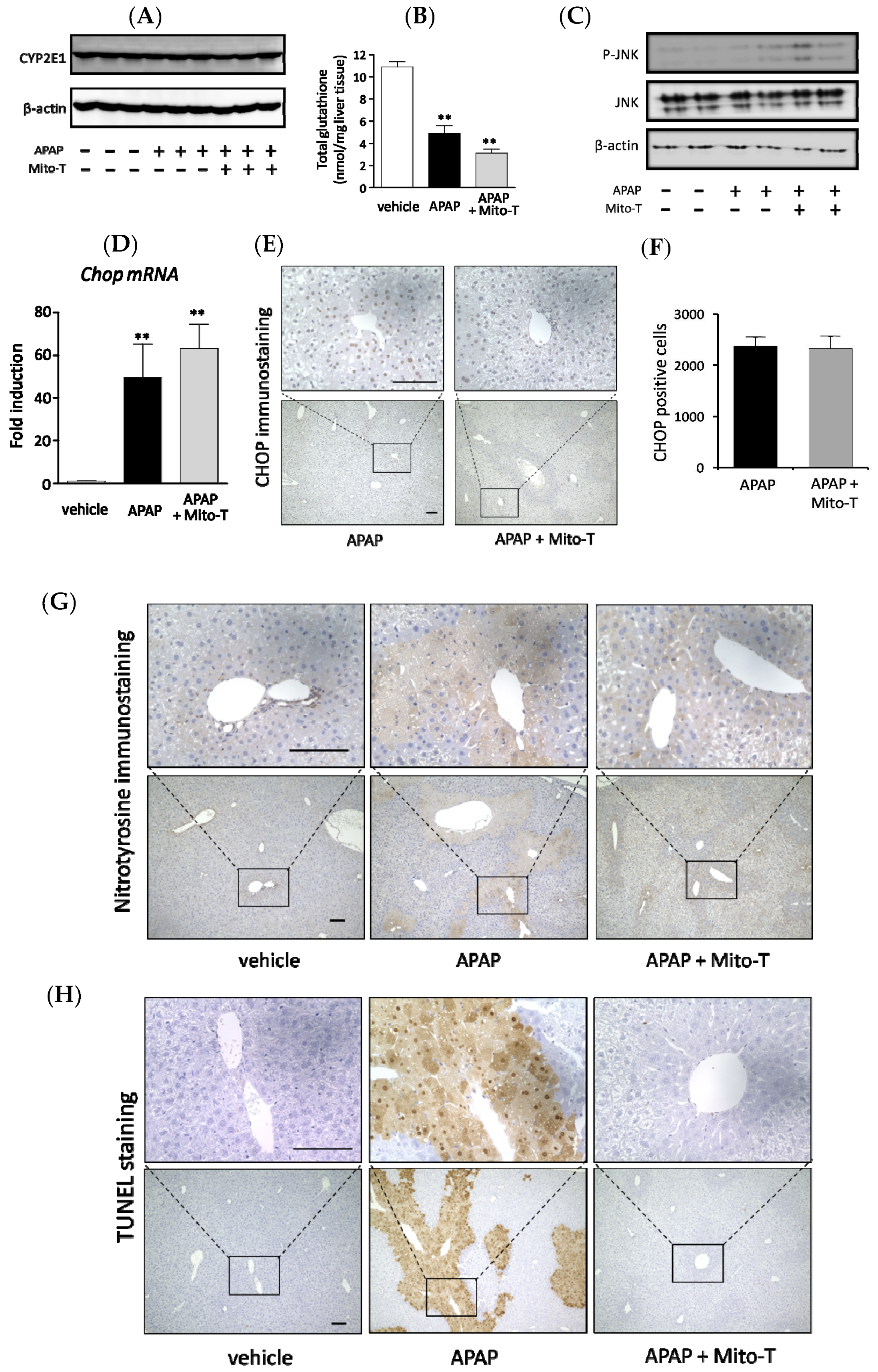
3.1. Protective Effect of Mito-T against APAP-Induced Liver Injury in C57BL/6J Mice
3.2. Efficacy of Mito-T on Liver Injury Progression in C57BL/6J Mice Following APAP Overdose
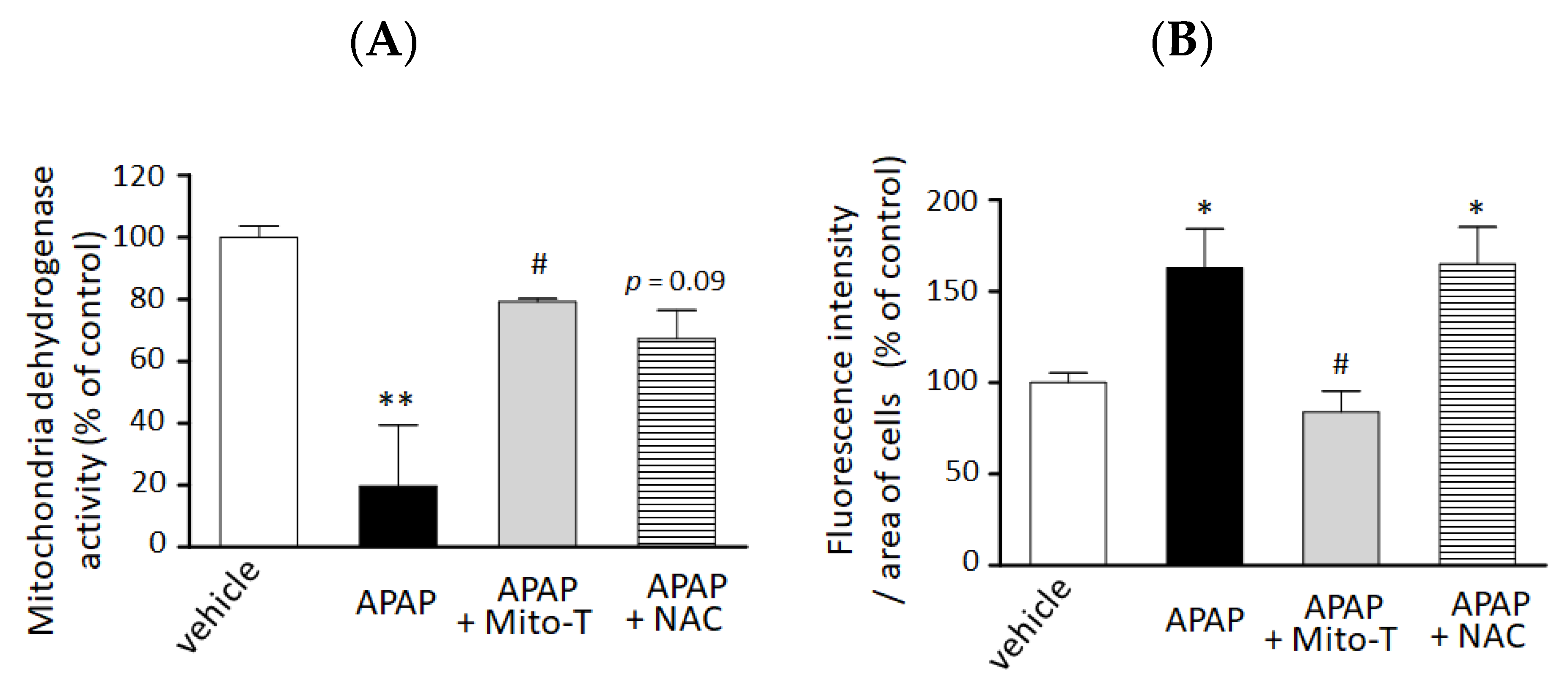
3.3. Mito-T Prevents Cellular Injury and Reduces Mitochondrial Oxidative Stress in APAP-Treated HepG2 Cells
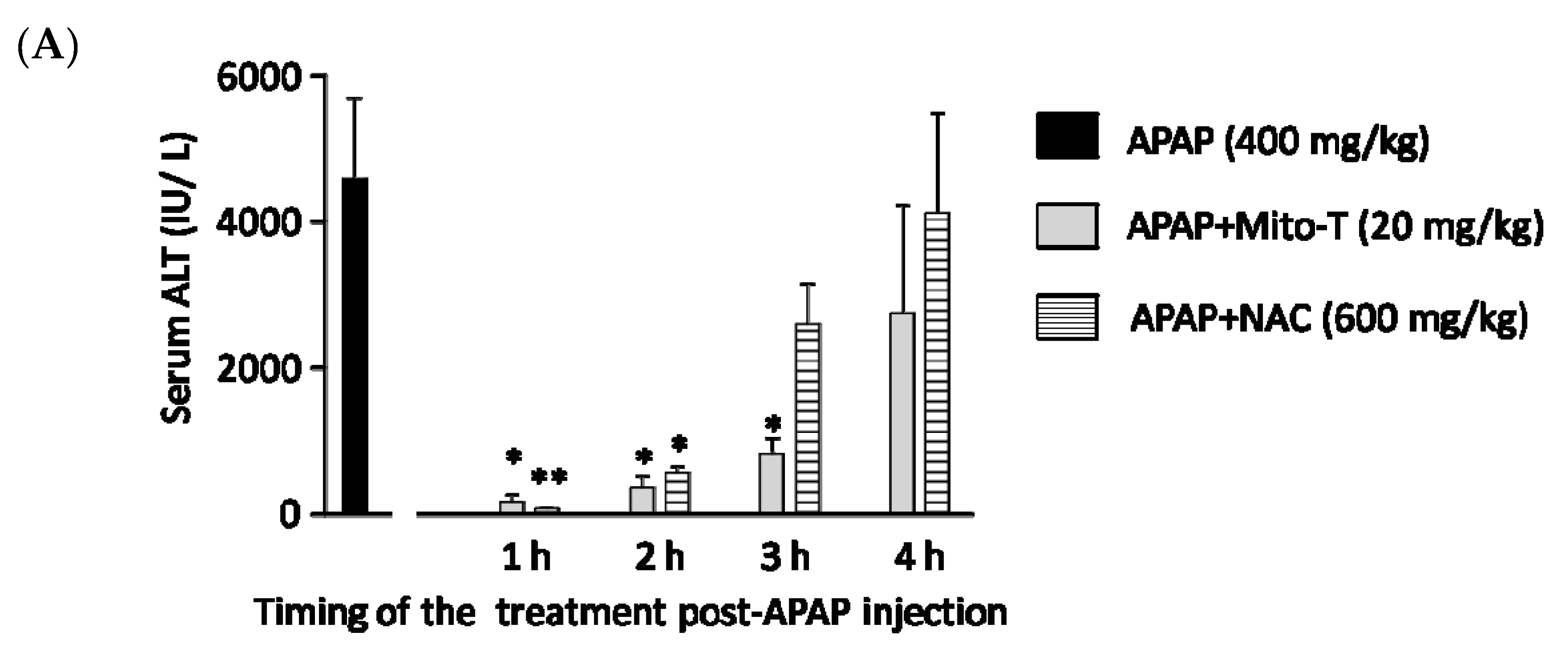
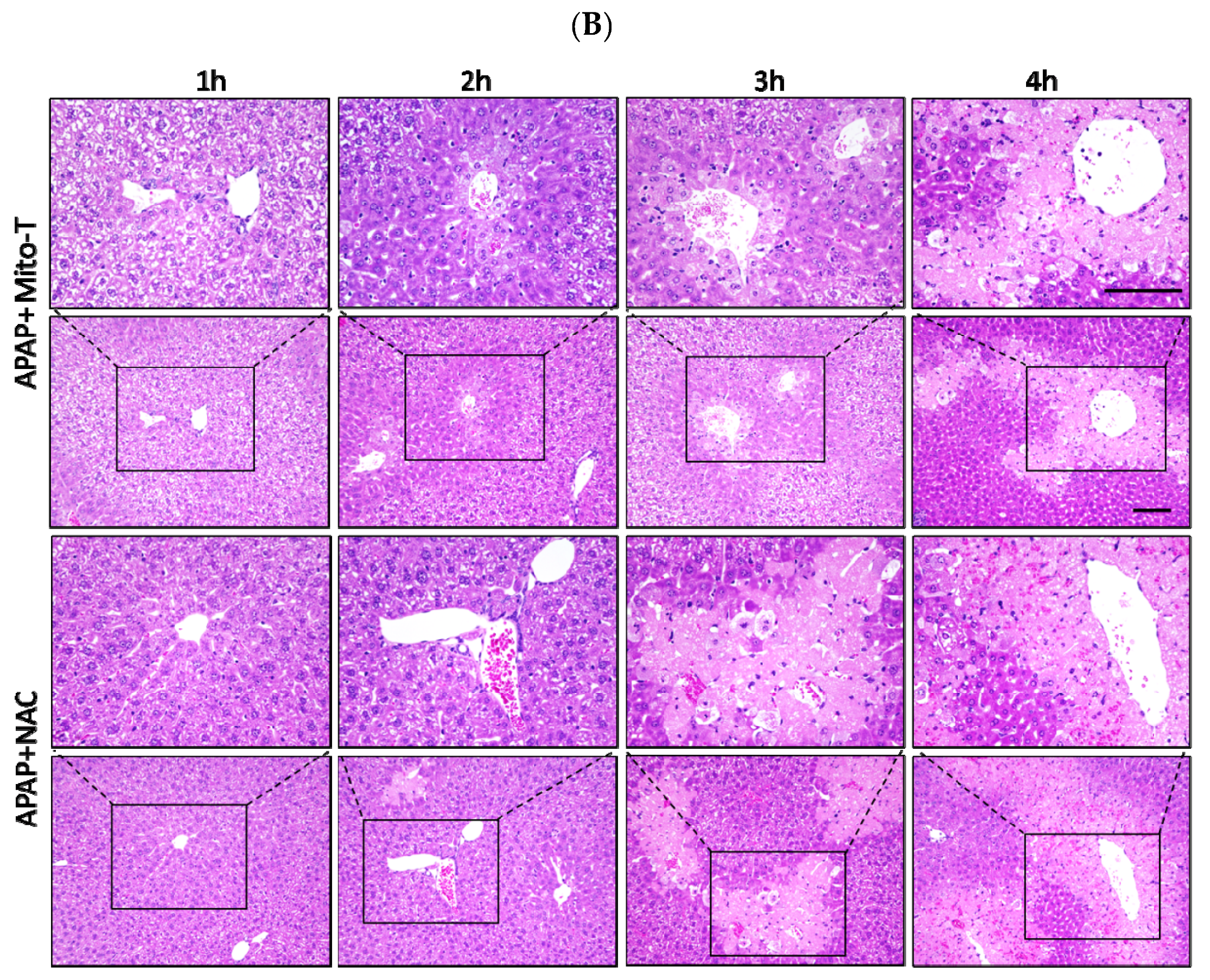
3.4. Therapeutic Time Window of Mito-T and Its Role in APAP-Induced Liver Injury
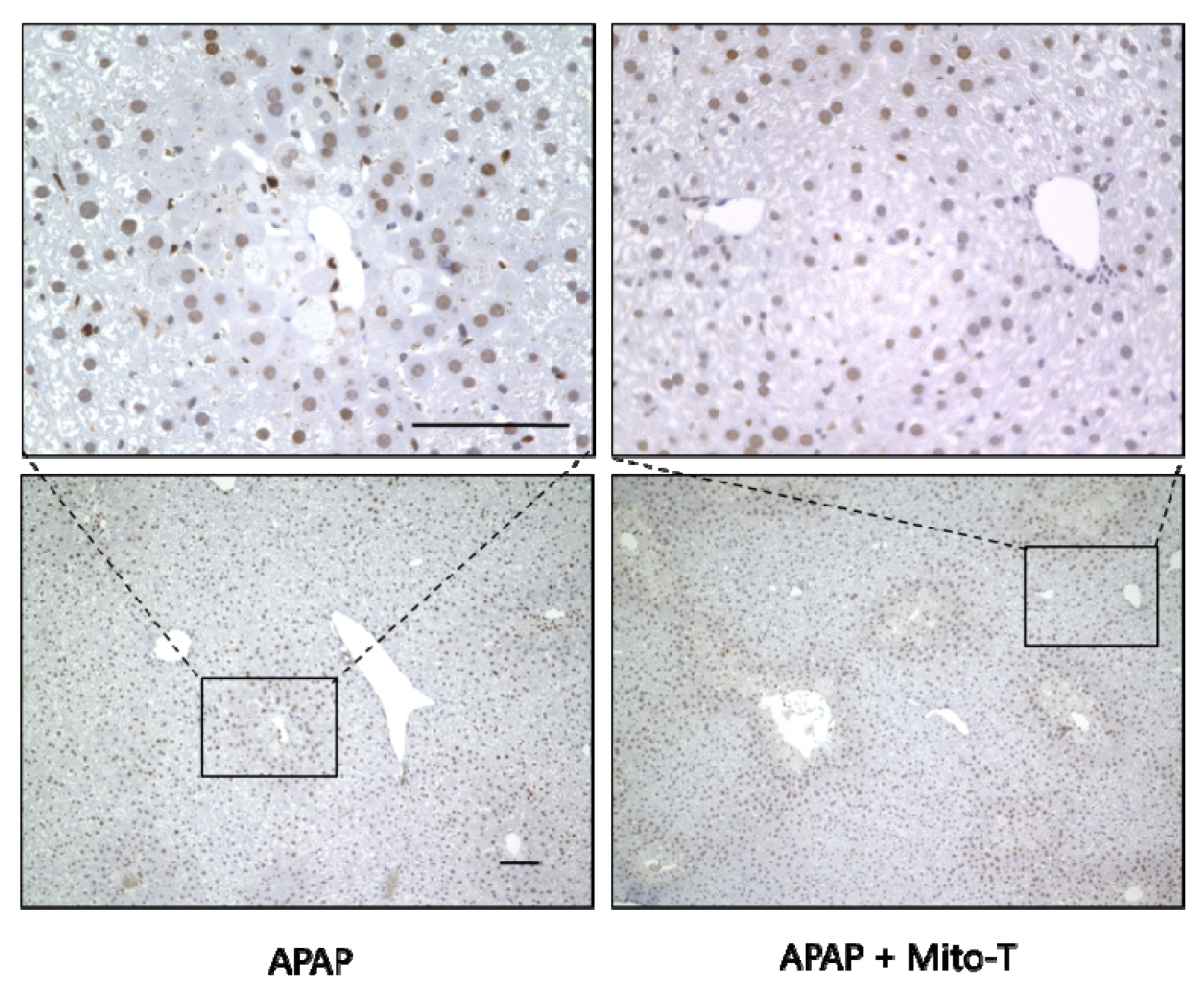
3.5. Effect of Mito-T on Liver Regeneration after APAP-Induced Liver Injury
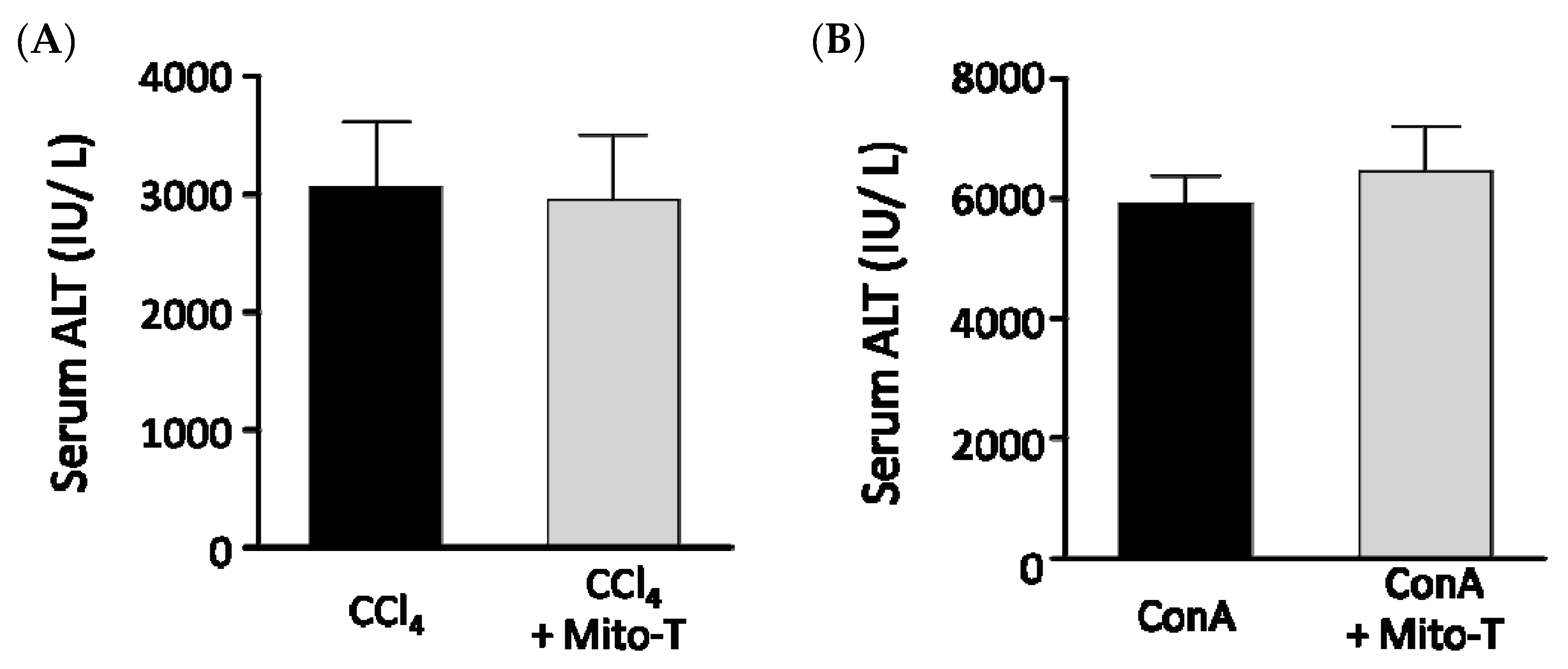
3.6. Effect of Mito-T on Other Drug-Induced Liver Injury Models
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Graham, G.G.; Scott, K.F.; Day, R.O. Tolerability of Paracetamol. Drug Saf. 2005, 28, 227–240. [Google Scholar] [CrossRef]
- Nourjah, P.; Ahmad, S.R.; Karwoski, C.; Willy, M. Estimates of acetaminophen (paracetomal)-associated overdoses in the United States. Pharmacoepidemiol. Drug Saf. 2006, 15, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Michna, E.; Duh, M.S.; Korves, C.; Dahl, J.L. Removal of Opioid/Acetaminophen Combination Prescription Pain Medications: Assessing the Evidence for Hepatotoxicity and Consequences of Removal of These Medications. Pain Med. 2010, 11, 369–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.M. Acetaminophen (APAP) hepatotoxicity—Isn’t it time for APAP to go away? J. Hepatol. 2017, 67, 1324–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, D.G.N.; Bates, C.M.; Davidson, J.S.; Martin, K.G.; Hayes, P.C.; Simpson, K.J. Overdose pattern and outcome in paracetamol-induced acute severe hepatotoxicity. Br. J. Clin. Pharmacol. 2011, 71, 273–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Martin, B.C. Trends in emergency department visits attributable to acetaminophen overdoses in the United States: 1993–2007. Pharmacoepidemiol. Drug Saf. 2011, 20, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Bernal, W.; Auzinger, G.; Dhawan, A.; Wendon, J. Acute liver failure. Lancet 2010, 376, 190–201. [Google Scholar] [CrossRef]
- Lee, S.S.T.; Buters, J.T.M.; Pineau, T.; Fernandez-Salguero, P.; Gonzalez, F.J. Role of CYP2E1 in the Hepatotoxicity of Acetaminophen. J. Biol. Chem. 1996, 271, 12063–12067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, F.J. Role of cytochromes P450 in chemical toxicity and oxidative stress: Studies with CYP2E1. Mutat. Res. Mol. Mech. Mutagen. 2005, 569, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.D.; Pumford, N.R.; Khairallah, E.A.; Boekelheide, K.; Pohl, L.R.; Amouzadeh, H.R.; Hinson, J.A. Selective Protein Covalent Binding and Target Organ Toxicity. Toxicol. Appl. Pharmacol. 1997, 143, 1–12. [Google Scholar] [CrossRef]
- McGill, M.R.; Jaeschke, H. Metabolism and Disposition of Acetaminophen: Recent Advances in Relation to Hepatotoxicity and Diagnosis. Pharm. Res. 2013, 30, 2174–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyers, L.L.; Beierschmitt, W.P.; Khairallah, E.A.; Cohen, S.D. Acetaminophen-induced inhibition of hepatic mitochondrial respiration in mice. Toxicol. Appl. Pharmacol. 1988, 93, 378–387. [Google Scholar] [CrossRef]
- Jaeschke, H. Glutathione disulfide formation and oxidant stress during acetaminophen-induced hepatotoxicity in mice in vivo: The protective effect of allopurinol. J. Pharmacol. Exp. Ther. 1990, 255, 935–941. [Google Scholar] [PubMed]
- Donnelly, P.J.; Walker, R.M.; Racz, W.J. Inhibition of mitochondrial respiration in vivo is an early event in acetaminophen-induced hepatotoxicity. Arch. Toxicol. 1994, 68, 110. [Google Scholar] [CrossRef] [PubMed]
- Hanawa, N.; Shinohara, M.; Saberi, B.; Gaarde, W.A.; Han, D.; Kaplowitz, N. Role of JNK Translocation to Mitochondria Leading to Inhibition of Mitochondria Bioenergetics in Acetaminophen-induced Liver Injury. J. Biol. Chem. 2008, 283, 13565–13577. [Google Scholar] [CrossRef] [Green Version]
- Du, K.; Ramachandran, A.; Jaeschke, H. Oxidative stress during acetaminophen hepatotoxicity: Sources, pathophysiological role and therapeutic potential. Redox Biol. 2016, 10, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Hinson, J.A.; Pike, S.L.; Pumford, N.R.; Mayeux, P.R. Nitrotyrosine−Protein Adducts in Hepatic Centrilobular Areas following Toxic Doses of Acetaminophen in Mice. Chem. Res. Toxicol. 1998, 11, 604–607. [Google Scholar] [CrossRef]
- LoGuidice, A.; Boelsterli, U.A. Acetaminophen overdose-induced liver injury in mice is mediated by peroxynitrite independently of the cyclophilin D-regulated permeability transition. Hepatology 2011, 54, 969–978. [Google Scholar] [CrossRef]
- Uzi, D.; Barda, L.; Scaiewicz, V.; Mills, M.; Mueller, T.; Gonzalez-Rodriguez, A.; Valverde, A.M.; Iwawaki, T.; Nahmias, Y.; Xavier, R.; et al. CHOP is a critical regulator of acetaminophen-induced hepatotoxicity. J. Hepatol. 2013, 59, 495–503. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Sugiyama, S.; Horie, T. Th1/Th2 cytokine balance as a determinant of acetaminophen-induced liver injury. Chem. Biol. Interact. 2009, 179, 273–279. [Google Scholar] [CrossRef]
- James, L.P.; McCullough, S.S.; Lamps, L.W.; Hinson, J.A. Effect of N-Acetylcysteine on Acetaminophen Toxicity in Mice: Relationship to Reactive Nitrogen and Cytokine Formation. Toxicol. Sci. 2003, 75, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Williams, C.D.; Ramachandran, A.; Bajt, M.L. Acetaminophen hepatotoxicity and repair: The role of sterile inflammation and innate immunity. Liver Int. Off. J. Int. Assoc. Study Liver 2012, 32, 8–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, E.; Nguyen, H.; Lippert, T.; Tuazon, J.; Borlongan, C.V.; Napoli, E. Mitochondrial targeting as a novel therapy for stroke. Brain Circ. 2018, 4, 84–94. [Google Scholar] [CrossRef]
- Teixeira, J.; Deus, C.M.; Borges, F.; Oliveira, P.J. Mitochondria: Targeting mitochondrial reactive oxygen species with mitochondriotropic polyphenolic-based antioxidants. Int. J. Biochem. Cell Biol. 2018, 97, 98–103. [Google Scholar] [CrossRef]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [Green Version]
- McGill, M.R.; Sharpe, M.R.; Williams, C.D.; Taha, M.; Curry, S.C.; Jaeschke, H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J. Clin. Investig. 2012, 122, 1574–1583. [Google Scholar] [CrossRef] [Green Version]
- Du, K.; Ramachandran, A.; Weemhoff, J.L.; Chavan, H.; Xie, Y.; Krishnamurthy, P.; Jaeschke, H. Editor’s Highlight: Metformin Protects Against Acetaminophen Hepatotoxicity by Attenuation of Mitochondrial Oxidant Stress and Dysfunction. Toxicol. Sci. 2016, 154, 214–226. [Google Scholar] [CrossRef]
- Lee, K.K.; Imaizumi, N.; Chamberland, S.R.; Alder, N.N.; Boelsterli, U.A. Targeting mitochondria with methylene blue protects mice against acetaminophen-induced liver injury. Hepatology 2015, 61, 326–336. [Google Scholar] [CrossRef]
- Shi, X.; Bai, H.; Zhao, M.; Li, X.; Sun, X.; Jiang, H.; Fu, A. Treatment of acetaminophen-induced liver injury with exogenous mitochondria in mice. Transl. Res. 2018, 196, 31–41. [Google Scholar] [CrossRef]
- Trnka, J.; Blaikie, F.H.; Smith, R.A.J.; Murphy, M.P. A mitochondria-targeted nitroxide is reduced to its hydroxylamine by ubiquinol in mitochondria. Free Radic. Biol. Med. 2008, 44, 1406–1419. [Google Scholar] [CrossRef]
- Du, K.; Farhood, A.; Jaeschke, H. Mitochondria-targeted antioxidant Mito-Tempo protects against acetaminophen hepatotoxicity. Arch. Toxicol. 2017, 91, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Ramachandran, A.; Weemhoff, J.L.; Woolbright, B.L.; Jaeschke, A.H.; Chao, X.; Ding, W.-X.; Jaeschke, H. Mito-tempo protects against acute liver injury but induces limited secondary apoptosis during the late phase of acetaminophen hepatotoxicity. Arch. Toxicol. 2019, 93, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, D.; Ishitsuka, Y.; Miyata, K.; Tomishima, Y.; Kondo, Y.; Irikura, M.; Iwawaki, T.; Oike, Y.; Irie, T. Protection afforded by pre- or post-treatment with 4-phenylbutyrate against liver injury induced by acetaminophen overdose in mice. Pharmacol. Res. 2014, 87, 26–41. [Google Scholar] [CrossRef]
- Aritomi, K.; Ishitsuka, Y.; Tomishima, Y.; Shimizu, D.; Abe, N.; Shuto, T.; Irikura, M.; Kai, H.; Irie, T. Evaluation of three-dimensional cultured HepG2 cells in a nano culture plate system: An in vitro human model of acetaminophen hepatotoxicity. J. Pharmacol. Sci. 2014, 124, 218–229. [Google Scholar] [CrossRef] [Green Version]
- Umezaki, Y.; Iohara, D.; Anraku, M.; Ishitsuka, Y.; Irie, T.; Uekama, K.; Hirayama, F. Preparation of hydrophilic C60(OH)10/2-hydroxypropyl-β-cyclodextrin nanoparticles for the treatment of a liver injury induced by an overdose of acetaminophen. Biomaterials 2015, 45, 115–123. [Google Scholar] [CrossRef]
- Tomishima, Y.; Ishitsuka, Y.; Matsunaga, N.; Nagatome, M.; Furusho, H.; Irikura, M.; Ohdo, S.; Irie, T. Ozagrel hydrochloride, a selective thromboxane A2 synthase inhibitor, alleviates liver injury induced by acetaminophen overdose in mice. BMC Gastroenterol. 2013, 13, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Ishitsuka, Y.; Hayasaka, M.; Yamada, Y.; Miyata, K.; Endo, M.; Kondo, Y.; Moriuchi, H.; Irikura, M.; Tanaka, K.; et al. The exacerbating roles of CCAAT/enhancer-binding protein homologous protein (CHOP) in the development of bleomycin-induced pulmonary fibrosis and the preventive effects of tauroursodeoxycholic acid (TUDCA) against pulmonary fibrosis in mice. Pharmacol. Res. 2015, 99, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Yan, D.; Zhang, Q.; Zhang, G.; Xia, M.; Li, J.; Zhan, W.; Shen, E.; Li, Z.; Lin, L.; et al. Treatment of acetaminophen-induced liver failure by blocking the death checkpoint protein TRAIL. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2020, 1866, 165583. [Google Scholar] [CrossRef] [PubMed]
- Endo, M.; Mori, M.; Akira, S.; Gotoh, T. C/EBP Homologous Protein (CHOP) Is Crucial for the Induction of Caspase-11 and the Pathogenesis of Lipopolysaccharide-Induced Inflammatio. J. Immunol. 2006, 176, 6245–6253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, F.-J.; Ma, L.-L.; Guo, J.-J.; Xu, L.-H.; Li, Y.; Qu, S. Endoplasmic reticulum stress/autophagy pathway is involved in diabetes-induced neuronal apoptosis and cognitive decline in mice. Clin. Sci. 2018, 132, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, K.; Zhang, S.; Shan, L.; Tang, J. Tetramethylpyrazine Showed Therapeutic Effects on Sepsis-Induced Acute Lung Injury in Rats by Inhibiting Endoplasmic Reticulum Stress Protein Kinase RNA-Like Endoplasmic Reticulum Kinase (PERK) Signaling-Induced Apoptosis of Pulmonary Microvascular Endothelial Cells. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 1225–1231. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, R.; Hennings, L.; Rafferty, T.M.; Letzig, L.G.; McCullough, S.; James, L.P.; MacMillan-Crow, L.A.; Hinson, J.A. Acetaminophen-Induced Hepatotoxicity and Protein Nitration in Neuronal Nitric-Oxide Synthase Knockout Mice. J. Pharmacol. Exp. Ther. 2012, 340, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Larson, A.M. Acetaminophen hepatotoxicity. Clin. Liver Dis. 2007, 11, 525–548. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Chu, S.; Zhang, Z.; Zuo, W.; Xia, C.; Ai, Q.; Luo, P.; Cao, P.; Chen, N. Early Stage Functions of Mitochondrial Autophagy and Oxidative Stress in Acetaminophen-Induced Liver Injury. J. Cell. Biochem. 2017, 118, 3130–3141. [Google Scholar] [CrossRef]
- Albrecht, J.H.; Poon, R.Y.; Ahonen, C.L.; Rieland, B.M.; Deng, C.; Crary, G.S. Involvement of p21 and p27 in the regulation of CDK activity and cell cycle progression in the regenerating liver. Oncogene 1998, 16, 2141–2150. [Google Scholar] [CrossRef] [Green Version]
- Slater, T.F.; Cheeseman, K.H.; Ingold, K.U. Carbon tetrachloride toxicity as a model for studying free-radical mediated liver injury. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1985, 311. [Google Scholar] [CrossRef]
- Tiegs, G.; Hentschel, J.; Wendel, A. A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J. Clin. Investig. 1992, 90, 196–203. [Google Scholar] [CrossRef]
- Itoh, A.; Isoda, K.; Kondoh, M.; Kawase, M.; Kobayashi, M.; Tamesada, M.; Yagi, K. Hepatoprotective Effect of Syringic Acid and Vanillic Acid on Concanavalin A-Induced Liver Injury. Biol. Pharm. Bull. 2009, 32, 1215–1219. [Google Scholar] [CrossRef] [Green Version]
- Seki, E.; Brenner, D.A.; Karin, M. A Liver Full of JNK: Signaling in Regulation of Cell Function and Disease Pathogenesis, and Clinical Approaches. Gastroenterology 2012, 143, 307–320. [Google Scholar] [CrossRef] [Green Version]
- Rumack, B.H.; Peterson, R.C.; Koch, G.G.; Amara, I.A. Acetaminophen Overdose: 662 Cases With Evaluation of Oral Acetylcysteine Treatment. JAMA Intern. Med. 1981, 141, 380–385. [Google Scholar] [CrossRef]
- Craig, D.G.N.; Bates, C.M.; Davidson, J.S.; Martin, K.G.; Hayes, P.C.; Simpson, K.J. Staggered overdose pattern and delay to hospital presentation are associated with adverse outcomes following paracetamol-induced hepatotoxicity. Br. J. Clin. Pharmacol. 2012, 73, 285–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.; Zou, X.; Koskinen, M.-L.; Tenhunen, J. Ethyl pyruvate reduces liver injury at early phase but impairs regeneration at late phase in acetaminophen overdose. Crit. Care 2012, 16, R9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhushan, B.; Chavan, H.; Borude, P.; Xie, Y.; Du, K.; McGill, M.R.; Lebofsky, M.; Jaeschke, H.; Krishnamurthy, P.; Apte, U. Dual Role of Epidermal Growth Factor Receptor in Liver Injury and Regeneration after Acetaminophen Overdose in Mice. Toxicol. Sci. 2016, 155, 363–378. [Google Scholar] [CrossRef]
- Gujral, J.S.; Knight, T.R.; Farhood, A.; Bajt, M.L.; Jaeschke, H. Mode of Cell Death after Acetaminophen Overdose in Mice: Apoptosis or Oncotic Necrosis? Toxicol. Sci. 2002, 67, 322–328. [Google Scholar] [CrossRef]
- Jaeschke, H.; Williams, C.D.; Farhood, A. No evidence for caspase-dependent apoptosis in acetaminophen hepatotoxicity. Hepatol. Baltim. Md. 2011, 53, 718–719. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Park, H.-J.; Park, J.; Song, H.-C.; Ryter, S.W.; Surh, Y.-J.; Kim, U.-H.; Joe, Y.; Chung, H.T. Carbon monoxide ameliorates acetaminophen-induced liver injury by increasing hepatic HO-1 and Parkin expression. FASEB J. 2019, 33, 13905–13919. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Hao, B.; Yang, Y.; Muhammad, I.; Zhang, Y.; Chang, Y.; Li, Y.; Li, C.; Li, R.; Liu, F. JNK Signaling Pathway Mediates Acetaminophen-Induced Hepatotoxicity Accompanied by Changes of Glutathione S-Transferase A1 Content and Expression. Front. Pharmacol. 2019, 10, 1092. [Google Scholar] [CrossRef]
- Liu, F.-C.; Lee, H.-C.; Liao, C.-C.; Li, A.H.; Yu, H.-P. Tropisetron Protects Against Acetaminophen-Induced Liver Injury via Suppressing Hepatic Oxidative Stress and Modulating the Activation of JNK/ERK MAPK Pathways. Available online: https://www.hindawi.com/journals/bmri/2016/1952947/ (accessed on 3 September 2020).
- Mobasher, M.A.; González-Rodriguez, Á.; Santamaría, B.; Ramos, S.; Martín, M.Á.; Goya, L.; Rada, P.; Letzig, L.; James, L.P.; Cuadrado, A.; et al. Protein tyrosine phosphatase 1B modulates GSK3 β /Nrf2 and IGFIR signaling pathways in acetaminophen-induced hepatotoxicity. Cell Death Dis. 2013, 4, e626. [Google Scholar] [CrossRef] [Green Version]
- Hur, K.Y.; So, J.-S.; Ruda, V.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Iwawaki, T.; Glimcher, L.H.; Lee, A.-H. IRE1α activation protects mice against acetaminophen-induced hepatotoxicity. J. Exp. Med. 2012, 209, 307–318. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Briedé, J.J.; Jennen, D.G.J.; Van Summeren, A.; Saritas-Brauers, K.; Schaart, G.; Kleinjans, J.C.S.; de Kok, T.M.C.M. Increased mitochondrial ROS formation by acetaminophen in human hepatic cells is associated with gene expression changes suggesting disruption of the mitochondrial electron transport chain. Toxicol. Lett. 2015, 234, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; McGill, M.R.; Ramachandran, A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: Lessons learned from acetaminophen hepatotoxicity. Drug Metab. Rev. 2012, 44, 88–106. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Miki, K.; He, X.; Killeen, M.E.; Fink, M.P. Prolonged treatment with N-acetylcystine delays liver recovery from acetaminophen hepatotoxicity. Crit. Care 2009, 13, R55. [Google Scholar] [CrossRef] [Green Version]
- Muldrew, K.L.; James, L.P.; Coop, L.; McCullough, S.S.; Hendrickson, H.P.; Hinson, J.A.; Mayeux, P.R. Determination of Acetaminophen-Protein Adducts in Mouse Liver and Serum and Human Serum after Hepatotoxic Doses of Acetaminophen Using High-Performance Liquid Chromatography with Electrochemical Detection. Drug Metab. Dispos. 2002, 30, 446–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; McGill, M.R.; Dorko, K.; Kumer, S.C.; Schmitt, T.M.; Forster, J.; Jaeschke, H. Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicol. Appl. Pharmacol. 2014, 279, 266–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGill, M.R.; Yan, H.-M.; Ramachandran, A.; Murray, G.J.; Rollins, D.E.; Jaeschke, H. HepaRG cells: A human model to study mechanisms of acetaminophen hepatotoxicity. Hepatology 2011, 53, 974–982. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.-H.; Lee, H.S.; Chung, C.-K.; Kim, E.J.; Kang, I.-J. Protective effects of an ethanol extract of Angelica keiskei against acetaminophen-induced hepatotoxicity in HepG2 and HepaRG cells. Nutr. Res. Pract. 2017, 11, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGill, M.R.; Lebofsky, M.; Norris, H.-R.K.; Slawson, M.H.; Bajt, M.L.; Xie, Y.; Williams, C.D.; Wilkins, D.G.; Rollins, D.E.; Jaeschke, H. Plasma and liver acetaminophen-protein adduct levels in mice after acetaminophen treatment: Dose–response, mechanisms, and clinical implications. Toxicol. Appl. Pharmacol. 2013, 269, 240–249. [Google Scholar] [CrossRef] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdullah-Al-Shoeb, M.; Sasaki, K.; Kikutani, S.; Namba, N.; Ueno, K.; Kondo, Y.; Maeda, H.; Maruyama, T.; Irie, T.; Ishitsuka, Y. The Late-Stage Protective Effect of Mito-TEMPO against Acetaminophen-Induced Hepatotoxicity in Mouse and Three-Dimensional Cell Culture Models. Antioxidants 2020, 9, 965. https://doi.org/10.3390/antiox9100965
Abdullah-Al-Shoeb M, Sasaki K, Kikutani S, Namba N, Ueno K, Kondo Y, Maeda H, Maruyama T, Irie T, Ishitsuka Y. The Late-Stage Protective Effect of Mito-TEMPO against Acetaminophen-Induced Hepatotoxicity in Mouse and Three-Dimensional Cell Culture Models. Antioxidants. 2020; 9(10):965. https://doi.org/10.3390/antiox9100965
Chicago/Turabian StyleAbdullah-Al-Shoeb, Mohammad, Kenta Sasaki, Saori Kikutani, Nanami Namba, Keiichi Ueno, Yuki Kondo, Hitoshi Maeda, Toru Maruyama, Tetsumi Irie, and Yoichi Ishitsuka. 2020. "The Late-Stage Protective Effect of Mito-TEMPO against Acetaminophen-Induced Hepatotoxicity in Mouse and Three-Dimensional Cell Culture Models" Antioxidants 9, no. 10: 965. https://doi.org/10.3390/antiox9100965
APA StyleAbdullah-Al-Shoeb, M., Sasaki, K., Kikutani, S., Namba, N., Ueno, K., Kondo, Y., Maeda, H., Maruyama, T., Irie, T., & Ishitsuka, Y. (2020). The Late-Stage Protective Effect of Mito-TEMPO against Acetaminophen-Induced Hepatotoxicity in Mouse and Three-Dimensional Cell Culture Models. Antioxidants, 9(10), 965. https://doi.org/10.3390/antiox9100965