The Peroxidatic Thiol of Peroxiredoxin 1 is Nitrosated by Nitrosoglutathione but Coordinates to the Dinitrosyl Iron Complex of Glutathione



Abstract
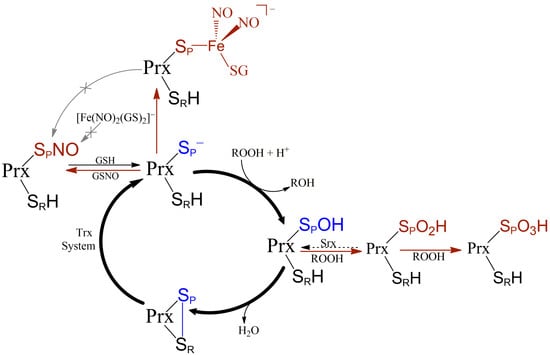
:
1. Introduction
2. Materials and Methods
2.1. Expression and Purification of Recombinant Proteins
2.2. Prx1 and Prx2 Thiol Reduction
2.3. Quantification of Prx1 Thiol Groups
2.4. Quantification of NO● and NO●-Derived Products
2.5. Kinetic Studies of Prx1 S-Nitrosation by GSNO
2.6. Kinetic Studies of Prx1 S-Denitrosation by GSH
2.7. Prx1 Thiol Alkylation and Prx1 His Ethoxyformylation
2.8. Prx1 Peroxidase Activity Assay
2.9. Dinitrosyl Iron Complex of GSH (DNIC-GS) Synthesis
2.10. Kinetics of Conversion of B-DNIC-GS into M-DNIC-GS
2.11. Electon Paramagnetic Ressonance (EPR)
3. Results
3.1. Kinetics and Products of the Reaction of Prx1 with GSNO
3.2. Kinetics of Prx1 S-Denitrosation
3.3. Interaction of Prx1 with DNIC-GS

3.4. Prx1 Residues Involved in the Coordination to DNIC-GS
3.5. Kinetics of the Cys52-Px1 Binding to DNIC-GS
3.6. Evaluation of DNIC-GS and DNIC-Prx1 as Nitrosating Agents
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hess, D.T.; Matsumoto, A.; Kim, S.-O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Stamler, J.S. Enzymatic mechanisms regulating protein S-nitrosylation: Implications in health and disease. J. Mol. Med. 2012, 90, 233–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Lipton, S.A. Protein S-Nitrosylation as a Therapeutic Target for Neurodegenerative Diseases. Trends Pharmacol. Sci. 2016, 37, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolhuter, K.; Whitwell, H.J.; Switzer, C.H.; Burgoyne, J.R.; Timms, J.F.; Eaton, P. Evidence against stable protein S-Nitrosylation as a widespread mechanism of post-translational regulation. Mol. Cell 2018, 69, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Toledo, J.C., Jr.; Augusto, O. Connecting the chemical and biological properties of nitric oxide. Chem. Res. Toxicol. 2012, 25, 975–989. [Google Scholar] [CrossRef]
- Broniowska, K.A.; Hogg, N. The chemical biology of S-nitrosothiols. Antioxid. Redox Signal. 2012, 17, 969–980. [Google Scholar] [CrossRef]
- Jia, L.; Bonaventura, C.; Bonaventura, J.; Stamler, J.S. S-nitrosohaemoglobin: A dynamic activity of blood involved in vascular control. Nature 1996, 380, 221–226. [Google Scholar] [CrossRef]
- Weichsel, A.; Maes, E.M.; Andersen, J.F.; Valenzuela, J.G.; Shokhireva, T.K.; Walker, F.A.; Montfort, W.R. Heme-assisted S-nitrosation of a proximal thiolate in a nitric oxide transport protein. Proc. Natl. Acad. Sci. USA 2005, 102, 594–599. [Google Scholar] [CrossRef] [Green Version]
- Boese, M.; Mordvintcev, P.; Vanin, A.; Busse, R.; Mulsch, A. S-nitrosation of serum-albumin by dinitrosyl-iron complex. J. Biol. Chem. 1995, 270, 29244–29249. [Google Scholar] [CrossRef] [Green Version]
- Bosworth, C.A.; Toledo, J.C.; Zmijewski, J.W.; Li, Q.; Lancaster, J.R. Dinitrosyliron complexes and the mechanism(s) of cellular protein nitrosothiol formation from nitric oxide. Proc. Natl. Acad. Sci. USA 2009, 106, 4671–4676. [Google Scholar] [CrossRef] [Green Version]
- Hickok, J.R.; Sahni, S.; Shen, H.; Arvind, A.; Antoniou, C.; Fung, L.W.M.; Thomas, D.D. Dinitrosyliron complexes are the most abundant nitric oxide-derived cellular adduct: Biological parameters of assembly and disappearance. Free Radic. Biol. Med. 2011, 51, 1558–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, S.G.; Woo, H.A.; Kil, I.S.; Bae, S.H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 2012, 287, 4403–4410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: Guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 2015, 40, 435–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netto, L.E.S.; Antunes, F. The Roles of Peroxiredoxin and Thioredoxin in Hydrogen Peroxide Sensing and in Signal Transduction. Mol. Cells 2016, 39, 65–71. [Google Scholar] [PubMed] [Green Version]
- Wood, Z.A.; Schröder, E.; Robin Harris, J.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef]
- Barranco-Medina, S.; Lázaro, J.-J.; Dietz, K.-J. The oligomeric conformation of peroxiredoxins links redox state to function. FEBS Lett. 2009, 583, 1809–1816. [Google Scholar] [CrossRef] [Green Version]
- Tairum, C.A.; Santos, M.C.; Breyer, C.A.; Geyer, R.R.; Nieves, C.J.; Portillo-Ledesma, S.; Ferrer-Sueta, G.; Toledo, J.C.; Toyama, M.H.; Augusto, O.; et al. Catalytic Thr or Ser Residue Modulates Structural Switches in 2-Cys Peroxiredoxin by Distinct Mechanisms. Sci. Rep. 2016, 6, 33133. [Google Scholar] [CrossRef] [Green Version]
- Bolduc, J.A.; Nelson, K.J.; Haynes, A.C.; Lee, J.; Reisz, J.A.; Graff, A.H.; Clodfelter, J.E.; Parsonage, D.; Poole, L.B.; Furdui, C.M.; et al. Novel hyperoxidation resistance motifs in 2-Cys peroxiredoxins. J. Biol. Chem. 2018, 293, 11901–11912. [Google Scholar] [CrossRef] [Green Version]
- Veal, E.A.; Underwood, Z.E.; Tomalin, L.E.; Morgan, B.A.; Pillay, C.S. Hyperoxidation of peroxiredoxins: Gain or loss of function? Antioxid. Redox Signal. 2018, 28, 574–590. [Google Scholar] [CrossRef]
- Martínez-Ruiz, A.; Lamas, S. Detection and proteomic identification of S-nitrosylated proteins in endothelial cells. Arch. Biochem. Biophys. 2004, 423, 192–199. [Google Scholar] [CrossRef]
- Forrester, M.T.; Thompson, J.W.; Foster, M.W.; Nogueira, L.; Moseley, M.A.; Stamler, J.S. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat. Biotechnol. 2009, 27, 557–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Liu, T.; Chen, W.; Oka, S.; Fu, C.; Jain, M.R.; Parrott, A.M.; Baykal, A.T.; Sadoshima, J.; Li, H. Redox Regulatory Mechanism of Transnitrosylation by Thioredoxin. Mol. Cell. Proteom. 2010, 9, 2262–2275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelman, R.; Weisman-Shomer, P.; Ziv, T.; Xu, J.; Arnér, E.S.J.; Benhar, M. Multilevel regulation of 2-Cys peroxiredoxin reaction cycle by S-nitrosylation. J. Biol. Chem. 2013, 288, 11312–11324. [Google Scholar] [CrossRef] [Green Version]
- Chung, M.-C.; Alem, F.; Hamer, S.G.; Narayanan, A.; Shatalin, K.; Bailey, C.; Nudler, E.; Hakami, R.M. S-nitrosylation of peroxiredoxin 1 contributes to viability of lung epithelial cells during Bacillus anthracis infection. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3019–3029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Nakamura, T.; Cho, D.-H.; Gu, Z.; Lipton, S.A. S-nitrosylation of peroxiredoxin 2 promotes oxidative stress-induced neuronal cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 18742–18747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broniowska, K.A.; Diers, A.R.; Hogg, N. S-nitrosoglutathione. Biochim. Biophys. Acta 2013, 1830, 3173–3181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogusucu, R.; Rettori, D.; Munhoz, D.C.; Netto, L.E.S.; Augusto, O. Reactions of yeast thioredoxin peroxidases I and II with hydrogen peroxide and peroxynitrite: Rate constants by competitive kinetics. Free Radic. Biol. Med. 2007, 42, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Chae, H.Z.; Seo, M.S.; Kim, K.; Baines, I.C.; Rhee, S.G. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-α. J. Biol. Chem. 1998, 273, 6297–6302. [Google Scholar] [CrossRef]
- Nagy, P.; Karton, A.; Betz, A.; Peskin, A.V.; Pace, P.; O’Reilly, R.J.; Hampton, M.B.; Radom, L.; Winterbourn, C.C. Model for the exceptional reactivity of peroxiredoxins 2 and 3 with hydrogen peroxide: A kinetic and computational study. J. Biol. Chem. 2011, 286, 18048–18055. [Google Scholar] [CrossRef] [Green Version]
- Truzzi, D.R.; Coelho, F.R.; Paviani, V.; Alves, S.V.; Netto, L.E.S.; Augusto, O. The bicarbonate/carbon dioxide pair increases hydrogen peroxide-mediated hyperoxidation of human peroxiredoxin 1. J. Biol. Chem. 2019, 294, 14055–14067. [Google Scholar] [CrossRef]
- de Oliveira, M.A.; Discola, K.F.; Alves, S.V.; Barbosa, J.A.R.G.; Medrano, F.J.; Netto, L.E.S.; Guimarães, B.G. Crystallization and preliminary X-ray diffraction analysis of NADPH-dependent thioredoxin reductase I from Saccharomyces cerevisiae. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2005, 61, 387–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassetti, D.R.; Murray, J.F. Determination of sulfhydryl groups with 2,2′- or 4,4′-dithiodipyridine. Arch. Biochem. Biophys. 1967, 119, 41–49. [Google Scholar] [CrossRef]
- Feelisch, M.; Rassaf, T.; Mnaimneh, S.; Singh, N.; Bryan, N.S.; Jourd’Heuil, D.; Kelm, M. Concomitant S-, N-, and heme-nitros(yl)ation in biological tissues and fluids: Implications for the fate of NO in vivo. FASEB J. 2002, 16, 1775–1785. [Google Scholar] [CrossRef] [PubMed]
- Keszler, A.; Diers, A.R.; Ding, Z.; Hogg, N. Thiolate-based dinitrosyl iron complexes: Decomposition and detection and differentiation from S-nitrosothiols. Nitric Oxide 2017, 65, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, L.C.; Wagner, D.A.; Glogowski, J.; Skipper, P.L.; Wishnok, J.S.; Tannenbaum, S.R. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal. Biochem. 1982, 126, 131–138. [Google Scholar] [CrossRef]
- Monteiro, G.; Horta, B.B.; Pimenta, D.C.; Augusto, O.; Netto, L.E.S. Reduction of 1-Cys peroxiredoxins by ascorbate changes the thiol-specific antioxidant paradigm, revealing another function of vitamin C. Proc. Natl. Acad. Sci. USA 2007, 104, 4886–4891. [Google Scholar] [CrossRef] [Green Version]
- Stamler, J.S.; Simon, D.I.; Osborne, J.A.; Mullins, M.E.; Jaraki, O.; Michel, T.; Singel, D.J.; Loscalzo, J. S-nitrosylation of proteins with nitric-oxide—Synthesis and characterization of biologically-active compounds. Proc. Natl. Acad. Sci. USA 1992, 89, 444–448. [Google Scholar] [CrossRef] [Green Version]
- Roosemont, J.L. Reaction of histidine residues in proteins with diethylpyrocarbonate—Differential molar absorptivities and reactivities. Anal. Biochem. 1978, 88, 314–320. [Google Scholar] [CrossRef]
- Dominici, P.; Valtattorni, C.B. Chemical modification of pig kidney 3,4-dihydroxyphenylalanine decarboxylase with diethyl pyrocarbonate. Evidence for an essential histidyl residue. J. Biol. Chem. 1985, 260, 10583–10589. [Google Scholar]
- Kim, J.A.; Park, S.; Kim, K.; Rhee, S.G.; Kang, S.W. Activity assay of mammalian 2-cys peroxiredoxins using yeast thioredoxin reductase system. Anal. Biochem. 2005, 338, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Borodulin, R.R.; Kubrina, L.N.; Shvydkiy, V.O.; Lakomkin, V.L.; Vanin, A.F. A simple protocol for the synthesis of dinitrosyl iron complexes with glutathione: EPR, optical, chromatographic and biological characterization of reaction products. Nitric Oxide-Biol. Chem. 2013, 35, 110–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, J.C.M.; Iretskii, A.V.; Han, R.-M.; Ford, P.C. Dinitrosyl iron complexes with cysteine. kinetics studies of the formation and reactions of DNICs in aqueous solution. J. Am. Chem. Soc. 2015, 137, 328–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trujillo, M.; Clippe, A.; Manta, B.; Ferrer-Sueta, G.; Smeets, A.; Declercq, J.-P.; Knoops, B.; Radi, R. Pre-steady state kinetic characterization of human peroxiredoxin 5: Taking advantage of Trp84 fluorescence increase upon oxidation. Arch. Biochem. Biophys. 2007, 467, 95–106. [Google Scholar] [CrossRef]
- Carvalho, L.A.C.; Truzzi, D.R.; Fallani, T.S.; Alves, S.V.; Toledo, J.C.; Augusto, O.; Netto, L.E.S.; Meotti, F.C. Urate hydroperoxide oxidizes human peroxiredoxin 1 and peroxiredoxin 2. J. Biol. Chem. 2017, 292, 8705–8715. [Google Scholar] [CrossRef] [Green Version]
- Parsonage, D.; Nelson, K.J.; Ferrer-Sueta, G.; Alley, S.; Karplus, P.A.; Furdui, C.M.; Poole, L.B. Dissecting peroxiredoxin catalysis: Separating binding, peroxidation, and resolution for a bacterial AhpC. Biochemistry 2015, 54, 1567–1575. [Google Scholar] [CrossRef] [Green Version]
- Balchin, D.; Wallace, L.; Dirr, H.W. S-nitrosation of glutathione transferase p1-1 is controlled by the conformation of a dynamic active site helix. J. Biol. Chem. 2013, 288, 14973–14984. [Google Scholar] [CrossRef] [Green Version]
- Saville, B. A scheme for the colorimetric determination of microgram amounts of thiols. Analyst 1958, 83, 670–672. [Google Scholar] [CrossRef]
- Peskin, A.V.; Dickerhof, N.; Poynton, R.A.; Paton, L.N.; Pace, P.E.; Hampton, M.B.; Winterbourn, C.C. Hyperoxidation of peroxiredoxins 2 and 3: Rate constants for the reactions of the sulfenic acid of the peroxidatic cysteine. J. Biol. Chem. 2013, 288, 14170–14177. [Google Scholar] [CrossRef] [Green Version]
- McDonald, C.C.; Phillips, W.D.; Mower, H.F. An electron spin resonance study of some complexes of iron, nitric oxide, and anionic ligands. J. Am. Chem. Soc. 1965, 87, 3319–3326. [Google Scholar] [CrossRef]
- Truzzi, D.R.; Augusto, O.; Iretskii, A.V.; Ford, P.C. Dynamics of dinitrosyl iron complex (DNIC) formation with low molecular weight thiols. Inorg. Chem. 2019, 58, 13446–13456. [Google Scholar] [CrossRef] [PubMed]
- Borodulin, R.R.; Kubrina, L.N.; Serezhenkov, V.A.; Burbaev, D.S.; Mikoyan, V.D.; Vanin, A.F. Redox conversions of dinitrosyl iron complexes with natural thiol-containing ligands. Nitric Oxide-Biol. Chem. 2013, 35, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Truzzi, D.R.; Augusto, O.; Ford, P.C. Thiyl radicals are co-products of dinitrosyl iron complex (DNIC) formation. Chem. Commun. 2019, 55, 9156–9159. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, R.J.; Martin, R.B. Interactions of histidine and other imidazole derivatives with transition metal ions in chemical and biological systems. Chem. Rev. 1974, 74, 471–517. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef]
- Borodulin, R.R.; Kubrina, L.N.; Mikoyan, V.D.; Poltorakov, A.P.; Shvydkiy, V.O.; Burbaev, D.S.; Serezhenkov, V.A.; Yakhontova, E.R.; Vanin, A.F. Dinitrosyl iron complexes with glutathione as NO and NO+ donors. Nitric Oxide-Biol. Chem. 2013, 29, 4–16. [Google Scholar] [CrossRef]
- Li, Q.; Li, C.; Mahtani, H.K.; Du, J.; Patel, A.R.; Lancaster, J.R. Nitrosothiol formation and protection against Fenton chemistry by nitric oxide-induced dinitrosyliron complex formation from anoxia-initiated cellular chelatable iron increase. J. Biol. Chem. 2014, 289, 19917–19927. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, D.A.; Marletta, M.A. Thioredoxin catalyzes the S-nitrosation of the caspase-3 active site cysteine. Nat. Chem. Biol. 2005, 1, 154–158. [Google Scholar] [CrossRef]
- Stoyanovsky, D.A.; Tyurina, Y.Y.; Tyurin, V.A.; Anand, D.; Mandavia, D.N.; Gius, D.; Ivanova, J.; Pitt, B.; Billiar, T.R.; Kagan, V.E. Thioredoxin and lipoic acid catalyze the denitrosation of low molecular weight and protein S-nitrosothiols. J. Am. Chem. Soc. 2005, 127, 15815–15823. [Google Scholar] [CrossRef]
- Bocedi, A.; Fabrini, R.; Farrotti, A.; Stella, L.; Ketterman, A.J.; Pedersen, J.Z.; Allocati, N.; Lau, P.C.K.; Grosse, S.; Eltis, L.D.; et al. The Impact of Nitric Oxide Toxicity on the Evolution of the Glutathione Transferase Superfamily. J. Biol. Chem. 2013, 288, 24936–24947. [Google Scholar] [CrossRef] [Green Version]
- Bocedi, A.; Fabrini, R.; Bello, M.L.; Caccuri, A.M.; Federici, G.; Mannervik, B.; Cornish-Bowden, A.; Ricci, G. Evolution of negative cooperativity in glutathione transferase enabled preservation of enzyme function. J. Biol. Chem. 2016, 291, 26739–26749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maria, F.D.; Pedersen, J.Z.; Caccuri, A.M.; Antonini, G.; Turella, P.; Stella, L.; Bello, M.L.; Federici, G.; Ricci, G. The specific interaction of dinitrosyl-diglutathionyl-iron complex, a natural no carrier, with the glutathione transferase superfamily suggestion for an evolutionary pressure in the direction of the storage of nitric oxide. J. Biol. Chem. 2003, 278, 42283–42293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, J.Z.; De Maria, F.; Turella, P.; Federici, G.; Mattei, M.; Fabrini, R.; Dawood, K.F.; Massimi, M.; Caccuri, A.M.; Ricci, G. Glutathione transferases sequester toxic dinitrosyl-iron complexes in cells. A protection mechanism against excess nitric oxide. J. Biol. Chem. 2007, 282, 6364–6371. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Reduced | Pre-Treated with GSNO | |
|---|---|---|---|
| R-SH/Protein | R-SH/Protein | R-SNO/Protein | |
| Prx1C83SC173S | 1.8 ± 0.2 | 1.0 ± 0.2 | 0.7 ± 0.1 |
| Prx1 | 3.5 ± 0.3 | 1.1 ± 0.2 | 1.8 ± 0.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Truzzi, D.R.; Alves, S.V.; Netto, L.E.S.; Augusto, O. The Peroxidatic Thiol of Peroxiredoxin 1 is Nitrosated by Nitrosoglutathione but Coordinates to the Dinitrosyl Iron Complex of Glutathione. Antioxidants 2020, 9, 276. https://doi.org/10.3390/antiox9040276
Truzzi DR, Alves SV, Netto LES, Augusto O. The Peroxidatic Thiol of Peroxiredoxin 1 is Nitrosated by Nitrosoglutathione but Coordinates to the Dinitrosyl Iron Complex of Glutathione. Antioxidants. 2020; 9(4):276. https://doi.org/10.3390/antiox9040276
Chicago/Turabian StyleTruzzi, Daniela R., Simone V. Alves, Luis E. S. Netto, and Ohara Augusto. 2020. "The Peroxidatic Thiol of Peroxiredoxin 1 is Nitrosated by Nitrosoglutathione but Coordinates to the Dinitrosyl Iron Complex of Glutathione" Antioxidants 9, no. 4: 276. https://doi.org/10.3390/antiox9040276
APA StyleTruzzi, D. R., Alves, S. V., Netto, L. E. S., & Augusto, O. (2020). The Peroxidatic Thiol of Peroxiredoxin 1 is Nitrosated by Nitrosoglutathione but Coordinates to the Dinitrosyl Iron Complex of Glutathione. Antioxidants, 9(4), 276. https://doi.org/10.3390/antiox9040276