Inhibition of HDAC6 Attenuates Diabetes-Induced Retinal Redox Imbalance and Microangiopathy


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Postmortem Samples
2.2. Animals and Treatment
2.3. Cells and Treatment
2.4. Histology
2.5. Immunofluorescence
2.6. Assessment of HDAC6 Activity
2.7. Assessment of Thioredoxin-1 Activity
2.8. Protein Analysis
2.9. Dot BlotAanalysis
2.10. Quantitative PCR Analysis
2.11. Reactive Oxygen Species Assays
2.12. Assessment of Senescence Markers
2.13. Statistical Analysis
3. Results
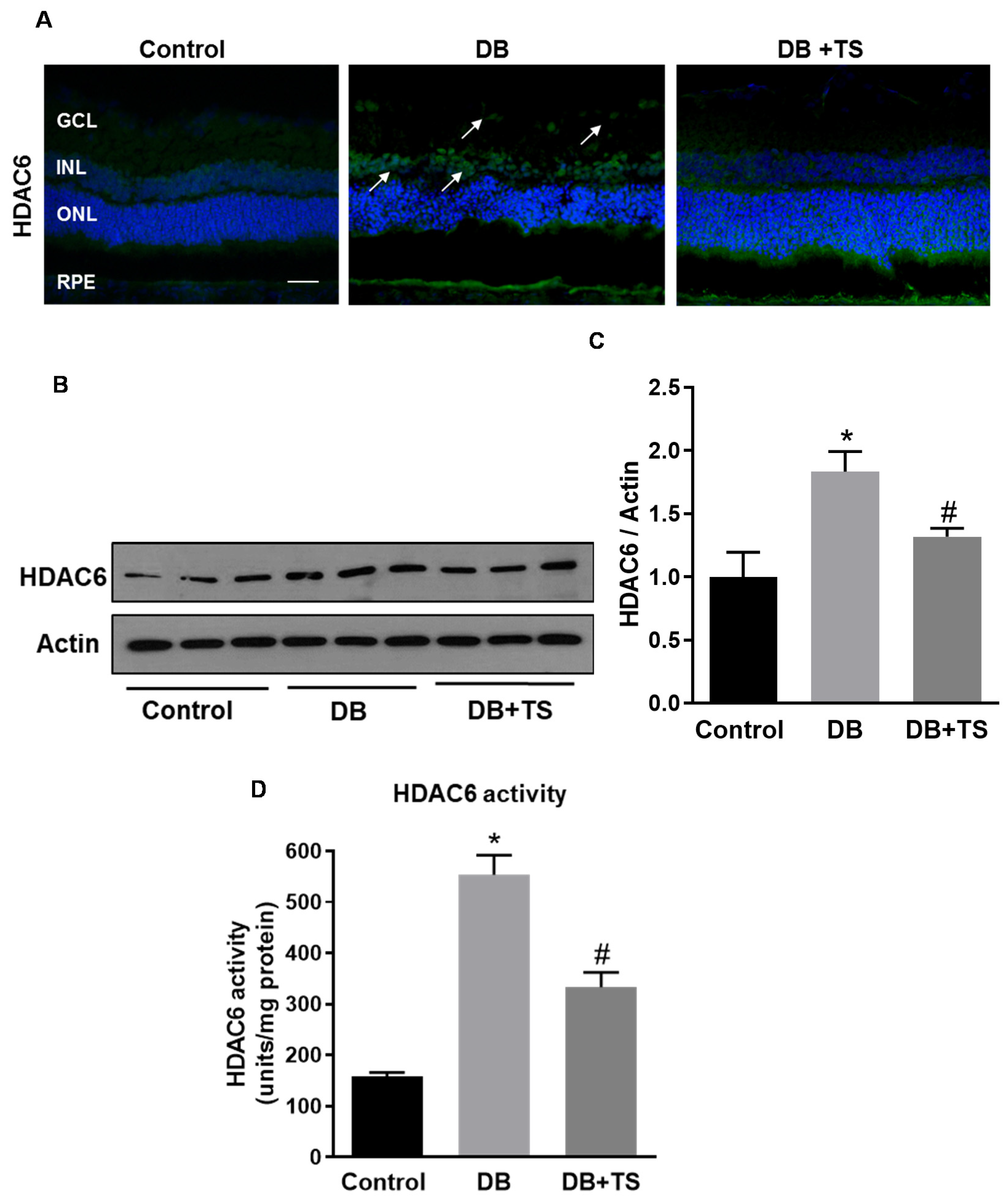
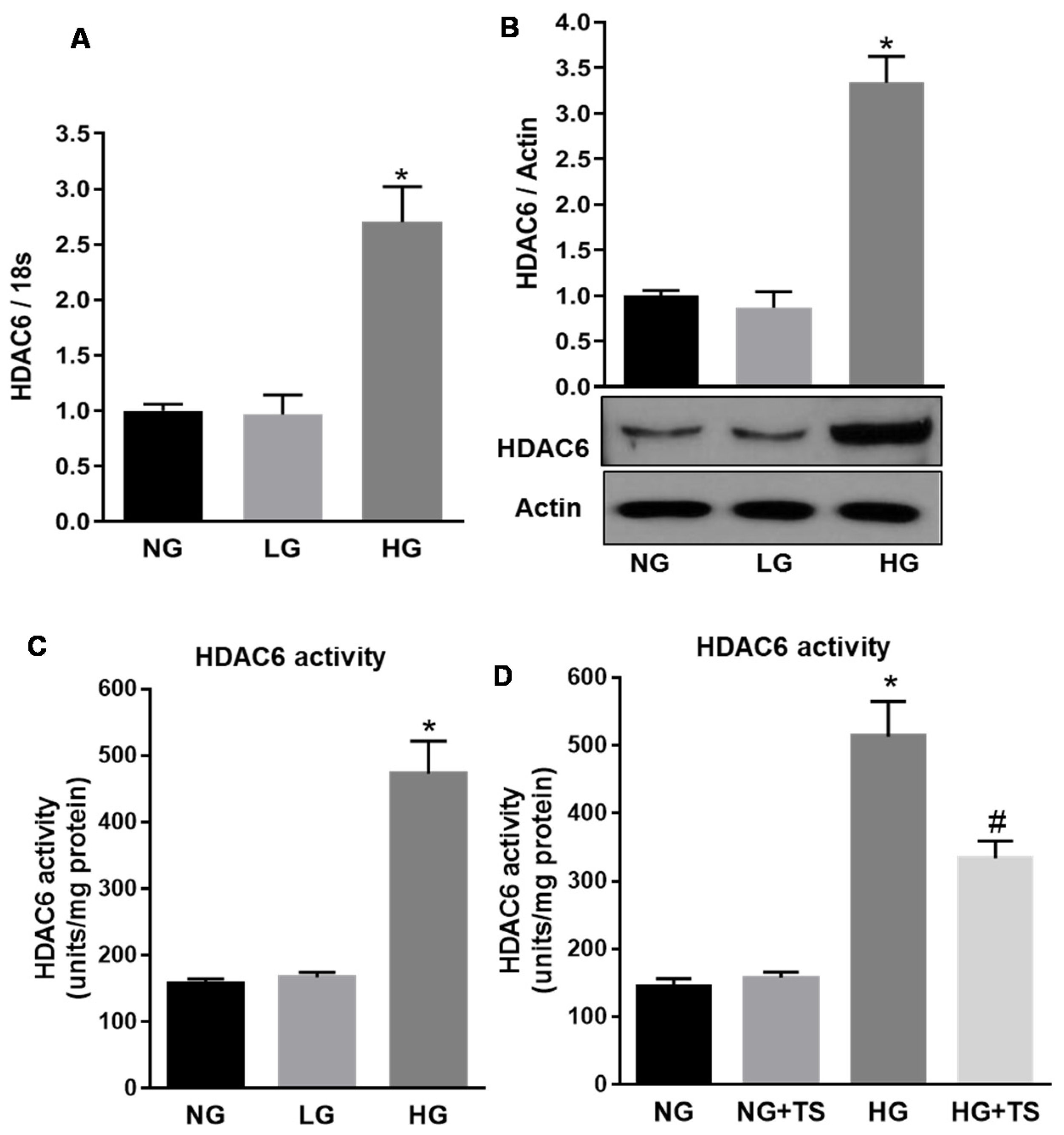
3.1. HDAC6 Expression and Activity are Increased in the Diabetic Retina
3.2. Tubastatin A Decreases the Expression and Activity of HDAC6 in the Diabetic Retina
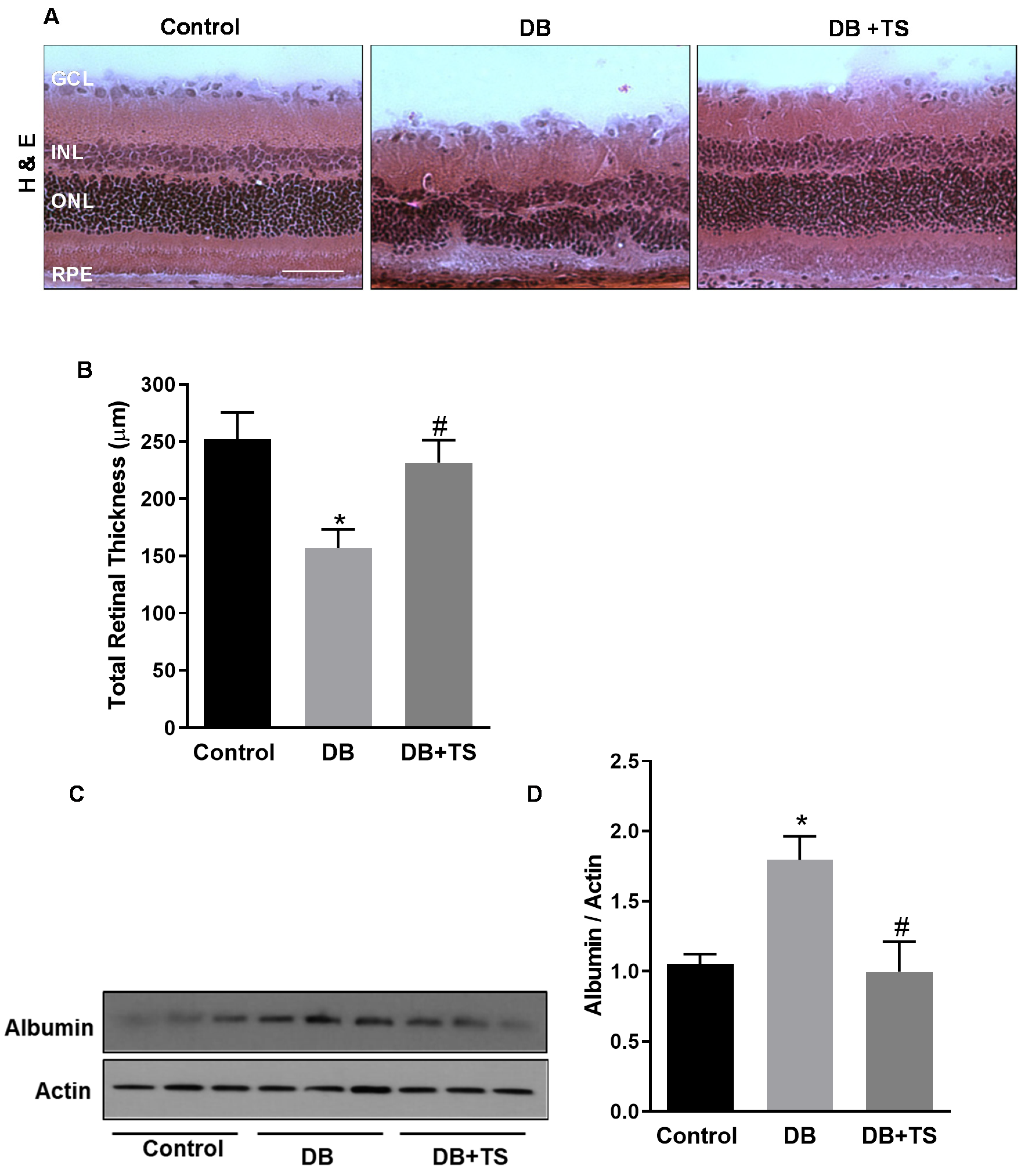
3.3. Tubastatin A Preserves Retinal Structural Morphology and Reduces Vascular Leakage in Diabetic Retina
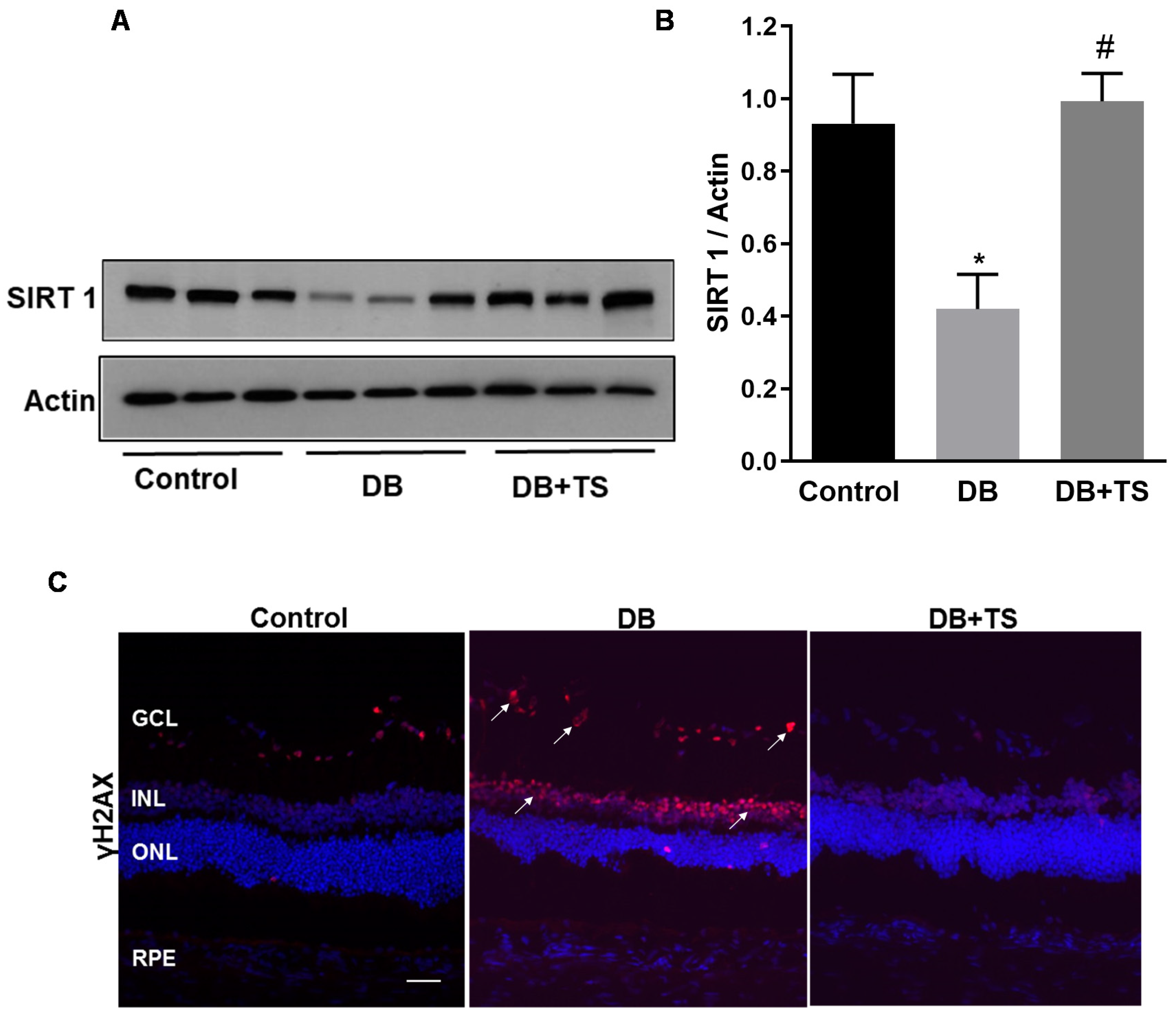
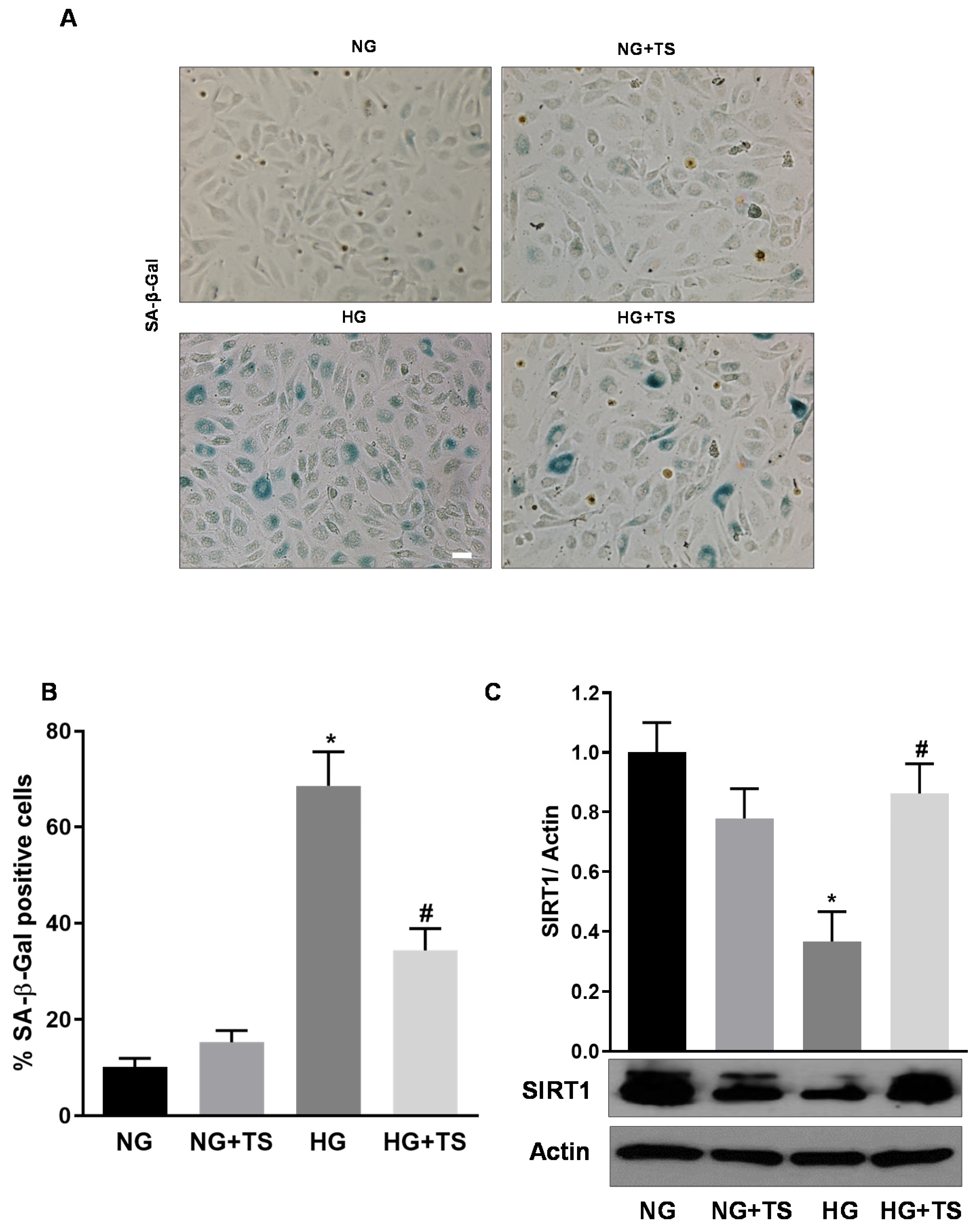
3.4. Tubastatin A Decreases the Levels of Senescence Markers in the Diabetic Retina
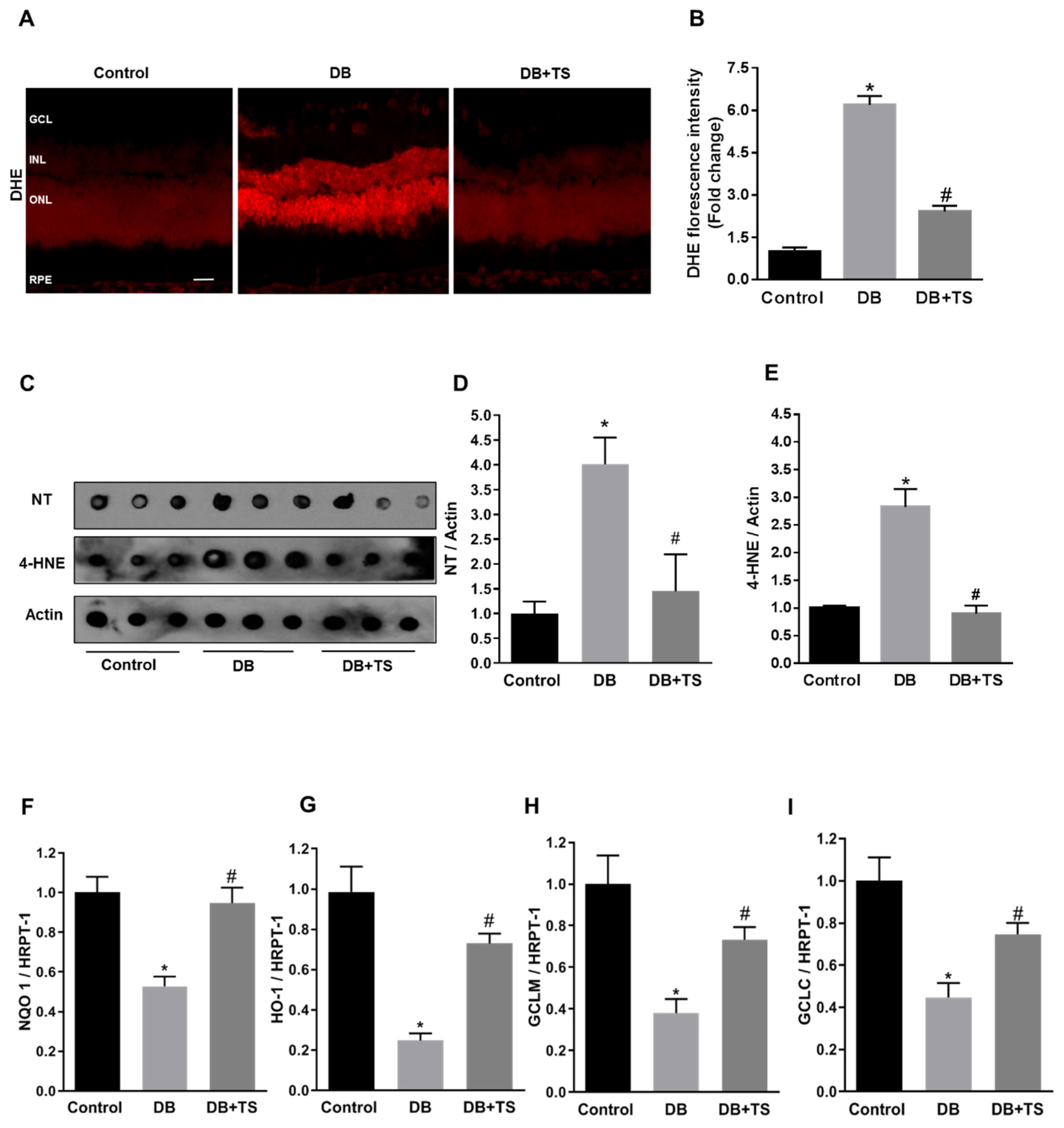
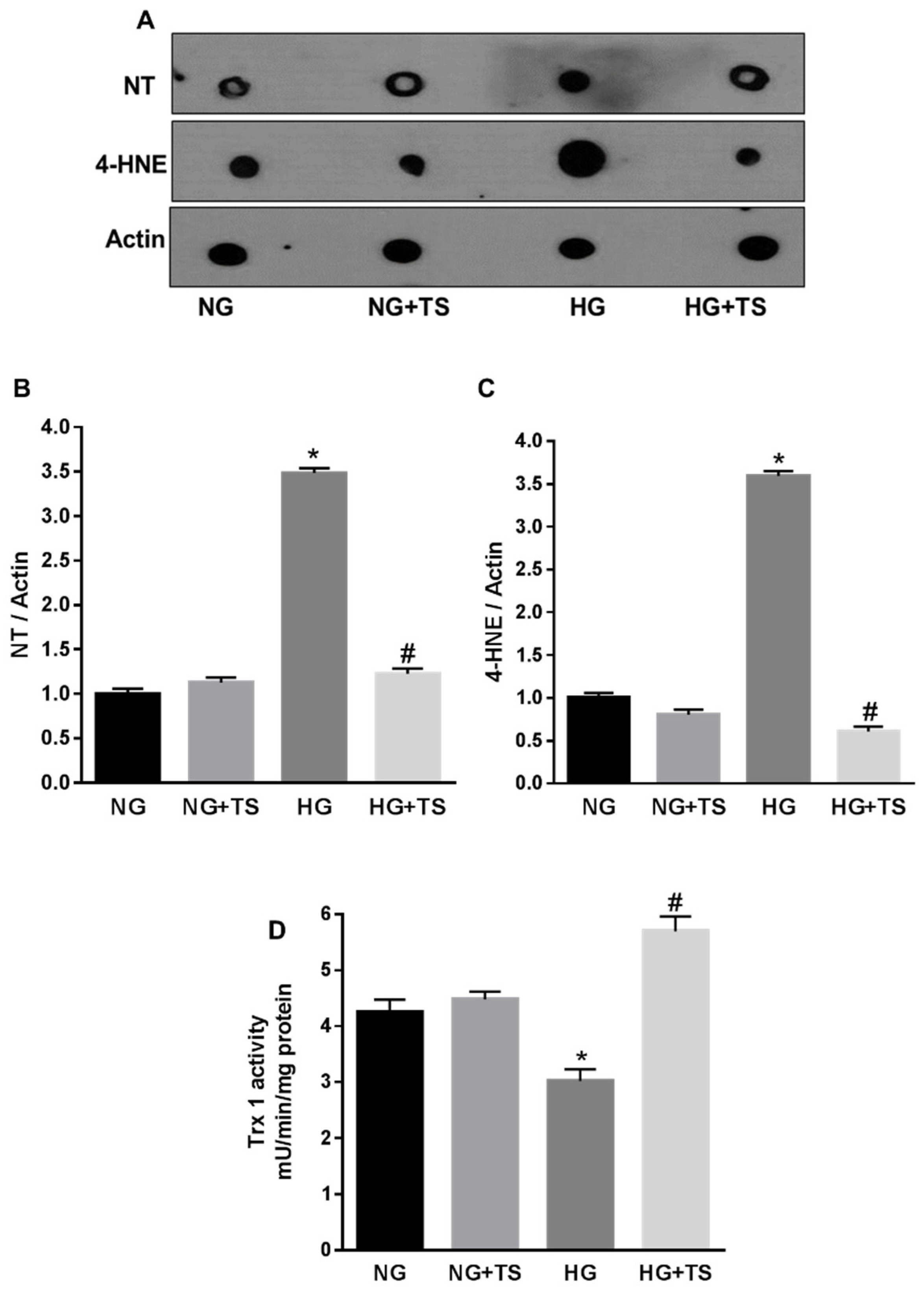
3.5. Tubastatin A Decreases Hyperglycemia-Induced Oxidative/Nitrative Stress in Retina
3.6. Tubastatin A Restores Antioxidant Activity in the Diabetic Retina
3.7. Effect of High Glucose and Tubastatin A on HDAC6 Expression and Activity in Human Retinal Endothelial Cells
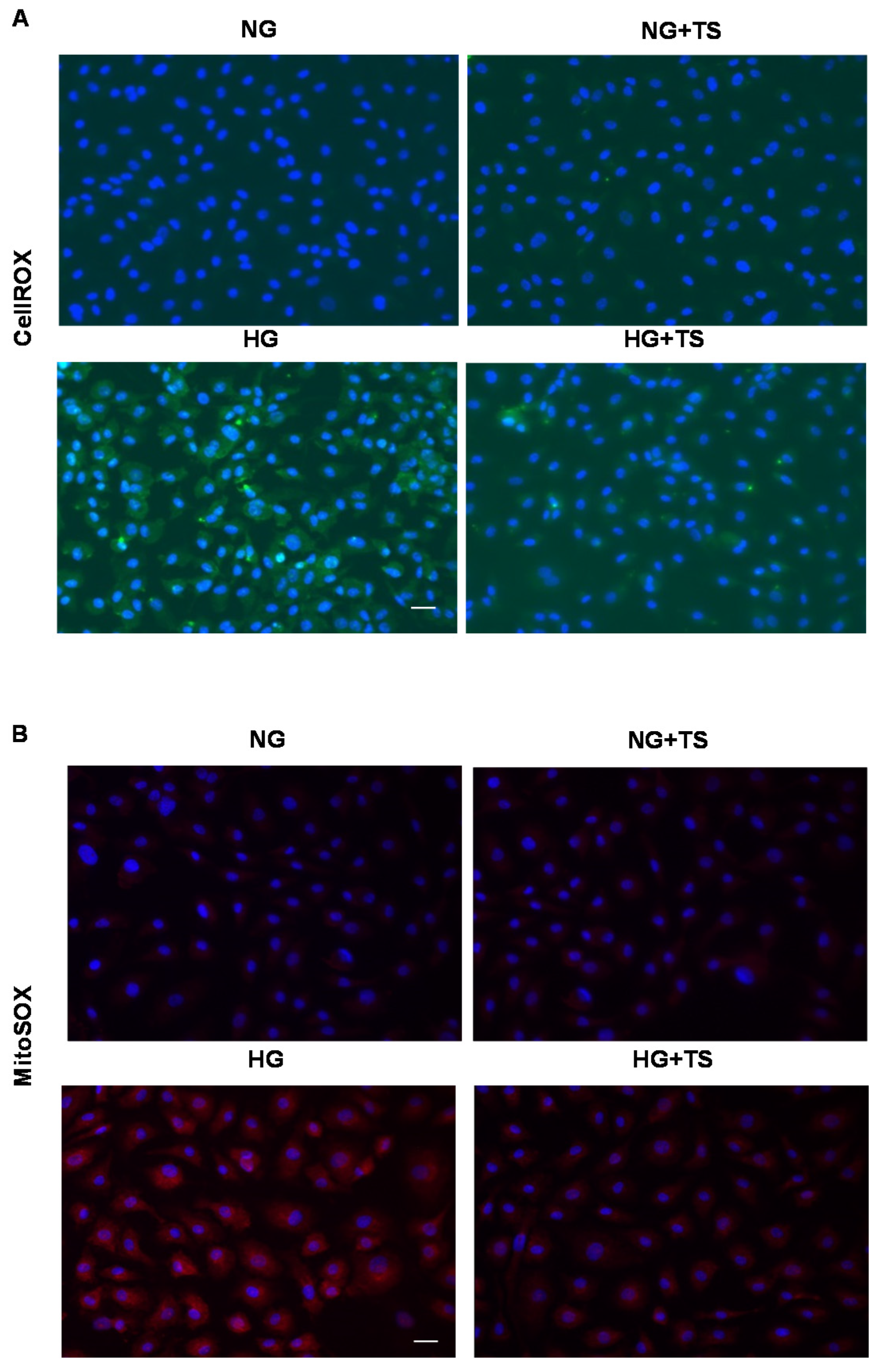
3.8. Effects of HDAC6 Inhibition on Oxidative/Nitrative Stress and Endogenous Antioxidants in HuREC
3.9. Effects of HDAC6 Inhibition on HG-induced HuREC Senescence
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ogurtsova, K.; da Rocha Fernandes, J.D.; Huang, Y.; Linnenkamp, U.; Guariguata, L.; Cho, N.H.; Cavan, D.; Shaw, J.E.; Makaroff, L.E. IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res.Clin. Pr. 2017, 128, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic retinopathy. New Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duh, E.J.; Sun, J.K.; Stitt, A.W. Diabetic retinopathy: Current understanding, mechanisms, and treatment strategies. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Lamoke, F.; Shaw, S.; Yuan, J.; Ananth, S.; Duncan, M.; Martin, P.; Bartoli, M. Increased oxidative and nitrative stress accelerates aging of the retinal vasculature in the diabetic retina. PLoS ONE 2015, 10, e0139664. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A. Mitochondrial stability in diabetic retinopathy: Lessons learned from epigenetics. Diabetes 2019, 68, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, R.A.; Mishra, M. Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim. Biophys. Acta 2015, 1852, 2474–2483. [Google Scholar] [CrossRef] [Green Version]
- Thounaojam, M.C.; Jadeja, R.N.; Warren, M.; Powell, F.L.; Raju, R.; Gutsaeva, D.; Khurana, S.; Martin, P.M.; Bartoli, M. MicroRNA-34a (miR-34a) mediates retinal endothelial cell premature senescence through mitochondrial dysfunction and loss of antioxidant activities. Antioxidants 2019, 8, 328. [Google Scholar] [CrossRef] [Green Version]
- Thounaojam, M.C.; Powell, F.L.; Patel, S.; Gutsaeva, D.R.; Tawfik, A.; Smith, S.B.; Nussbaum, J.; Block, N.L.; Martin, P.M.; Schally, A.V.; et al. Protective effects of agonists of growth hormone-releasing hormone (GHRH) in early experimental diabetic retinopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 13248–13253. [Google Scholar] [CrossRef] [Green Version]
- Liang, T.; Fang, H. Structure, functions and selective inhibitors of HDAC6. Curr. Top. Med. Chem. 2018, 18, 2429–2447. [Google Scholar] [CrossRef]
- Choi, H.; Kim, H.J.; Kim, J.; Kim, S.; Yang, J.; Lee, W.; Park, Y.; Hyeon, S.J.; Lee, D.S.; Ryu, H.; et al. Increased acetylation of Peroxiredoxin1 by HDAC6 inhibition leads to recovery of Abeta-induced impaired axonal transport. Mol. Neurodegener. 2017, 12, 23. [Google Scholar] [CrossRef] [Green Version]
- Parmigiani, R.B.; Xu, W.S.; Venta-Perez, G.; Erdjument-Bromage, H.; Yaneva, M.; Tempst, P.; Marks, P.A. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc. Natl. Acad. Sci. USA 2008, 105, 9633–9638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simoes-Pires, C.; Zwick, V.; Nurisso, A.; Schenker, E.; Carrupt, P.A.; Cuendet, M. HDAC6 as a target for neurodegenerative diseases: What makes it different from the other HDACs? Mol. Neurodegener. 2013, 8, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, B.S.; McKinsey, T.A. Non-sirtuin histone deacetylases in the control of cardiac aging. J. Mol. Cell. Cardiol. 2015, 83, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Zhang, C.; Hassan, S.; Liu, X.; Song, F.; Chen, K.; Zhang, W.; Yang, J. Histone deacetylase 6 in cancer. J. Hematol. Oncol. 2018, 11, 111. [Google Scholar] [CrossRef] [PubMed]
- Brijmohan, A.S.; Batchu, S.N.; Majumder, S.; Alghamdi, T.A.; Thieme, K.; McGaugh, S.; Liu, Y.; Advani, S.L.; Bowskill, B.B.; Kabir, M.G.; et al. HDAC6 inhibition promotes transcription factor EB activation and is protective in experimental kidney disease. Front. Pharmacol. 2018, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- Leng, Y.; Wu, Y.; Lei, S.; Zhou, B.; Qiu, Z.; Wang, K.; Xia, Z. Inhibition of HDAC6 activity alleviates myocardial ischemia/reperfusion injury in diabetic rats: Potential role of peroxiredoxin 1 acetylation and redox regulation. Oxidative Med. Cell. Longev. 2018, 2018, 9494052. [Google Scholar] [CrossRef] [Green Version]
- Leyk, J.; Daly, C.; Janssen-Bienhold, U.; Kennedy, B.N.; Richter-Landsberg, C. HDAC6 inhibition by tubastatin A is protective against oxidative stress in a photoreceptor cell line and restores visual function in a zebrafish model of inherited blindness. Cell Death Dis. 2017, 8, e3028. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Li, H.; Yu, P.; Fan, Q.; Zhang, X.; Huang, W.; Shen, J.; Cui, Y.; Zhou, W. Involvement of HDAC6 in ischaemia and reperfusion-induced rat retinal injury. BMC Ophthalmol. 2018, 18, 300. [Google Scholar] [CrossRef]
- Semeraro, F.; Morescalchi, F.; Cancarini, A.; Russo, A.; Rezzola, S.; Costagliola, C. Diabetic retinopathy, a vascular and inflammatory disease: Therapeutic implications. Diabetes Metab. 2019, 45, 517–527. [Google Scholar] [CrossRef]
- Shin, E.S.; Sorenson, C.M.; Sheibani, N. Diabetes and retinal vascular dysfunction. J. Ophthalmic Vis. Res. 2014, 9, 362–373. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Chan, P.S. Oxidative stress and diabetic retinopathy. Exp. Diabetes Res. 2007, 2007, 43603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francisqueti-Ferron, F.V.; Ferron, A.J.T.; Garcia, J.L.; Silva, C.; Costa, M.R.; Gregolin, C.S.; Moreto, F.; Ferreira, A.L.A.; Minatel, I.O.; Correa, C.R. Basic concepts on the role of nuclear factor erythroid-derived 2-like 2 (Nrf2) in age-related diseases. Int. J. Mol. Sci. 2019, 20, 3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Q.; Kowluru, R.A. Role of histone acetylation in the development of diabetic retinopathy and the metabolic memory phenomenon. J. Cell. Biochem. 2010, 110, 1306–1313. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Li, J.; Wang, M.; She, M.; Tang, Y.; Li, J.; Li, H.; Hui, H. GLP-1 treatment improves diabetic retinopathy by alleviating autophagy through GLP-1R-ERK1/2-HDAC6 signaling pathway. Int. J. Med. Sci. 2017, 14, 1203–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, M.; Gerbeth, L.; Gerling, M.; Rosenthal, R.; Steiger, K.; Weidinger, C.; Keye, J.; Wu, H.; Schmidt, F.; Weichert, W.; et al. HDAC inhibitors promote intestinal epithelial regeneration via autocrine TGFbeta1 signalling in inflammation. Mucosal Immunol. 2019, 12, 656–667. [Google Scholar] [CrossRef]
- Borgas, D.; Chambers, E.; Newton, J.; Ko, J.; Rivera, S.; Rounds, S.; Lu, Q. Cigarette smoke disrupted lung endothelial barrier integrity and increased susceptibility to acute lung injury via histone deacetylase 6. Am. J. Respir. Cell Mol. Biol. 2016, 54, 683–696. [Google Scholar] [CrossRef]
- Lu, Q.; Sakhatskyy, P.; Grinnell, K.; Newton, J.; Ortiz, M.; Wang, Y.; Sanchez-Esteban, J.; Harrington, E.O.; Rounds, S. Cigarette smoke causes lung vascular barrier dysfunction via oxidative stress-mediated inhibition of RhoA and focal adhesion kinase. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L847–L857. [Google Scholar] [CrossRef] [Green Version]
- Siwak, M.; Maslankiewicz, M.; Nowak-Zdunczyk, A.; Rozpedek, W.; Wojtczak, R.; Szymanek, K.; Szaflik, M.; Szaflik, J.; Szaflik, J.P.; Majsterek, I. The relationship between HDAC6, CXCR3, and SIRT1 genes expression levels with progression of primary open-angle glaucoma. Ophthalmic Genet. 2018, 39, 325–331. [Google Scholar] [CrossRef]
- Bai, J.; Lei, Y.; An, G.L.; He, L. Down-regulation of deacetylase HDAC6 inhibits the melanoma cell line A375.S2 growth through ROS-dependent mitochondrial pathway. PLoS ONE 2015, 10, e0121247. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Li, S.; Zhou, Z.; Fu, M.; Yang, X.; Hao, K.; Liu, Y. HDAC6 inhibitor Cay10603 inhibits high glucose-induced oxidative stress, inflammation and apoptosis in retinal pigment epithelial cells via regulating NF-kappaB and NLRP3 inflammasome pathway. Gen. Physiol. Biophys. 2020, 39, 169–177. [Google Scholar] [CrossRef]
- Guedes-Dias, P.; Oliveira, J.M. Lysine deacetylases and mitochondrial dynamics in neurodegeneration. Biochim. Biophys. Acta 2013, 1832, 1345–1359. [Google Scholar] [CrossRef] [PubMed]
- Yan, J. Interplay between HDAC6 and its interacting partners: Essential roles in the aggresome-autophagy pathway and neurodegenerative diseases. DNA Cell Biol. 2014, 33, 567–580. [Google Scholar] [CrossRef] [PubMed]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abouhish, H.; Thounaojam, M.C.; Jadeja, R.N.; Gutsaeva, D.R.; Powell, F.L.; Khriza, M.; Martin, P.M.; Bartoli, M. Inhibition of HDAC6 Attenuates Diabetes-Induced Retinal Redox Imbalance and Microangiopathy. Antioxidants 2020, 9, 599. https://doi.org/10.3390/antiox9070599
Abouhish H, Thounaojam MC, Jadeja RN, Gutsaeva DR, Powell FL, Khriza M, Martin PM, Bartoli M. Inhibition of HDAC6 Attenuates Diabetes-Induced Retinal Redox Imbalance and Microangiopathy. Antioxidants. 2020; 9(7):599. https://doi.org/10.3390/antiox9070599
Chicago/Turabian StyleAbouhish, Hossameldin, Menaka C. Thounaojam, Ravirajsinh N. Jadeja, Diana R. Gutsaeva, Folami L. Powell, Mohamed Khriza, Pamela M. Martin, and Manuela Bartoli. 2020. "Inhibition of HDAC6 Attenuates Diabetes-Induced Retinal Redox Imbalance and Microangiopathy" Antioxidants 9, no. 7: 599. https://doi.org/10.3390/antiox9070599
APA StyleAbouhish, H., Thounaojam, M. C., Jadeja, R. N., Gutsaeva, D. R., Powell, F. L., Khriza, M., Martin, P. M., & Bartoli, M. (2020). Inhibition of HDAC6 Attenuates Diabetes-Induced Retinal Redox Imbalance and Microangiopathy. Antioxidants, 9(7), 599. https://doi.org/10.3390/antiox9070599