Antioxidant, Anti-inflammatory and Neuroprotective Profiles of Novel 1,4-Dihydropyridine Derivatives for the Treatment of Alzheimer’s Disease

 ,
,  ,
,  and
and 
Abstract
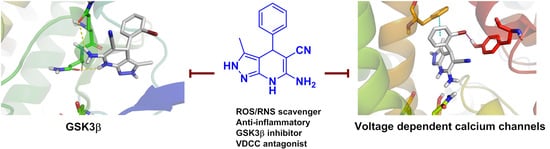
:
1. Introduction
2. Materials and Methods
2.1. Synthesis
2.2. Oxygen Radical Absorbance Capacity (ORAC) Assay
2.3. DPPH Reduction Assay
2.4. Mixed Primary Glial Culture
2.5. Measurement of the Anti-inflammatory Capacity Measured as Nitrite Production Reduction in Mixed Primary Glial Cultures
2.6. GSK-3β Inhibitory Capacity
2.7. SH-SY5Y Neuroblastoma Cell Line Culture
2.8. Voltage Dependent Calcium Channels Blockade Assay
2.9. Blood-Brain Barrier Permeation Assay (PAMPA)
2.10. Molecular Docking Calculations
2.11. Neuroprotection Studies in the SH-SY5Y Human Neuroblastoma Cell Line
2.12. Acute Treatment of Rat Hippocampal Slices
2.13. Measurement of ROS Production in Rat Hippocampal Slices
2.14. Statistical Analysis
3. Results and Discussion
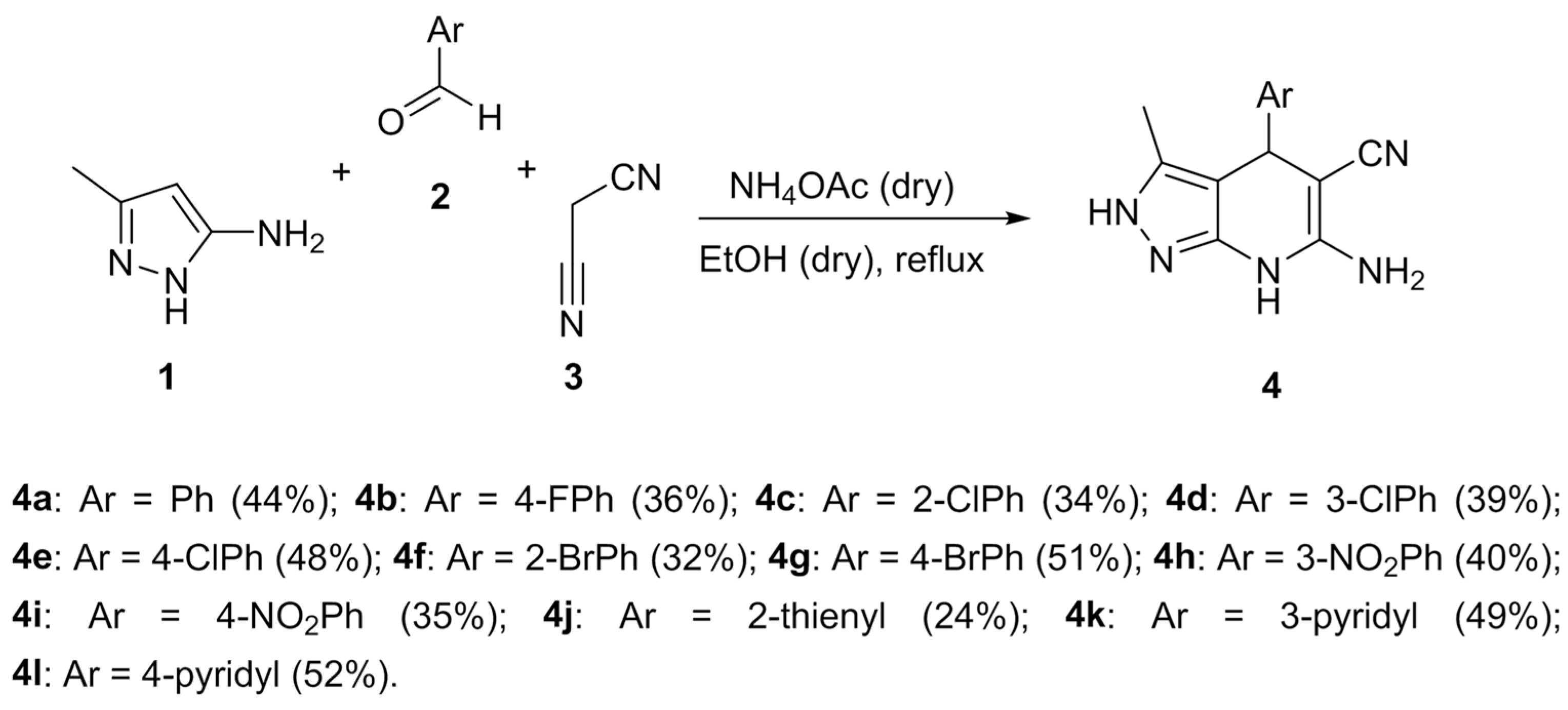
3.1. Synthesis of 4,7-Dihydro-2H-Pyrazolo[3,4-b]Pyridine Derivatives
3.2. Pharmacological Evaluation
3.2.1. Antioxidant and Anti-inflammatory Properties of Novel 4,7-Dihydro-2H-Pyrazolo[3,4-b]Pyridine Derivatives 4a–l
3.2.2. GSK-3β Inhibition, Voltage-Dependent Ca2+ Channels Blockade and BBB Permeation Properties
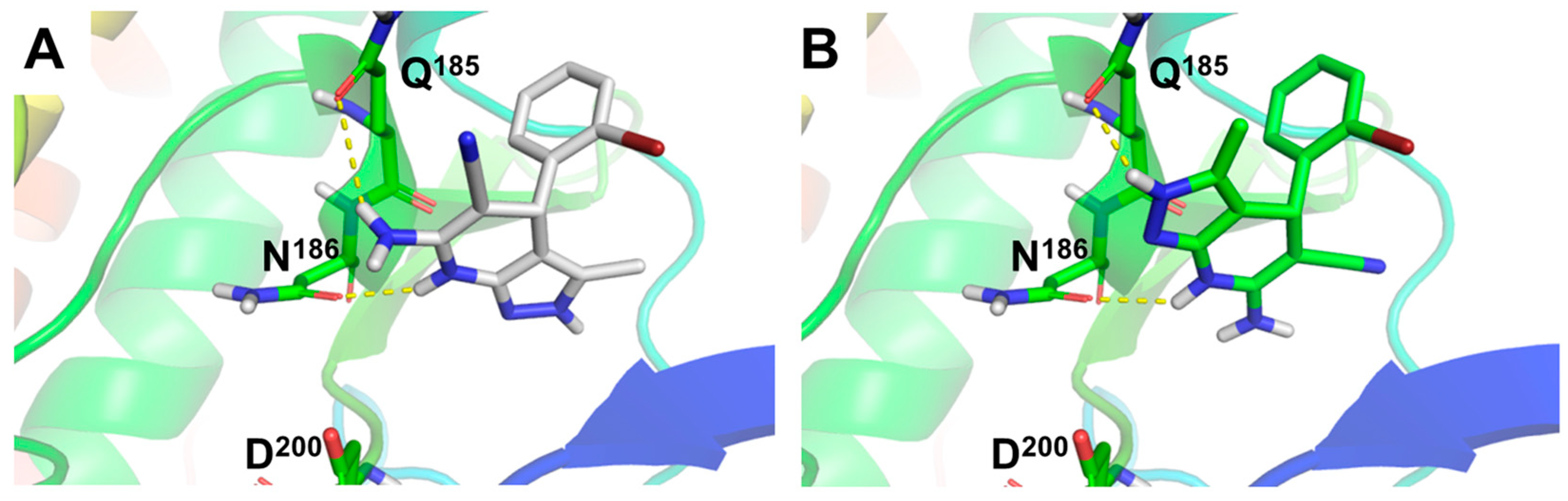
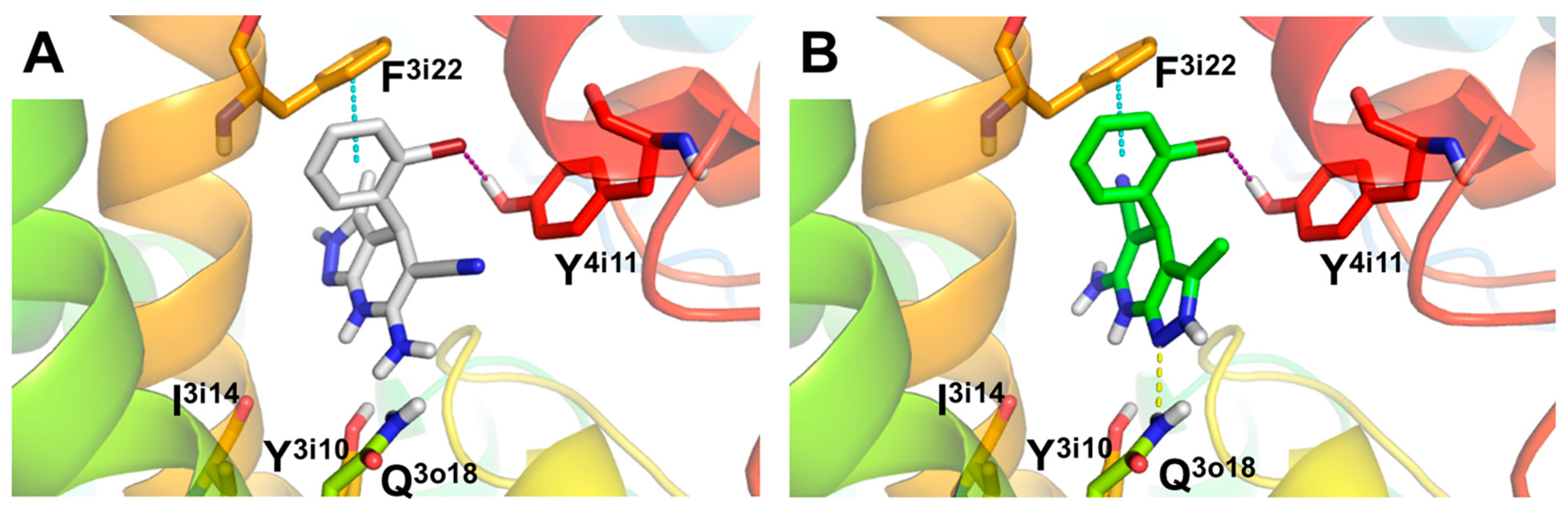
3.3. Molecular Docking: Proposed Binding Mode of Compound 4f at the ATP Binding Site of GSK-3β and the 1,4-DHP Binding Site of L-Type VDCC
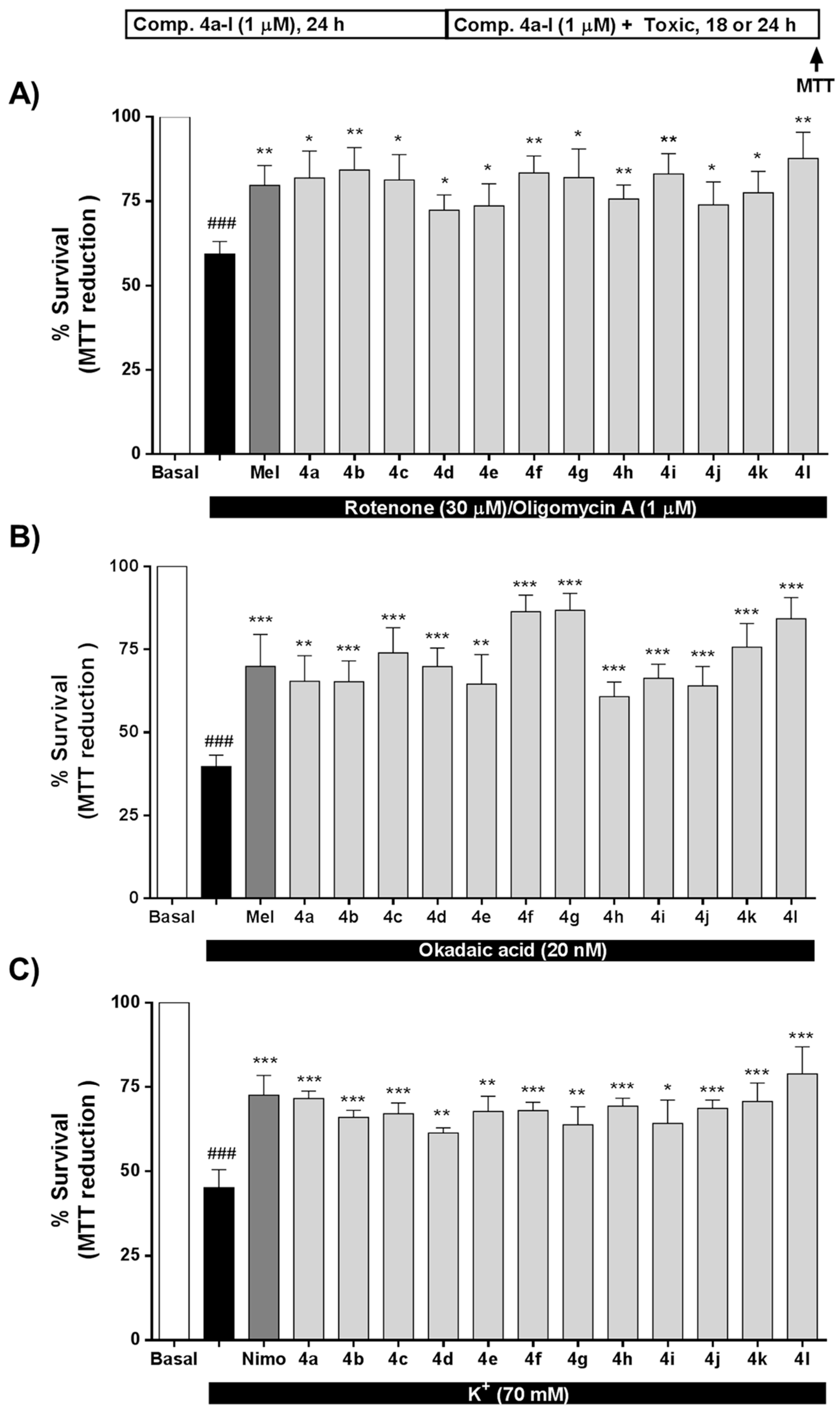
3.4. Neuroprotective Properties of Derivatives 4a–l towards Tau-Hyperphosphorylation, [Ca2+]c Overload and Oxidative Stress
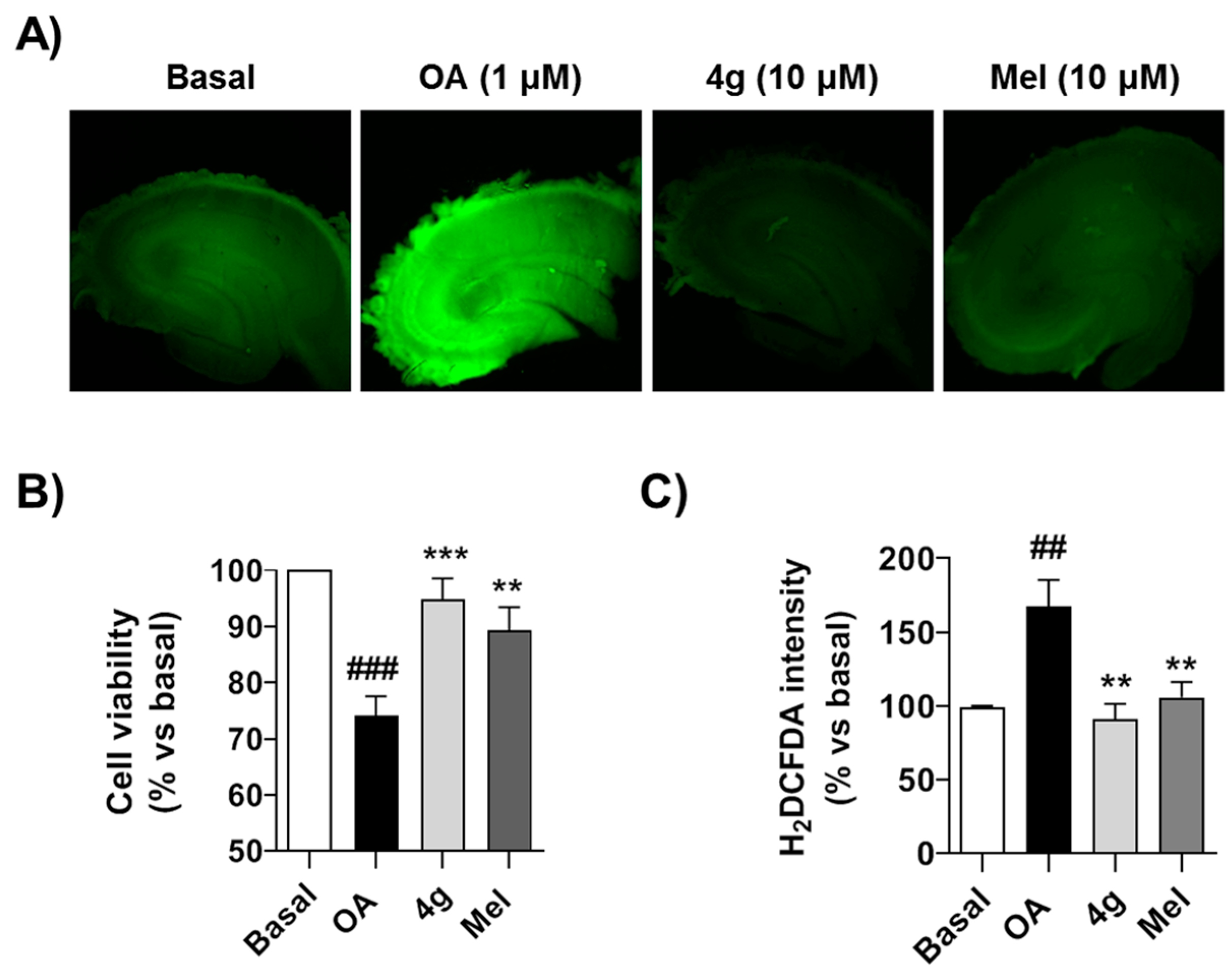
3.5. Compound 4g Reduces Cellular Death Induced by Okadaic Acid in Hippocampal Slices by Reducing Oxidative Stress
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Alzheimer Report 2019 Attitudes to Dementia. 2019. Available online: https://www.alz.co.uk/research/WorldAlzheimerReport2019.pdf (accessed on 16 June 2020).
- Lee, V.M.Y.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative Tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef] [PubMed]
- Vinters, H.V. Cerebral amyloid angiopathy. A critical review. Stroke 1987, 18, 311–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinuevo, J.L.; Sanchez-Valle, R.; Lladó, A.; Fortea, J.; Bartres-Faz, D.; Rami, L. Identifying Earlier Alzheimer’s Disease: Insights from the Preclinical and Prodromal Phases. Neurodegener. Dis. 2012, 10, 158–160. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A.; Eckert, A. Brain aging and neurodegeneration: From a mitochondrial point of view. J. Neurochem. 2017, 143, 418–431. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative Stress, Mitochondrial Dysfunction, and Aging. J. Signal Transduct. 2012, 2012, 646354. [Google Scholar] [CrossRef] [Green Version]
- Mecocci, P.; Boccardi, V.; Cecchetti, R.; Bastiani, P.; Scamosci, M.; Ruggiero, C.; Baroni, M. A Long Journey into Aging, Brain Aging, and Alzheimer’s Disease Following the Oxidative Stress Tracks1. J. Alzheimer’s Dis. 2018, 62, 1319–1335. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [Green Version]
- Sheehan, J.P.; Swerdlow, R.H.; Miller, S.W.; Davis, R.E.; Parks, J.K.; Parker, W.D.; Tuttle, J.B. Calcium Homeostasis and Reactive Oxygen Species Production in Cells Transformed by Mitochondria from Individuals with Sporadic Alzheimer’s Disease. J. Neurosci. 1997, 17, 4612–4622. [Google Scholar] [CrossRef]
- Lévy, E.; El Banna, N.; Baïlle, D.; Heneman-Masurel, A.; Truchet, S.; Rezaei, H.; Huang, M.-E.; Beringue, V.; Martin, D.; Vernis, L. Causative Links between Protein Aggregation and Oxidative Stress: A Review. Int. J. Mol. Sci. 2019, 20, 3896. [Google Scholar] [CrossRef] [Green Version]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Du Yan, S. Mitochondrial Aβ: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar] [CrossRef]
- Canevari, L.; Clark, J.B.; Bates, T.E. β-Amyloid fragment 25–35 selectively decreases complex IV activity in isolated mitochondria. FEBS Lett. 1999, 457, 131–134. [Google Scholar] [CrossRef] [Green Version]
- Cenini, G.; Rüb, C.; Bruderek, M.; Voos, W. Amyloid β-peptides interfere with mitochondrial preprotein import competence by a coaggregation process. Mol. Biol. Cell 2016, 27, 3257–3272. [Google Scholar] [CrossRef] [PubMed]
- Lloret, A.; Badia, M.-C.; Giraldo, E.; Ermak, G.; Alonso, M.-D.; Pallardó, F.V.; Davies, K.E.; Viña, J. Amyloid-β Toxicity and Tau Hyperphosphorylation are Linked Via RCAN1 in Alzheimer’s Disease. J. Alzheimer’s Dis. 2011, 27, 701–709. [Google Scholar] [CrossRef] [Green Version]
- Leroy, K.; Yılmaz, Z.; Brion, J.-P.; Yilmaz, Z. Increased level of active GSK-3? In Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 2007, 33, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Kanamaru, T.; Kamimura, N.; Yokota, T.; Iuchi, K.; Nishimaki, K.; Takami, S.; Akashiba, H.; Shitaka, Y.; Katsura, K.-I.; Kimura, K.; et al. Oxidative stress accelerates amyloid deposition and memory impairment in a double-transgenic mouse model of Alzheimer’s disease. Neurosci. Lett. 2015, 587, 126–131. [Google Scholar] [CrossRef]
- Coma, M.; Guix, F.X.; Ill-Raga, G.; Uribesalgo, I.; Alameda, F.; Valverde, M.A.; Muñoz, F.J. Oxidative stress triggers the amyloidogenic pathway in human vascular smooth muscle cells. Neurobiol. Aging 2008, 29, 969–980. [Google Scholar] [CrossRef]
- Mondragón-Rodríguez, S.; Perry, G.; Luna-Muñoz, J.; Acevedo-Aquino, M.C.; Williams, S. Phosphorylation of tau protein at sites Ser396–404 is one of the earliest events in Alzheimer’s disease and Down syndrome. Neuropathol. Appl. Neurobiol. 2014, 40, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Himmelstein, D.S.; Ward, S.M.; Lancia, J.K.; Patterson, K.R.; Binder, L.I. Tau as a therapeutic target in neurodegenerative disease. Pharmacol. Ther. 2012, 136, 8–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohman, T.J.; Koran, M.E.I.; Thornton-Wells, T.A.; Initiative, F.T.A.N. Interactions between GSK3β and amyloid genes explain variance in amyloid burden. Neurobiol. Aging 2014, 35, 460–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2007, 104, 1433–1439. [Google Scholar] [CrossRef] [Green Version]
- Mines, M.A.; Beurel, E.; Jope, R.S. Regulation of Cell Survival Mechanisms in Alzheimer’s Disease by Glycogen Synthase Kinase-3. Int. J. Alzheimer’s Dis. 2011, 2011. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, G.; Muñoz-Montaño, J.R.; Satrústegui, J.; Avila, J.; Bogonez, E.; Díaz-Nido, J. Lithium protects cultured neurons against β-amyloid-induced neurodegeneration. FEBS Lett. 1999, 453, 260–264. [Google Scholar] [CrossRef] [Green Version]
- Salazar-Roa, M.; Rojo, A.I.; Velasco, D.; De Sagarra, R.M.; Cuadrado, A. Glycogen Synthase Kinase-3β Inhibits the Xenobiotic and Antioxidant Cell Response by Direct Phosphorylation and Nuclear Exclusion of the Transcription Factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.J.; Hayes, J.D.; Cuadrado, A. SCF/β-TrCP Promotes Glycogen Synthase Kinase 3-Dependent Degradation of the Nrf2 Transcription Factor in a Keap1-Independent Manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampa, A.; Gobbi, S.; Di Martino, R.M.C.; Belluti, F.; Bisi, A. Dual BACE-1/GSK-3β Inhibitors to Combat Alzheimer’s Disease: A Focused Review. Curr. Top. Med. Chem. 2017, 17, 3361–3369. [Google Scholar] [CrossRef]
- Gandini, A.; Bartolini, M.; Tedesco, D.; Martínez-González, L.; Roca, C.; Campillo, N.E.; Zaldivar-Diez, J.; Perez, C.; Zuccheri, G.; Miti, A.; et al. Tau-Centric Multitarget Approach for Alzheimer’s Disease: Development of First-in-Class Dual Glycogen Synthase Kinase 3β and Tau-Aggregation Inhibitors. J. Med. Chem. 2018, 61, 7640–7656. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, I.; Michalska, P.; Tenti, G.; Cores, Á.; Buendía, I.; Rojo, A.I.; Georgakopoulos, N.D.; Guijo, J.M.H.; Ramos, M.T.; Wells, G.; et al. Discovery of the first dual GSK3β inhibitor/Nrf2 inducer. A new multitarget therapeutic strategy for Alzheimer’s disease. Sci. Rep. 2017, 7, 45701. [Google Scholar] [CrossRef]
- Jankowska, A.; Satała, G.; Bojarski, A.J.; Pawłowski, M.; Chłoń-Rzepa, G. Multifunctional Ligands with Glycogen Synthase Kinase 3 Inhibitory Activity as a New Direction in Drug Research for Alzheimer’s Disease. Curr. Med. Chem. 2020, 27. [Google Scholar] [CrossRef]
- Kummer, M.; Hermes, M.; Delekarte, A.; Hammerschmidt, T.; Kumar, S.; Terwel, D.; Walter, J.; Pape, H.-C.; König, S.; Roeber, S.; et al. Nitration of Tyrosine 10 Critically Enhances Amyloid β Aggregation and Plaque Formation. Neuron 2011, 71, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Hensleya, K.; Maidt, M.L.; Yu, Z.; Sang, H.; Markesbery, W.R.; Floyd, R.A. Electrochemical Analysis of Protein Nitrotyrosine and Dityrosine in the Alzheimer Brain Indicates Region-Specific Accumulation. J. Neurosci. 1998, 18, 8126–8132. [Google Scholar] [CrossRef]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; Van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2009, 11, 155–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheedy, F.J.; Grebe, A.; Rayner, K.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, U.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, V.T.; Benveniste, E.N. Critical role of tumor necrosis factor-alpha and NF-kappa B in interferon-gamma-induced CD40 expression in microglia/macrophages. J. Biol. Chem. 2002, 277, 13796–13803. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, L.; Barger, S.W.; Griffin, W.S.T. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J. Neurosci. 2003, 23, 1605–1611. [Google Scholar] [CrossRef] [Green Version]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef] [Green Version]
- Praticò, D.; Clark, C.M.; Liun, F.; Lee, V.Y.-M.; Trojanowski, J.Q. Increase of Brain Oxidative Stress in Mild Cognitive Impairment. Arch. Neurol. 2002, 59, 972–976. [Google Scholar] [CrossRef] [Green Version]
- Popugaeva, E.; Pchitskaya, E.; Bezprozvanny, I.B. Dysregulation of Intracellular Calcium Signaling in Alzheimer’s Disease. Antioxid. Redox Signal. 2018, 29, 1176–1188. [Google Scholar] [CrossRef]
- Tong, B.C.-K.; Wu, A.J.; Li, M.; Cheung, K.-H. Calcium signaling in Alzheimer’s disease & therapies. Biochimica et Biophysica Acta BBA Bioenerg. 2018, 1865, 1745–1760. [Google Scholar] [CrossRef]
- Hayley, M.; Perspicace, S.; Schulthess, T.; Seelig, J. Calcium enhances the proteolytic activity of BACE1: An in vitro biophysical and biochemical characterization of the BACE1–calcium interaction. Biochimica et Biophysica Acta BBA Biomembr. 2009, 1788, 1933–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, N.; Yin, X.; Yu, D.; Cao, M.; Gong, C.-X.; Iqbal, K.; Ding, F.; Gu, X.; Liu, F. Truncation and activation of GSK-3β by calpain I: A molecular mechanism links to tau hyperphosphorylation in Alzheimer’s disease. Sci. Rep. 2015, 5, 8187. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.S.; Guzman, J.N.; Ilijić, E.; Mercer, J.N.; Rick, C.; Tkatch, T.; Meredith, G.E.; Surmeier, D.J. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 2007, 447, 1081–1086. [Google Scholar] [CrossRef]
- Zhivotovsky, B.; Orrenius, S. Calcium and cell death mechanisms: A perspective from the cell death community. Cell Calcium 2011, 50, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, E.; De Oliveira, C.R.; Pereira, C.M. The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol. Dis. 2008, 30, 331–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurley, M.J.; Brandon, B.; Gentleman, S.M.; Dexter, D.T. Parkinson’s disease is associated with altered expression of CaV1 channels and calcium-binding proteins. Brain 2013, 136, 2077–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, L.B.; Schmeidler, J.; Lesser, G.T.; Beeri, M.S.; Purohit, D.P.; Grossman, H.T.; Haroutunian, V. Less Alzheimer disease neuropathology in medicated hypertensive than nonhypertensive persons. Neurology 2009, 72, 1720–1726. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, K.; Grundke-Iqbal, I. Alzheimer disease is multifactorial and heterogeneous. Neurobiol. Aging 2000, 21, 901–902. [Google Scholar] [CrossRef]
- Velēna, A.; Zarkovic, N.; Trošelj, K.G.; Bisenieks, E.; Krauze, A.; Poikāns, J.; Duburs, G. 1,4-Dihydropyridine Derivatives: Dihydronicotinamide Analogues—Model Compounds Targeting Oxidative Stress. Oxidative Med. Cell. Longev. 2016, 2016, 1892412. [Google Scholar] [CrossRef] [Green Version]
- Cao, G.; Alessio, H.M.; Cutler, R.G. Oxygen-radical absorbance capacity assay for antioxidants. Free. Radic. Biol. Med. 1993, 14, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Ou, B.; Hampsch-Woodill, M.; Prior, R.L. Development and validation of an improved oxygen radical absorbance capacity assay using fluorescein as the fluorescent probe. J. Agric. Food Chem. 2001, 49, 4619–4626. [Google Scholar] [CrossRef]
- Dudonné, S.; Vitrac, X.; Coutière, P.; Woillez, M.; Mérillon, J.-M. Comparative Study of Antioxidant Properties and Total Phenolic Content of 30 Plant Extracts of Industrial Interest Using DPPH, ABTS, FRAP, SOD, and ORAC Assays. J. Agric. Food Chem. 2009, 57, 1768–1774. [Google Scholar] [CrossRef]
- Baki, A.; Bielik, A.; Molnár, L.; Szendrei, G.; Keserü, G.M. A High Throughput Luminescent Assay for Glycogen Synthase Kinase-3β Inhibitors. ASSAY Drug Dev. Technol. 2007, 5, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tikhonov, D.B.; Zhorov, B.S. Structural Model for Dihydropyridine Binding to L-type Calcium Channels. J. Biol. Chem. 2009, 284, 19006–19017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef]
- Golonka, I.; Oleksy, M.; Junka, A.; Matera-Witkiewicz, A.; Bartoszewicz, M.; Musiał, W. Selected Physicochemical and Biological Properties of Ethyl Ascorbic Acid Compared to Ascorbic Acid. Biol. Pharm. Bull. 2017, 40, 1199–1206. [Google Scholar] [CrossRef] [Green Version]
- Vijesh, A.; Isloor, A.M.; Peethambar, S.; Shivananda, K.; Arulmoli, T.; Isloor, N.A. Hantzsch reaction: Synthesis and characterization of some new 1,4-dihydropyridine derivatives as potent antimicrobial and antioxidant agents. Eur. J. Med. Chem. 2011, 46, 5591–5597. [Google Scholar] [CrossRef]
- Mulder, P.; Litwinienko, G.; Lin, S.; MacLean, P.D.; Barclay, L.R.C.; Ingold, K.U. The L-Type Calcium Channel Blockers, Hantzsch 1,4-Dihydropyridines, Are Not Peroxyl Radical-Trapping, Chain-Breaking Antioxidants. Chem. Res. Toxicol. 2006, 19, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Brooks, D.J.; Okello, A.; Edison, P. An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain 2017, 140, 792–803. [Google Scholar] [CrossRef]
- Guerreiro, R.; Bras, J.; Hardy, J. SnapShot: Genetics of Alzheimer’s Disease. Cell 2013, 155, 968. [Google Scholar] [CrossRef] [Green Version]
- Ko, C.-Y.; Wang, W.-L.; Wang, S.-M.; Chu, Y.-Y.; Chang, W.-C.; Wang, J.-M. Glycogen synthase kinase-3β–mediated CCAAT/enhancer-binding protein delta phosphorylation in astrocytes promot es migration and activation of microglia/macrophages. Neurobiol. Aging 2014, 35, 24–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llorens-Martín, M.; Jurado-Arjona, J.; Fuster-Matanzo, A.; Hernandez, F.; Rabano, A.; Avila, J. Peripherally triggered and GSK-3β-driven brain inflammation differentially skew adult hippocampal neurogenesis, behavioral pattern separation and microglial activation in response to ibuprofen. Transl. Psychiatry 2014, 4, e463. [Google Scholar] [CrossRef] [PubMed]
- Yung-Chi, C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Gaisina, I.; Yuan, H.; Petukhov, P.A.; Blond, S.Y.; Fedolak, A.; Caldarone, B.; Mcgonigle, P. Structure-Based Design Leads to the Identification of Lithium Mimetics That Block Mania-like Effects in Rodents. Possible New GSK-3β Therapies for Bipolar Disorders. J. Am. Chem. Soc. 2007, 129, 8328–8332. [Google Scholar] [CrossRef] [PubMed]
- Tiwaskar, M.; Langote, A.; Kashyap, R.; Toppo, A. Amlodipine in the Era of New Generation Calcium Channel Blockers. J. Assoc. Physicians India 2018, 66, 64–69. [Google Scholar] [PubMed]
- Wappl, E.; Mitterdorfer, J.; Glossmann, H.; Striessnig, J. Mechanism of Dihydropyridine Interaction with Critical Binding Residues of L-type Ca2+ Channel α1 Subunits. J. Biol. Chem. 2001, 276, 12730–12735. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Kamat, P.K.; Rai, S.; Nath, C. Okadaic acid induced neurotoxicity: An emerging tool to study Alzheimer’s disease pathology. Neurotoxicology 2013, 37, 163–172. [Google Scholar] [CrossRef]
- Pérez, M.; Hernandez, F.; Gómez-Ramos, A.; Smith, M.; Perry, G.; Avila, J. Formation of aberrant phosphotau fibrillar polymers in neural cultured cells. JBIC J. Biol. Inorg. Chem. 2002, 269, 1484–1489. [Google Scholar] [CrossRef]
- Kamat, P.K.; Rai, S.; Swarnkar, S.; Shukla, R.; Nath, C. Mechanism of synapse redox stress in Okadaic acid (ICV) induced memory impairment: Role of NMDA receptor. Neurochem. Int. 2014, 76, 32–41. [Google Scholar] [CrossRef]
- Tenti, G.; Parada, E.; León, R.; Egea, J.; Martínez-Revelles, S.; Briones, A.M.; Sridharan, V.; Lopez, M.G.; Ramos, M.T.; Menéndez, J.C. New 5-Unsubstituted Dihydropyridines with Improved CaV1.3 Selectivity as Potential Neuroprotective Agents against Ischemic Injury. J. Med. Chem. 2014, 57, 4313–4323. [Google Scholar] [CrossRef] [PubMed]
- Khaliq, Z.M.; Bean, B.P. Pacemaking in dopaminergic ventral tegmental area neurons: Depolarizing drive from background and voltage-dependent sodium conductances. J. Neurosci. 2010, 30, 7401–7413. [Google Scholar] [CrossRef] [PubMed]
- Luengo, E.; Buendia, I.; Fernández-Mendívil, C.; Trigo-Alonso, P.; Negredo, P.; Michalska, P.; Hernández-García, B.; Sánchez-Ramos, C.; Bernal, J.A.; Ikezu, T.; et al. Pharmacological doses of melatonin impede cognitive decline in tau-related Alzheimer models, once tauopathy is initiated, by restoring the autophagic flux. J. Pineal Res. 2019, 67, e12578. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ar | ORAC (T. Eq.) | IC50 DPPH (µM) | EC50 Nitrite Reduction (μM) |
|---|---|---|---|---|
| Melatonin | - | 2.14 ± 0.12 | >5000 | - |
| Ascorbic ac. | - | - | 61.9 ± 4.96 | - |
| Sulforaphane | - | - | - | 1.40 ± 0.30 [28] |
| 4a | Ph | 0.69 ± 0.04 | 25.2 ± 7.91 | 5.20 ± 1.89 |
| 4b | 4-FPh | 1.86 ± 0.08 | 22.8 ± 6.68 | 5.70 ± 0.62 |
| 4c | 2-ClPh | 0.94 ± 0.04 | 23.3 ± 2.60 | 14.4 ± 2.19 |
| 4d | 3-ClPh | 0.63 ± 0.09 | 22.8 ± 4.63 | 0.78 ± 1.98 |
| 4e | 4-ClPh | 0.66 ± 0.07 | 20.9 ± 5.91 | 4.40 ± 0.35 |
| 4f | 2-BrPh | 1.00 ± 0.16 | 21.6 ± 3.99 | 10.1 ± 0.72 |
| 4g | 4-BrPh | 0.96 ± 0.05 | 24.1 ± 3.74 | 4.50 ± 0.60 |
| 4h | 3-NO2Ph | 0.73 ± 0.09 | 24.7 ± 4.03 | 3.50 ± 0.29 |
| 4i | 4-NO2Ph | 0.96 ± 0.05 | 23.2 ± 4.31 | 6.10 ± 1.84 |
| 4j | 2-Thienyl | 2.93 ± 0.15 | 20.9 ± 4.23 | 5.40 ± 0.27 |
| 4k | 3-Pyridyl | 1.03 ± 0.16 | 17.1 ± 2.18 | 22.3 ± 3.42 |
| 4l | 4-Pyridyl | 1.10 ± 0.13 | 22.9 ± 4.23 | 15.6 ± 3.35 |
| Compound | Ar | IC50 GSK3β (µM) | Ki (µM) | VDCC % Response | PAMPA Pe (10−6 cm s−1) | PAMPA Prediction |
|---|---|---|---|---|---|---|
| SB216763 | - | 0.034 ± 0.01 [28] | 0.009 | - | - | - |
| Nimodipine | - | - | - | 51.0 ± 2.6 | - | - |
| 4a | Ph | 18.6 ± 0.4 [28] | 13.1 | 72.1 ± 5.8 | 8.00 ± 1.5 | CNS+ |
| 4b | 4-FPh | 2.14 ± 1.1 | 1.50 | 48.5 ± 7.6 | 4.80 ± 2.6 | CNS+ |
| 4c | 2-ClPh | 1.51 ± 1.1 | 1.06 | 58.7 ± 6.8 | 9.40 ± 3.5 | CNS+ |
| 4d | 3-ClPh | 1.05 ± 0.3 | 0.74 | 76.0 ± 6.5 | 11.8 ± 3.0 | CNS+ |
| 4e | 4-ClPh | 1.94 ± 0.5 | 1.36 | 6.46 ± 1.6 | 12.9 ± 3.6 | CNS+ |
| 4f | 2-BrPh | 0.83 ± 0.3 | 0.58 | 7.39 ± 2.0 | 10.1 ± 5.4 | CNS+ |
| 4g | 4-BrPh | 2.35 ± 1.1 | 1.65 | 62.1 ± 1.6 | 7.20 ± 2.4 | CNS+ |
| 4h | 3-NO2Ph | 0.82 ± 0.2 | 0.57 | 75.5 ± 2.7 | 2.10 ± 1.0 | CNS± |
| 4i | 4-NO2Ph | 3.09 ± 0.4 | 2.17 | 73.2 ± 2.9 | 3.10 ± 1.0 | CNS± |
| 4j | 2-Thienyl | 1.74 ± 1.0 | 1.22 | 60.6 ± 6.8 | 8.80 ± 4.4 | CNS+ |
| 4k | 3-Pyridyl | 4.53 ± 2.1 | 3.18 | 85.6 ± 5.3 | 0.70 ± 0.9 | CNS- |
| 4l | 4-Pyridyl | 10.2 ± 3.2 | 7.14 | 68.9 ± 4.5 | 5.00 ± 2.9 | CNS+ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michalska, P.; Mayo, P.; Fernández-Mendívil, C.; Tenti, G.; Duarte, P.; Buendia, I.; Ramos, M.T.; López, M.G.; Menéndez, J.C.; León, R. Antioxidant, Anti-inflammatory and Neuroprotective Profiles of Novel 1,4-Dihydropyridine Derivatives for the Treatment of Alzheimer’s Disease. Antioxidants 2020, 9, 650. https://doi.org/10.3390/antiox9080650
Michalska P, Mayo P, Fernández-Mendívil C, Tenti G, Duarte P, Buendia I, Ramos MT, López MG, Menéndez JC, León R. Antioxidant, Anti-inflammatory and Neuroprotective Profiles of Novel 1,4-Dihydropyridine Derivatives for the Treatment of Alzheimer’s Disease. Antioxidants. 2020; 9(8):650. https://doi.org/10.3390/antiox9080650
Chicago/Turabian StyleMichalska, Patrycja, Paloma Mayo, Cristina Fernández-Mendívil, Giammarco Tenti, Pablo Duarte, Izaskun Buendia, María Teresa Ramos, Manuela G. López, J. Carlos Menéndez, and Rafael León. 2020. "Antioxidant, Anti-inflammatory and Neuroprotective Profiles of Novel 1,4-Dihydropyridine Derivatives for the Treatment of Alzheimer’s Disease" Antioxidants 9, no. 8: 650. https://doi.org/10.3390/antiox9080650
APA StyleMichalska, P., Mayo, P., Fernández-Mendívil, C., Tenti, G., Duarte, P., Buendia, I., Ramos, M. T., López, M. G., Menéndez, J. C., & León, R. (2020). Antioxidant, Anti-inflammatory and Neuroprotective Profiles of Novel 1,4-Dihydropyridine Derivatives for the Treatment of Alzheimer’s Disease. Antioxidants, 9(8), 650. https://doi.org/10.3390/antiox9080650