ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease



Abstract
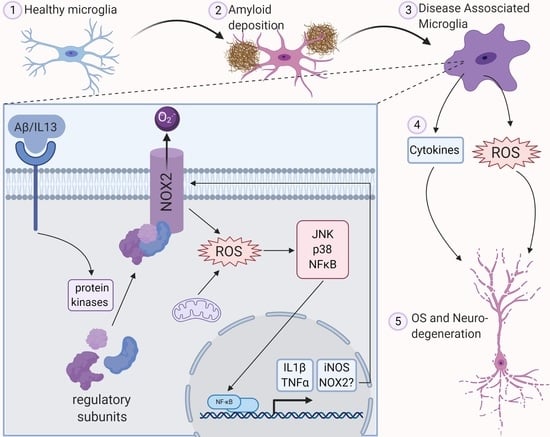
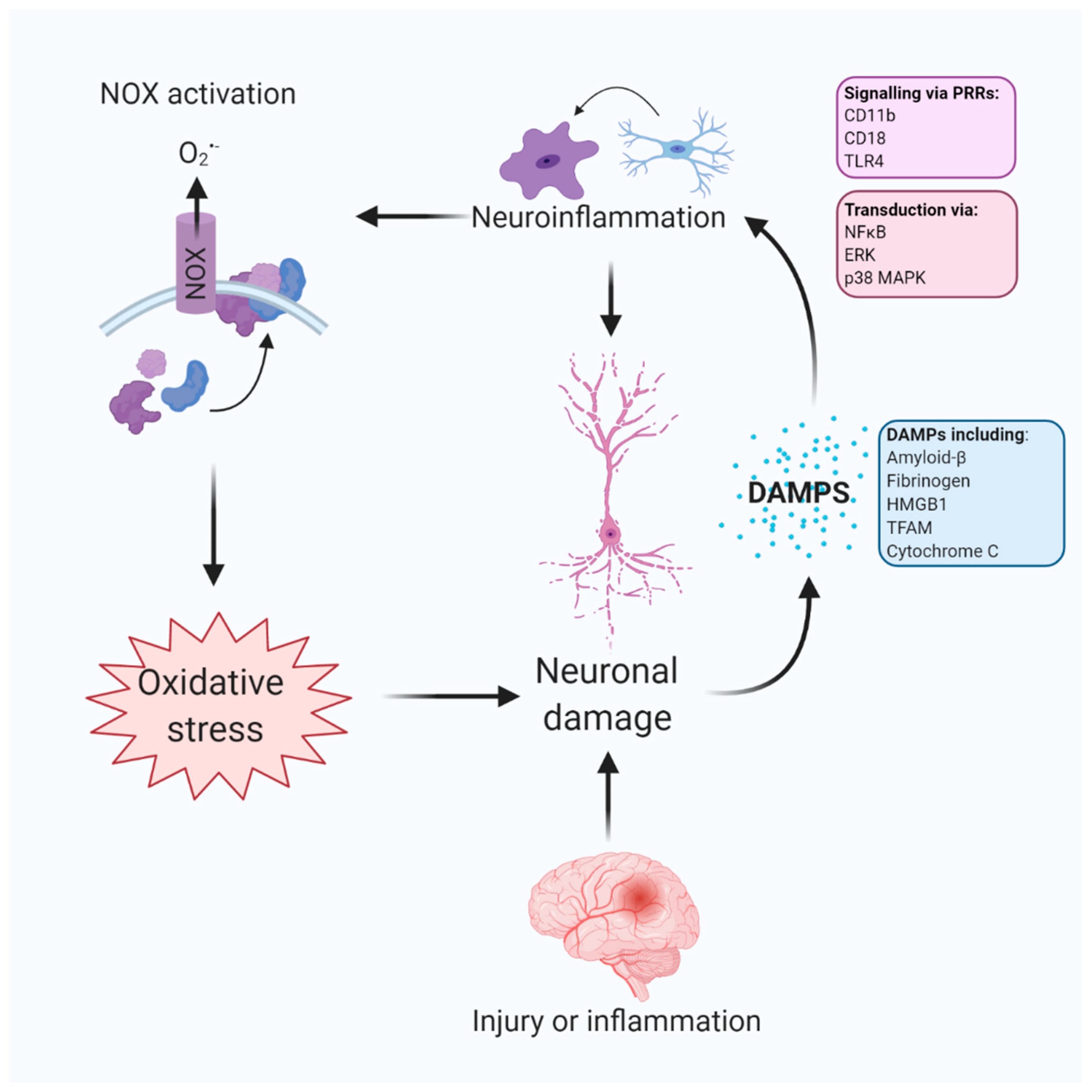
:
{kind=link}
{kind=link}
{kind=link}
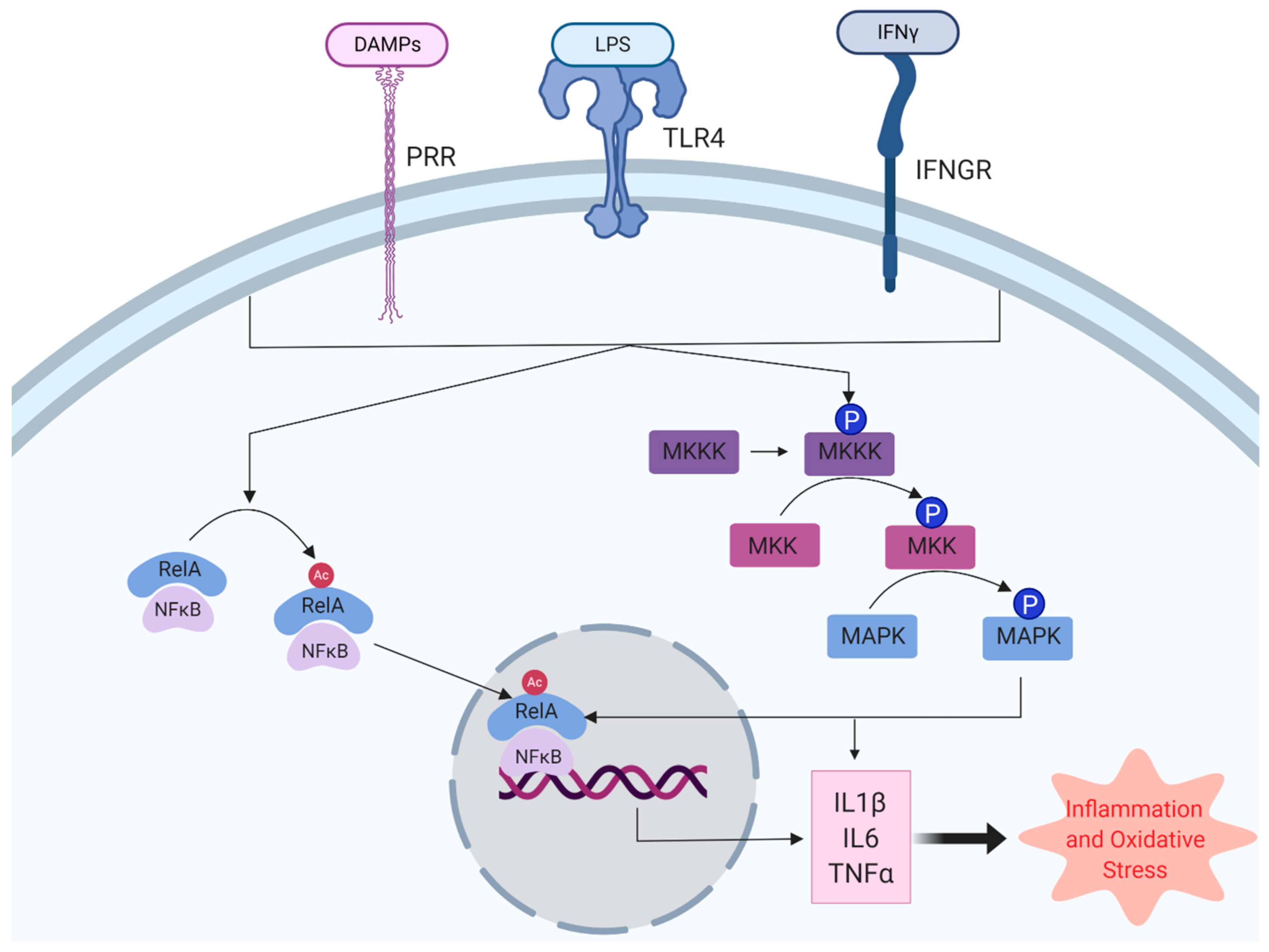{kind=link}
1. Introduction
1.1. Neurodegenerative Disease Is a Global Public Health Challenge
1.2. Microglial Activation and Oxidative Stress Are Hallmarks of Neurodegenerative Disease
1.3. The Brain Is Especially Susceptible to Oxidative Stress
2. Inflammatory-Related ROS Production—NOX Mediates Production of ROS in Microglia
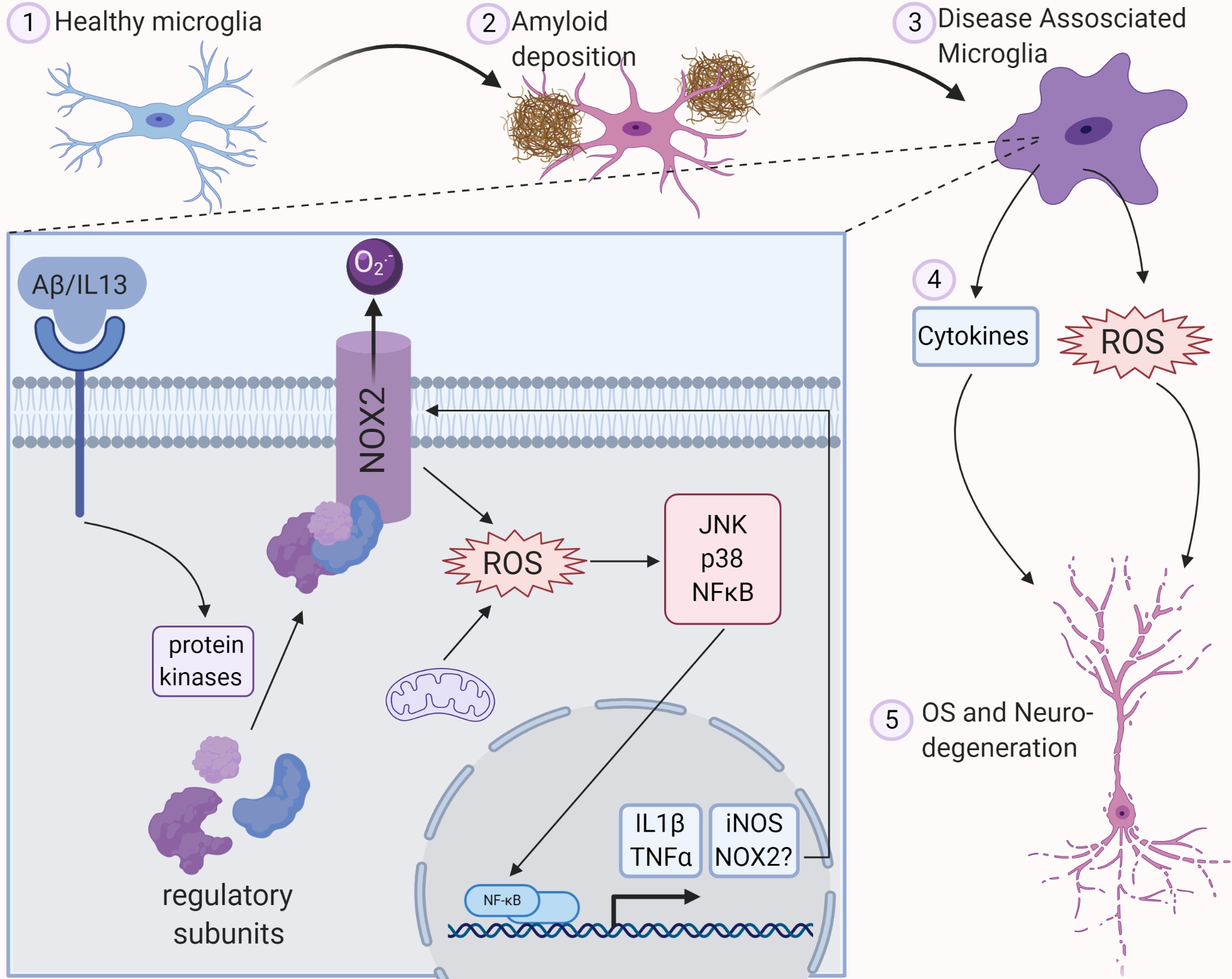
2.1. NOX Are a Family of Enzyme Subunits that Mediate Deliberate Production of ROS in Inflammation
2.2. NOX Enzymes Can Be Subgrouped Based on Their Homology to gp91phox and Regulation of Activity
2.3. Phagocytic NOX (NOX1-3) Are Regulated by the Assembly of Cytosolic Subunits
3. Expression and Regulation of NOX in Microglia
Microglia Express NOX2 and NOX4
4. Microglial NOX Is Activated in Inflammation and Neurodegeneration
4.1. NOX2 and NOX4 Are Activated by Acute, Pro-Inflammatory Stimulation of Microglia
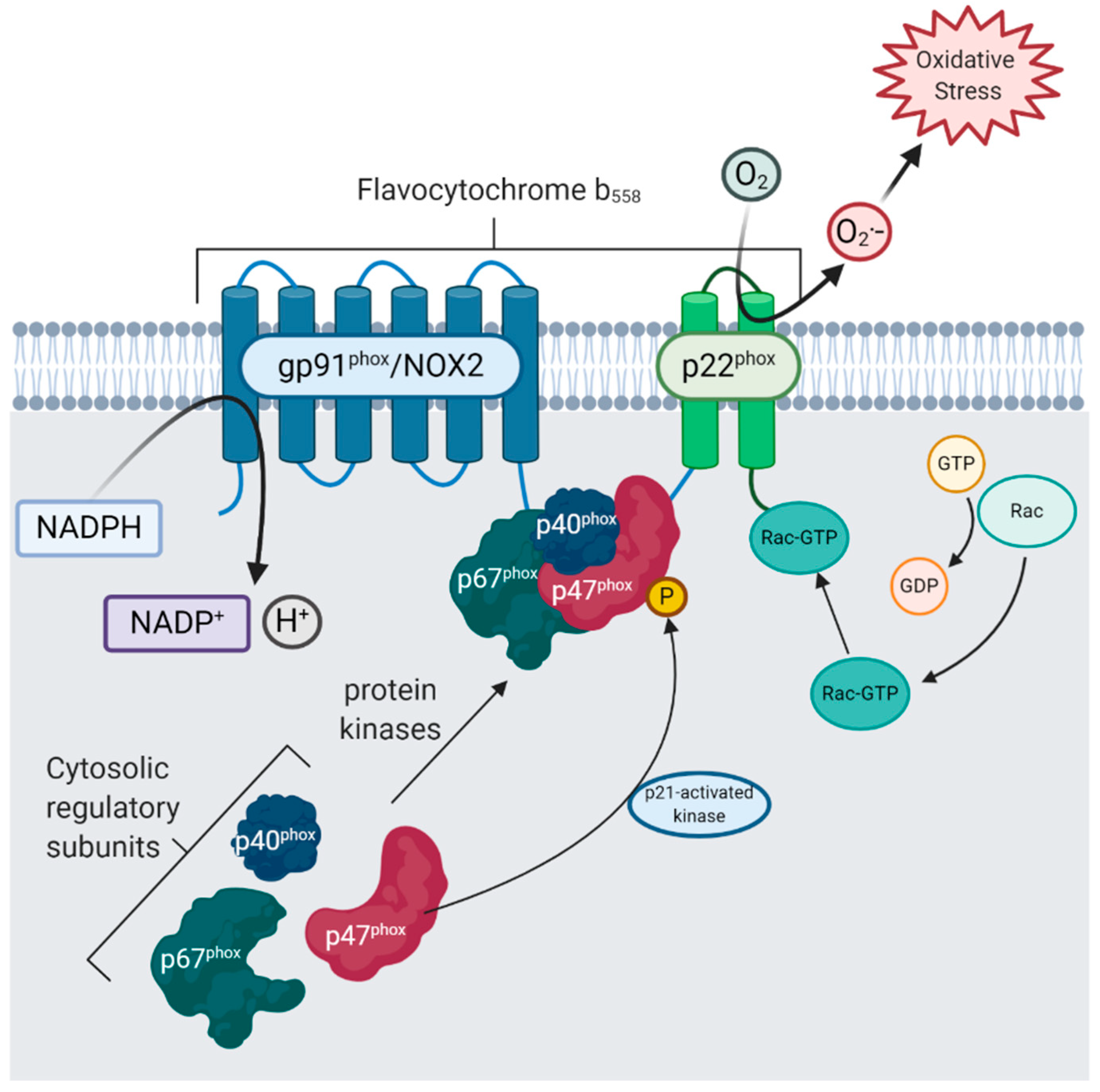
4.2. Damage-Associated Molecular Patterns (DAMPs) Originating from Neurons Mediate NOX Activation via Pattern Recognition Receptors CR3 and TLR4
4.3. NOX Is Activated in Chronic Disease-Associated Microglia (DAM)—A Focus on Alzheimer’s Disease
4.3.1. Alzheimer’s as a Chronic Inflammatory Disease
4.3.2. NOX Is Activated in the Human AD Brain
4.3.3. Mammalian Models of AD Indicate NOX Activation Contributes to Neuronal Loss
4.3.4. NOX Is Activated in AD-Associated Microglia
4.3.5. Further Investigation of Microglial NOX Should Focus on Tau Pathology and Cell-Specific Manipulation of NOX
5. ROS Are Secondary Messengers Activating Pro-inflammatory Pathways in Microglia
5.1. NFκB, a Master Regulator of Inflammation in Microglia, Is Associated with NOX Expression
5.1.1. H2O2 Activates NFκB Signalling in TLR4-Activated Macrophages
5.1.2. There Is a Relationship between NOX Expression and NFκB Activation in Microglia
5.2. ROS Serves to Activate the MAPK Family Resulting in Microglial Activation
5.2.1. JNK and p38 MAPK Activation Is Associated with NOX4
5.2.2. ROS of Mitochondrial Origin Contribute to MAPK Activation
6. Lipid Droplet-Accumulating Microglia as a Source of OS in Neurodegenerative Disease
6.1. Lipid Droplet Accumulation Is Stimulated by ROS and Correlated with Neuronal Death
6.2. Lipid-Droplet Accumulating Microglia May Represent a Double-Edged Sword in Neurodegeneration
7. Balancing the Scales—Antioxidant Enzymes Limit Microglial Activation
7.1. Expression of Classical Antioxidant Proteins Are Controlled by Nrf2 in Microglia
7.2. A Role for TLDc Proteins in Neuroinflammation
8. Conclusions and Future Directions
8.1. Understanding the Molecular Mechanisms of Neuroinflammation Is Key to Developing Effective Antioxidant Therapies
8.2. At the Forefront of Drug Discovery: ToxSeq Analysis of ROS-Generating Microglia
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Erkkinen, M.G.; Kim, M.-O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Partridge, L.; Deelen, J.; Slagboom, P.E. Facing up to the global challenges of ageing. Nature 2018, 561, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Garre-Olmo, J. Epidemiology of Alzheimer’s disease and other dementias. Rev. Neurol. 2018, 66, 377–386. [Google Scholar] [PubMed]
- Winblad, I.; Viramo, P.; Remes, A.; Manninen, M.; Jokelainen, J. Prevalence of dementia—A rising challenge among ageing populations. Eur. Geriatr. Med. 2010, 1, 330–333. [Google Scholar] [CrossRef]
- Luengo-Fernandez, R.; Leal, J.; Gray, A. UK research spend in 2008 and 2012: Comparing stroke, cancer, coronary heart disease and dementia. BMJ Open 2015, 5, e006648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prince, M.; Wimo, A.; Guerchet, M.; Ali, G.-C.; Wu, Y.-T.; Prina, M. World Alzheimer Report 2015, the Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends; Alzheimer’s Disease International: London, UK, 2015; p. 87. [Google Scholar]
- Louveau, A.; Harris, T.H.; Kipnis, J. Revisiting the concept of CNS immune privilege. Trends Immunol. 2015, 36, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Louveau, A.; Plog, B.A.; Antila, S.; Alitalo, K.; Nedergaard, M.; Kipnis, J. Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J. Clin. Investig. 2017, 127, 3210–3219. [Google Scholar] [CrossRef] [Green Version]
- Sankowski, R.; Böttcher, C.; Masuda, T.; Geirsdottir, L.; Sindram, E.; Seredenina, T.; Muhs, A.; Scheiwe, C.; Shah, M.J. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 2019, 22, 2098–2110. [Google Scholar] [CrossRef]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Mielke, M.L.; Gómez-Isla, T.; Betensky, R.A.; Growdon, J.H.; Frosch, M.P.; Hyman, B.T. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am. J. Pathol. 2011, 179, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Allison, E.K.; Khoury, J.E. Microglial Dysfunction and Defective β-Amyloid Clearance Pathways in Aging Alzheimer’s Disease Mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Hu, W.; Tsai, J.; Li, W.; Gan, W.-B. Microglia limit the expansion of β-amyloid plaques in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 47. [Google Scholar] [CrossRef]
- Zhang, B.; Gaiteri, C.; Bodea, L.-G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takatori, S.; Wang, W.; Iguchi, A.; Tomita, T. Genetic Risk Factors for Alzheimer Disease: Emerging Roles of Microglia in Disease Pathomechanisms. Adv. Exp. Med. Biol. 2019, 1118, 83–116. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Gratuze, M.; Leyns, C.E.G.; Holtzman, D.M. New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 66. [Google Scholar] [CrossRef] [Green Version]
- Eriksen, J.L.; Mackenzie, I.R.A. Progranulin: Normal function and role in neurodegeneration. J. Neurochem. 2008, 104, 287–297. [Google Scholar] [CrossRef]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef]
- Bolós, M.; Llorens-Martín, M.; Jurado-Arjona, J.; Hernández, F.; Rábano, A.; Avila, J. Direct Evidence of Internalization of Tau by Microglia In Vitro and In Vivo. J. Alzheimers Dis. JAD 2016, 50, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.C.; Rizer, J.; Selenica, M.-L.B.; Reid, P.; Kraft, C.; Johnson, A.; Blair, L.; Gordon, M.N.; Dickey, C.A.; Morgan, D. LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J. Neuroinflamm. 2010, 7, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Croisier, E.; Moran, L.B.; Dexter, D.T.; Pearce, R.K.; Graeber, M.B. Microglial inflammation in the parkinsonian substantia nigra: Relationship to alpha-synuclein deposition. J. Neuroinflamm. 2005, 2, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, X.; Maguire-Zeiss, K.A.; Giuliano, R.; Prifti, L.; Venkatesh, K.; Federoff, H.J. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol. Aging 2008, 29, 1690–1701. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Ho, D.-H.; Suk, J.-E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.-J.; et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [Green Version]
- Van Muiswinkel, F.L.; Veerhuis, R.; Eikelenboom, P. Amyloid beta protein primes cultured rat microglial cells for an enhanced phorbol 12-myristate 13-acetate-induced respiratory burst activity. J. Neurochem. 1996, 66, 2468–2476. [Google Scholar] [CrossRef]
- Klegeris, A.; McGeer, P.L. beta-amyloid protein enhances macrophage production of oxygen free radicals and glutamate. J. Neurosci. Res. 1997, 49, 229–235. [Google Scholar] [CrossRef]
- Schilling, T.; Eder, C. Amyloid-β-induced reactive oxygen species production and priming are differentially regulated by ion channels in microglia. J. Cell. Physiol. 2011, 226, 3295–3302. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D.; Sung, S. Lipid peroxidation and oxidative imbalance: Early functional events in Alzheimer’s disease. J. Alzheimers Dis. JAD 2004, 6, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D.; Clark, C.M.; Lee, V.M.-Y.; Trojanowski, J.Q.; Rokach, J.; FitzGerald, G.A. Increased 8,12-iso-iPF2α-VI in Alzheimer’s disease: Correlation of a noninvasive index of lipid peroxidation with disease severity. Ann. Neurol. 2000, 48, 809–812. [Google Scholar] [CrossRef]
- Lovell, M.A.; Ehmann, W.D.; Butler, S.M.; Markesbery, W.R. Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer’s disease. Neurology 1995, 45, 1594–1601. [Google Scholar] [CrossRef]
- Subbarao, K.V.; Richardson, J.S.; Ang, L.C. Autopsy samples of Alzheimer’s cortex show increased peroxidation in vitro. J. Neurochem. 1990, 55, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Hall, N.; Subramaniam, R.; Cole, P.; Harris, M.; Aksenov, M.; Aksenova, M.; Gabbita, S.P.; Wu, J.F.; Carney, J.M. Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J. Neurochem. 1995, 65, 2146–2156. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.D.; Carney, J.M.; Starke-Reed, P.E.; Oliver, C.N.; Stadtman, E.R.; Floyd, R.A.; Markesbery, W.R. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1991, 88, 10540–10543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mecocci, P.; MacGarvey, U.; Beal, M.F. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol. 1994, 36, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; Khoury, J.E. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359. [Google Scholar] [CrossRef]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Claude, J.; Linnartz-Gerlach, B.; Kudin, A.P.; Kunz, W.S.; Neumann, H. Microglial CD33-related Siglec-E inhibits neurotoxicity by preventing the phagocytosis-associated oxidative burst. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 18270–18276. [Google Scholar] [CrossRef]
- Herzog, C.; Pons Garcia, L.; Keatinge, M.; Greenald, D.; Moritz, C.; Peri, F.; Herrgen, L. Rapid clearance of cellular debris by microglia limits secondary neuronal cell death after brain injury in vivo. Dev. Camb. Engl. 2019, 146. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimers Dis. JAD 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiss, A.B.; Arain, H.A.; Stecker, M.M.; Siegart, N.M.; Kasselman, L.J. Amyloid toxicity in Alzheimer’s disease. Rev. Neurosci. 2018, 29, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.F.; Tsunawaki, S. Secretion of toxic oxygen products by macrophages: Regulatory cytokines and their effects on the oxidase. Ciba Found. Symp. 1986, 118, 211–230. [Google Scholar] [CrossRef] [PubMed]
- Berendes, H.; Bridges, R.A.; Good, R.A. A fatal granulomatosus of childhood: The clinical study of a new syndrome. Minn. Med. 1957, 40, 309–312. [Google Scholar]
- Quie, P.G.; White, J.G.; Holmes, B.; Good, R.A. In vitro bactericidal capacity of human polymorphonuclear leukocytes: Diminished activity in chronic granulomatous disease of childhood. J. Clin. Investig. 1967, 46, 668–679. [Google Scholar] [CrossRef]
- Baehner, R.L.; Nathan, D.G. Leukocyte oxidase: Defective activity in chronic granulomatous disease. Science 1967, 155, 835–836. [Google Scholar] [CrossRef]
- Babior, B.M.; Curnutte, J.T.; Kipnes, B.S. Pyridine nucleotide-dependent superoxide production by a cell-free system from human granulocytes. J. Clin. Investig. 1975, 56, 1035–1042. [Google Scholar] [CrossRef]
- Segal, A.W.; Jones, O.T. Novel cytochrome b system in phagocytic vacuoles of human granulocytes. Nature 1978, 276, 515–517. [Google Scholar] [CrossRef]
- Segal, A.W.; West, I.; Wientjes, F.; Nugent, J.H.; Chavan, A.J.; Haley, B.; Garcia, R.C.; Rosen, H.; Scrace, G. Cytochrome b-245 is a flavocytochrome containing FAD and the NADPH-binding site of the microbicidal oxidase of phagocytes. Biochem. J. 1992, 284, 781–788. [Google Scholar] [CrossRef] [Green Version]
- Royer-Pokora, B.; Kunkel, L.M.; Monaco, A.P.; Goff, S.C.; Newburger, P.E.; Baehner, R.L.; Cole, F.S.; Curnutte, J.T.; Orkin, S.H. Cloning the gene for an inherited human disorder–chronic granulomatous disease–on the basis of its chromosomal location. Nature 1986, 322, 32–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teahan, C.; Rowe, P.; Parker, P.; Totty, N.; Segal, A.W. The X-linked chronic granulomatous disease gene codes for the beta-chain of cytochrome b-245. Nature 1987, 327, 720–721. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.-A.; Arnold, R.S.; Lassegue, B.; Shi, J.; Xu, X.; Sorescu, D.; Chung, A.B.; Griendling, K.K.; Lambeth, J.D. Cell transformation by the superoxide-generating oxidase Mox1. Nature 1999, 401, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Nisimoto, Y.; Diebold, B.A.; Cosentino-Gomes, D.; Constentino-Gomes, D.; Lambeth, J.D. Nox4: A hydrogen peroxide-generating oxygen sensor. Biochemistry 2014, 53, 5111–5120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiszt, M.; Kopp, J.B.; Várnai, P.; Leto, T.L. Identification of renox, an NAD(P)H oxidase in kidney. Proc. Natl. Acad. Sci. USA 2000, 97, 8010–8014. [Google Scholar] [CrossRef] [Green Version]
- Shiose, A.; Kuroda, J.; Tsuruya, K.; Hirai, M.; Hirakata, H.; Naito, S.; Hattori, M.; Sakaki, Y.; Sumimoto, H. A novel superoxide-producing NAD(P)H oxidase in kidney. J. Biol. Chem. 2001, 276, 1417–1423. [Google Scholar] [CrossRef] [Green Version]
- Darrah, P.A.; Hondalus, M.K.; Chen, Q.; Ischiropoulos, H.; Mosser, D.M. Cooperation between reactive oxygen and nitrogen intermediates in killing of Rhodococcus equi by activated macrophages. Infect. Immun. 2000, 68, 3587–3593. [Google Scholar] [CrossRef] [Green Version]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef]
- Edens, W.A.; Sharling, L.; Cheng, G.; Shapira, R.; Kinkade, J.M.; Lee, T.; Edens, H.A.; Tang, X.; Sullards, C.; Flaherty, D.B.; et al. Tyrosine cross-linking of extracellular matrix is catalyzed by Duox, a multidomain oxidase/peroxidase with homology to the phagocyte oxidase subunit gp91phox. J. Cell Biol. 2001, 154, 879–891. [Google Scholar] [CrossRef]
- Diebold, B.A.; Bokoch, G.M. Molecular basis for Rac2 regulation of phagocyte NADPH oxidase. Nat. Immunol. 2001, 2, 211–215. [Google Scholar] [CrossRef]
- Roepstorff, K.; Rasmussen, I.; Sawada, M.; Cudre-Maroux, C.; Salmon, P.; Bokoch, G.; van Deurs, B.; Vilhardt, F. Stimulus-dependent Regulation of the Phagocyte NADPH Oxidase by a VAV1, Rac1, and PAK1 Signaling Axis. J. Biol. Chem. 2008, 283, 7983–7993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayernia, Z.; Jaquet, V.; Krause, K.-H. New Insights on NOX Enzymes in the Central Nervous System. Antioxid. Redox Signal. 2013, 20, 2815–2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooney, S.J.; Bermudez-Sabogal, S.L.; Byrnes, K.R. Cellular and temporal expression of NADPH oxidase (NOX) isotypes after brain injury. J. Neuroinflamm. 2013, 10, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorce, S.; Krause, K.-H. NOX Enzymes in the Central Nervous System: From Signaling to Disease. Antioxid. Redox Signal. 2009, 11, 2481–2504. [Google Scholar] [CrossRef] [PubMed]
- Altenhöfer, S.; Kleikers, P.W.M.; Radermacher, K.A.; Scheurer, P.; Rob Hermans, J.J.; Schiffers, P.; Ho, H.; Wingler, K.; Schmidt, H.H.H.W. The NOX toolbox: Validating the role of NADPH oxidases in physiology and disease. Cell. Mol. Life Sci. 2012, 69, 2327–2343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrander, L.; Cartier, L.; Bedard, K.; Banfi, B.; Lardy, B.; Plastre, O.; Sienkiewicz, A.; Fórró, L.; Schlegel, W.; Krause, K.-H. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem. J. 2007, 406, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Ansari, M.A.; Scheff, S.W. NADPH-oxidase activation and cognition in Alzheimer disease progression. Free Radic. Biol. Med. 2011, 51, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M.V. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef]
- Nauseef, W.M. Biological Roles for the NOX Family NADPH Oxidases. J. Biol. Chem. 2008, 283, 16961–16965. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, C.J.; Leung, Y.F.; Suter, D.M. Expression dynamics of NADPH oxidases during early zebrafish development. J. Comp. Neurol. 2016, 524, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Vallet, P.; Charnay, Y.; Steger, K.; Ogier-Denis, E.; Kovari, E.; Herrmann, F.; Michel, J.-P.; Szanto, I. Neuronal expression of the NADPH oxidase NOX4, and its regulation in mouse experimental brain ischemia. Neuroscience 2005, 132, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Bedard, K.; Sorce, S.; Hinz, B.; Dubois-Dauphin, M.; Krause, K.-H. NOX4 expression in human microglia leads to constitutive generation of reactive oxygen species and to constitutive IL-6 expression. J. Innate Immun. 2009, 1, 570–581. [Google Scholar] [CrossRef]
- Chéret, C.; Gervais, A.; Lelli, A.; Colin, C.; Amar, L.; Ravassard, P.; Mallet, J.; Cumano, A.; Krause, K.-H.; Mallat, M. Neurotoxic Activation of Microglia Is Promoted by a Nox1-Dependent NADPH Oxidase. J. Neurosci. 2008, 28, 12039–12051. [Google Scholar] [CrossRef]
- Gyoneva, S.; Hosur, R.; Gosselin, D.; Zhang, B.; Ouyang, Z.; Cotleur, A.C.; Peterson, M.; Allaire, N.; Challa, R.; Cullen, P.; et al. Cx3cr1-deficient microglia exhibit a premature aging transcriptome. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [Green Version]
- Hickman, S.E.; Allison, E.K.; Coleman, U.; Kingery-Gallagher, N.D.; El Khoury, J. Heterozygous CX3CR1 Deficiency in Microglia Restores Neuronal β-Amyloid Clearance Pathways and Slows Progression of Alzheimer’s Like-Disease in PS1-APP Mice. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Varvel, N.H.; Konerth, M.E.; Xu, G.; Cardona, A.E.; Ransohoff, R.M.; Lamb, B.T. CX3CR1 Deficiency Alters Microglial Activation and Reduces Beta-Amyloid Deposition in Two Alzheimer’s Disease Mouse Models. Am. J. Pathol. 2010, 177, 2549–2562. [Google Scholar] [CrossRef]
- Tan, Y.-L.; Yuan, Y.; Tian, L. Microglial regional heterogeneity and its role in the brain. Mol. Psychiatry 2020, 25, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Böttcher, C.; Amann, L.; Sagar, N.; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.W.; Wang, J.; Zhang, Q.; Wang, R.; Dhandapani, K.M.; Vadlamudi, R.K.; Brann, D.W. NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 2017, 12, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Gao, J.; Yan, Z.; Huang, X.; Guo, P.; Sun, L.; Liu, Z.; Hu, Y.; Zuo, L.; Yu, S.; et al. Minimally Toxic Dose of Lipopolysaccharide and α-Synuclein Oligomer Elicit Synergistic Dopaminergic Neurodegeneration: Role and Mechanism of Microglial NOX2 Activation. Mol. Neurobiol. 2018, 55, 619–632. [Google Scholar] [CrossRef]
- Fu, H.; Liu, B.; Frost, J.L.; Hong, S.; Jin, M.; Ostaszewski, B.; Shankar, G.M.; Costantino, I.M.; Carroll, M.C.; Mayadas, T.N.; et al. Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Aβ by microglia. Glia 2012, 60, 993–1003. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Wang, K.; Zhang, C.; Sun, F.; Che, Y.; Zhao, X.; Zhang, D.; Li, H.; Wang, Q. Complement receptor 3 mediates NADPH oxidase activation and dopaminergic neurodegeneration through a Src-Erk-dependent pathway. Redox Biol. 2018, 14, 250–260. [Google Scholar] [CrossRef]
- Bermudez, S.; Khayrullina, G.; Zhao, Y.; Byrnes, K.R. NADPH oxidase isoform expression is temporally regulated and may contribute to microglial/macrophage polarization after spinal cord injury. Mol. Cell. Neurosci. 2016, 77, 53–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Barrett, J.P.; Alvarez-Croda, D.-M.; Stoica, B.A.; Faden, A.I.; Loane, D.J. NOX2 drives M1-like microglial/macrophage activation and neurodegeneration following experimental traumatic brain injury. Brain. Behav. Immun. 2016, 58, 291–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gülke, E.; Gelderblom, M.; Magnus, T. Danger signals in stroke and their role on microglia activation after ischemia. Ther. Adv. Neurol. Disord. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Rubartelli, A. DAMP-Mediated Activation of NLRP3-Inflammasome in Brain Sterile Inflammation: The Fine Line between Healing and Neurodegeneration. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef]
- Savage, C.D.; Lopez-Castejon, G.; Denes, A.; Brough, D. NLRP3-Inflammasome Activating DAMPs Stimulate an Inflammatory Response in Glia in the Absence of Priming Which Contributes to Brain Inflammation after Injury. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Revilla-Sanchez, R.; Moss, S.; Haydon, P.G. Multiphoton in vivo imaging of amyloid in animal models of Alzheimer’s disease. Neuropharmacology 2010, 59, 268–275. [Google Scholar] [CrossRef] [Green Version]
- Damisah, E.C.; Hill, R.A.; Rai, A.; Chen, F.; Rothlin, C.V.; Ghosh, S.; Grutzendler, J. Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Sci. Adv. 2020, 6, eaba3239. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.T.; Puskas, F.; Agoston, V.A.; Cleveland, J.C.; Freeman, K.A.; Gamboni, F.; Herson, P.S.; Meng, X.; Smith, P.D.; Weyant, M.J.; et al. Toll-like receptor 4-dependent microglial activation mediates spinal cord ischemia-reperfusion injury. Circulation 2013, 128, S152–S156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajwa, E.; Pointer, C.B.; Klegeris, A. The Role of Mitochondrial Damage-Associated Molecular Patterns in Chronic Neuroinflammation. Available online: https://www.hindawi.com/journals/mi/2019/4050796/ (accessed on 29 July 2020).
- Zhang, D.; Hu, X.; Qian, L.; Chen, S.-H.; Zhou, H.; Wilson, B.; Miller, D.S.; Hong, J.-S. Microglial MAC1 receptor and PI3K are essential in mediating β-amyloid peptide-induced microglial activation and subsequent neurotoxicity. J. Neuroinflamm. 2011, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Zhang, D.; Pang, H.; Caudle, W.M.; Li, Y.; Gao, H.; Liu, Y.; Qian, L.; Wilson, B.; Di Monte, D.A.; et al. Macrophage antigen complex-1 mediates reactive microgliosis and progressive dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. J. Immunol. 2008, 181, 7194–7204. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, H.; McGeer, P.L. Brain microglia constitutively express beta-2 integrins. J. Neuroimmunol. 1990, 30, 81–93. [Google Scholar] [CrossRef]
- Gao, H.-M.; Zhou, H.; Zhang, F.; Wilson, B.C.; Kam, W.; Hong, J.-S. HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 1081–1092. [Google Scholar] [CrossRef]
- Levesque, S.; Taetzsch, T.; Lull, M.E.; Johnson, J.A.; McGraw, C.; Block, M.L. The role of MAC1 in diesel exhaust particle-induced microglial activation and loss of dopaminergic neuron function. J. Neurochem. 2013, 125, 756–765. [Google Scholar] [CrossRef] [Green Version]
- Pei, Z.; Pang, H.; Qian, L.; Yang, S.; Wang, T.; Zhang, W.; Wu, X.; Dallas, S.; Wilson, B.; Reece, J.M.; et al. MAC1 mediates LPS-induced production of superoxide by microglia: The role of pattern recognition receptors in dopaminergic neurotoxicity. Glia 2007, 55, 1362–1373. [Google Scholar] [CrossRef] [PubMed]
- Merlini, M.; Rafalski, V.A.; Rios Coronado, P.E.; Gill, T.M.; Ellisman, M.; Muthukumar, G.; Subramanian, K.S.; Ryu, J.K.; Syme, C.A.; Davalos, D.; et al. Fibrinogen Induces Microglia-Mediated Spine Elimination and Cognitive Impairment in an Alzheimer’s Disease Model. Neuron 2019, 101, 1099–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, I.C. The Contribution and Therapeutic Potential of Epigenetic Modifications in Alzheimer’s Disease. Front. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, E.; Szoeke, C.E.I.; Vollset, S.E.; Abbasi, N.; Abd-Allah, F.; Abdela, J.; Aichour, M.T.E.; Akinyemi, R.O.; Alahdab, F.; Asgedom, S.W.; et al. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Disease International. World Alzheimer Report 2019: Attitudes to Dementia; Alzheimer’s Disease International: London, UK, 2019. [Google Scholar]
- Alzheimer, A.; Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. N. Y. NY 1995, 8, 429–431. [Google Scholar] [CrossRef]
- Mangialasche, F.; Solomon, A.; Winblad, B.; Mecocci, P.; Kivipelto, M. Alzheimer’s disease: Clinical trials and drug development. Lancet Neurol. 2010, 9, 702–716. [Google Scholar] [CrossRef]
- Hillen, H. The Beta Amyloid Dysfunction (BAD) Hypothesis for Alzheimer’s Disease. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [Green Version]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin. Investig. Drugs 2017, 26, 735–739. [Google Scholar] [CrossRef]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Shimohama, S.; Tanino, H.; Kawakami, N.; Okamura, N.; Kodama, H.; Yamaguchi, T.; Hayakawa, T.; Nunomura, A.; Chiba, S.; Perry, G.; et al. Activation of NADPH oxidase in Alzheimer’s disease brains. Biochem. Biophys. Res. Commun. 2000, 273, 5–9. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Wands, J.R. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J. Alzheimers Dis. JAD 2006, 9, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Han, B.H.; Zhou, M.-L.; Johnson, A.W.; Singh, I.; Liao, F.; Vellimana, A.K.; Nelson, J.W.; Milner, E.; Cirrito, J.R.; Basak, J.; et al. Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction, and microhemorrhage in aged Tg2576 mice. Proc. Natl. Acad. Sci. USA 2015, 112, E881–E890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, L.; Anrather, J.; Zhou, P.; Frys, K.; Pitstick, R.; Younkin, S.; Carlson, G.A.; Iadecola, C. NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 1769–1777. [Google Scholar] [CrossRef]
- Bruce-Keller, A.J.; Gupta, S.; Knight, A.G.; Beckett, T.L.; McMullen, J.M.; Davis, P.R.; Murphy, M.P.; Van Eldik, L.J.; St Clair, D.; Keller, J.N. Cognitive impairment in humanized APP×PS1 mice is linked to Aβ(1-42) and NOX activation. Neurobiol. Dis. 2011, 44, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumont, M.; Stack, C.; Elipenhali, C.; Calingasan, N.Y.; Wille, E.; Beal, M.F. Apocynin administration does not improve behavioral and neuropathological deficits in a transgenic mouse model of Alzheimer’s disease. Neurosci. Lett. 2011, 492, 150–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce-Keller, A.J.; Gupta, S.; Parrino, T.E.; Knight, A.G.; Ebenezer, P.J.; Weidner, A.M.; LeVine, H.; Keller, J.N.; Markesbery, W.R. NOX activity is increased in mild cognitive impairment. Antioxid. Redox Signal. 2010, 12, 1371–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.-H.; Aid, S.; Kim, H.-W.; Jackson, S.H.; Bosetti, F. Inhibition of NADPH oxidase promotes alternative and anti-inflammatory microglial activation during neuroinflammation. J. Neurochem. 2012, 120, 292–301. [Google Scholar] [CrossRef]
- Bianca, V.D.; Dusi, S.; Bianchini, E.; Prà, I.D.; Rossi, F. β-Amyloid Activates the O⨪2 Forming NADPH Oxidase in Microglia, Monocytes, and Neutrophils A POSSIBLE INFLAMMATORY MECHANISM OF NEURONAL DAMAGE IN ALZHEIMER’S DISEASE. J. Biol. Chem. 1999, 274, 15493–15499. [Google Scholar] [CrossRef] [Green Version]
- Carrano, A.; Hoozemans, J.J.M.; van der Vies, S.M.; Rozemuller, A.J.M.; van Horssen, J.; de Vries, H.E. Amyloid Beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid. Redox Signal. 2011, 15, 1167–1178. [Google Scholar] [CrossRef]
- Carrano, A.; Hoozemans, J.J.M.; van der Vies, S.M.; van Horssen, J.; de Vries, H.E.; Rozemuller, A.J.M. Neuroinflammation and blood-brain barrier changes in capillary amyloid angiopathy. Neurodegener. Dis. 2012, 10, 329–331. [Google Scholar] [CrossRef]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, L.; Fan, L.M.; Liu, F.; Smith, C.; Li, J.-M. Nox2 dependent redox-regulation of microglial response to amyloid-β stimulation and microgliosis in aging. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.W.; Baik, H.H.; Jin, B.K. IL-13-induced oxidative stress via microglial NADPH oxidase contributes to death of hippocampal neurons in vivo. J. Immunol. 2009, 183, 4666–4674. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Kundel, F.; Amodeo, G.F.; Pavlov, E.V.; Klenerman, D.; Abramov, A.Y. Insoluble tau aggregates induce neuronal death through modification of membrane ion conductance, activation of voltage-gated calcium channels and NADPH oxidase. FEBS J. 2020. [Google Scholar] [CrossRef]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, H.-U.; McCabe, C.; Gjoneska, E.; Sullivan, S.E.; Kaskow, B.J.; Tang, A.; Smith, R.V.; Xu, J.; Pfenning, A.R.; Bernstein, B.E.; et al. Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in the aging and Alzheimer human brain. Nat. Neurosci. 2019, 22, 37–46. [Google Scholar] [CrossRef]
- Dujardin, S.; Commins, C.; Lathuiliere, A.; Beerepoot, P.; Fernandes, A.R.; Kamath, T.V.; De Los Santos, M.B.; Klickstein, N.; Corjuc, D.L.; Corjuc, B.T.; et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat. Med. 2020, 1–8. [Google Scholar] [CrossRef]
- Sheng, W.S.; Hu, S.; Feng, A.; Rock, R.B. Reactive Oxygen Species From Human Astrocytes Induced Functional Impairment and Oxidative Damage. Neurochem. Res. 2013, 38, 2148–2159. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, T.; Feng, G. Tmem119-EGFP and Tmem119-CreERT2 Transgenic Mice for Labeling and Manipulating Microglia. eNeuro 2019, 6. [Google Scholar] [CrossRef] [Green Version]
- Ruan, C.; Sun, L.; Kroshilina, A.; Beckers, L.; De Jager, P.; Bradshaw, E.M.; Hasson, S.A.; Yang, G.; Elyaman, W. A novel Tmem119-tdTomato reporter mouse model for studying microglia in the central nervous system. Brain. Behav. Immun. 2020, 83, 180–191. [Google Scholar] [CrossRef]
- Caruso, G.; Fresta, C.G.; Musso, N.; Giambirtone, M.; Grasso, M.; Spampinato, S.F.; Merlo, S.; Drago, F.; Lazzarino, G.; Sortino, M.A.; et al. Carnosine Prevents Aβ-Induced Oxidative Stress and Inflammation in Microglial Cells: A Key Role of TGF-β1. Cells 2019, 8, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, S.A.; Sood, A.; Shukla, R.; Hanif, K. AT2R Activation Prevents Microglia Pro-inflammatory Activation in a NOX-Dependent Manner: Inhibition of PKC Activation and p47phox Phosphorylation by PP2A. Mol. Neurobiol. 2019, 56, 3005–3023. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Available online: https://www.hindawi.com/journals/omcl/2016/4350965/ (accessed on 2 May 2020).
- Murphy, M.P.; Holmgren, A.; Larsson, N.-G.; Halliwell, B.; Chang, C.J.; Kalyanaraman, B.; Rhee, S.G.; Thornalley, P.J.; Partridge, L.; Gems, D.; et al. Unraveling the Biological Roles of Reactive Oxygen Species. Cell Metab. 2011, 13, 361–366. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.-Y.; Tan, M.-S.; Yu, J.-T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3. [Google Scholar] [CrossRef]
- Zheng, C.; Yin, Q.; Wu, H. Structural studies of NF-κB signaling. Cell Res. 2011, 21, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Dresselhaus, E.C.; Meffert, M.K. Cellular Specificity of NF-κB Function in the Nervous System. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, V.Y.-F.; Huang, W.; Asagiri, M.; Spann, N.; Hoffmann, A.; Glass, C.; Ghosh, G. The transcriptional specificity of NF-κB dimers is coded within the κB DNA response elements. Cell Rep. 2012, 2, 824–839. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Bi, W.; Xiao, S.; Lan, X.; Cheng, X.; Zhang, J.; Lu, D.; Wei, W.; Wang, Y.; Li, H.; et al. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 2019, 9, 5790. [Google Scholar] [CrossRef] [Green Version]
- Schoonbroodt, S.; Ferreira, V.; Best-Belpomme, M.; Boelaert, J.R.; Legrand-Poels, S.; Korner, M.; Piette, J. Crucial Role of the Amino-Terminal Tyrosine Residue 42 and the Carboxyl-Terminal PEST Domain of IκBα in NF-κB Activation by an Oxidative Stress. J. Immunol. 2000, 164, 4292–4300. [Google Scholar] [CrossRef]
- Takada, Y.; Mukhopadhyay, A.; Kundu, G.C.; Mahabeleshwar, G.H.; Singh, S.; Aggarwal, B.B. Hydrogen Peroxide Activates NF-κB through Tyrosine Phosphorylation of IκBα and Serine Phosphorylation of p65 evidence for the involvement of IκBα kinase and Syk protein-tyrosine kinase. J. Biol. Chem. 2003, 278, 24233–24241. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Engelhardt, J.F. Interleukin-1β Induction of NFκB Is Partially Regulated by H2O2-mediated Activation of NFκB-inducing Kinase. J. Biol. Chem. 2006, 281, 1495–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-H.; Na, H.-J.; Kim, C.-K.; Kim, J.-Y.; Ha, K.-S.; Lee, H.; Chung, H.-T.; Kwon, H.J.; Kwon, Y.-G.; Kim, Y.-M. The non-provitamin A carotenoid, lutein, inhibits NF-κB-dependent gene expression through redox-based regulation of the phosphatidylinositol 3-kinase/PTEN/Akt and NF-κB-inducing kinase pathways: Role of H2O2 in NF-κB activation. Free Radic. Biol. Med. 2008, 45, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Luo, Y.-F.; Wang, Y.-S.; Yang, Q.; Xiao, Y.-L.; Cai, H.-R.; Xie, C.-M. Using ROS as a Second Messenger, NADPH Oxidase 2 Mediates Macrophage Senescence via Interaction with NF-κB during Pseudomonas aeruginosa Infection. Available online: https://www.hindawi.com/journals/omcl/2018/9741838/ (accessed on 16 June 2020).
- Delgado, M. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit the MEKK1/MEK4/JNK signaling pathway in endotoxin-activated microglia. Biochem. Biophys. Res. Commun. 2002, 293, 771–776. [Google Scholar] [CrossRef]
- Barata, A.G.; Dick, T.P. A role for peroxiredoxins in H2O2- and MEKK-dependent activation of the p38 signaling pathway. Redox Biol. 2020, 28, 101340. [Google Scholar] [CrossRef]
- Qin, L.; Crews, F.T. NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. J. Neuroinflamm. 2012, 9, 5. [Google Scholar] [CrossRef] [Green Version]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S.T. Microglia and neuroinflammation: A pathological perspective. J. Neuroinflamm. 2004, 1, 14. [Google Scholar] [CrossRef] [Green Version]
- Kaminska, B.; Gozdz, A.; Zawadzka, M.; Ellert-Miklaszewska, A.; Lipko, M. MAPK signal transduction underlying brain inflammation and gliosis as therapeutic target. Anat. Rec. 2009, 292, 1902–1913. [Google Scholar] [CrossRef]
- McDermott, E.P.; O’Neill, L.A.J. Ras Participates in the Activation of p38 MAPK by Interleukin-1 by Associating with IRAK, IRAK2, TRAF6, and TAK-1. J. Biol. Chem. 2002, 277, 7808–7815. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Liu, S.; Wang, C.; Wang, F.; Song, Y.; Yan, N.; Xi, S.; Liu, Z.; Sun, G. JNK and NADPH oxidase involved in fluoride-induced oxidative stress in BV-2 microglia cells. Mediat. Inflamm. 2013, 2013, 895975. [Google Scholar] [CrossRef]
- Liu, Z.; Yao, X.; Jiang, W.; Li, W.; Zhu, S.; Liao, C.; Zou, L.; Ding, R.; Chen, J. Advanced oxidation protein products induce microglia-mediated neuroinflammation via MAPKs-NF-κB signaling pathway and pyroptosis after secondary spinal cord injury. J. Neuroinflamm. 2020, 17, 90. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Wind, S.; Beuerlein, K.; Eucker, T.; Müller, H.; Scheurer, P.; Armitage, M.; Ho, H.; Schmidt, H.; Wingler, K. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br. J. Pharmacol. 2010, 161, 885–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Min, J.-S.; Kim, B.; Chae, U.-B.; Yun, J.W.; Choi, M.-S.; Kong, I.-K.; Chang, K.-T.; Lee, D.-S. Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-κB pathways. Neurosci. Lett. 2015, 584, 191–196. [Google Scholar] [CrossRef]
- Bordt, E.A.; Polster, B.M. NADPH oxidase- and mitochondria-derived reactive oxygen species in proinflammatory microglial activation: A bipartisan affair? Free Radic. Biol. Med. 2014, 76, 34–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, C.L. Lipid droplet biogenesis. Curr. Opin. Cell Biol. 2019, 59, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Khatchadourian, A.; Bourque, S.D.; Richard, V.R.; Titorenko, V.I.; Maysinger, D. Dynamics and regulation of lipid droplet formation in lipopolysaccharide (LPS)-stimulated microglia. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2012, 1821, 607–617. [Google Scholar] [CrossRef] [Green Version]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 2020, 23, 194–208. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, K.; Sandoval, H.; Yamamoto, S.; Jaiswal, M.; Sanz, E.; Li, Z.; Hui, J.; Graham, B.H.; Quintana, A.; et al. Glial Lipid Droplets and ROS Induced by Mitochondrial Defects Promote Neurodegeneration. Cell 2015, 160, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; MacKenzie, K.R.; Putluri, N.; Maletić-Savatić, M.; Bellen, H.J. The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell Metab. 2017, 26, 719–737. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.-E.; Bird, T.D.; Bekris, L.M.; Montine, T.J.; Leverenz, J.B.; Steinbart, E.; Galloway, N.M.; Feldman, H.; Woltjer, R.; Miller, C.A.; et al. The Spectrum of Mutations in Progranulin. Arch. Neurol. 2010, 67, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Finelli, M.J.; Oliver, P.L. TLDc proteins: New players in the oxidative stress response and neurological disease. Mamm. Genome 2017, 28, 395–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Innamorato, N.G.; Rojo, A.I.; García-Yagüe, A.J.; Yamamoto, M.; de Ceballos, M.L.; Cuadrado, A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J. Immunol. Baltim. 2008, 181, 680–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojo, A.I.; Innamorato, N.G.; Martín-Moreno, A.M.; De Ceballos, M.L.; Yamamoto, M.; Cuadrado, A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia 2010, 58, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Jazwa, A.; Rojo, A.I.; Innamorato, N.G.; Hesse, M.; Fernández-Ruiz, J.; Cuadrado, A. Pharmacological targeting of the transcription factor Nrf2 at the basal ganglia provides disease modifying therapy for experimental parkinsonism. Antioxid. Redox Signal. 2011, 14, 2347–2360. [Google Scholar] [CrossRef] [Green Version]
- Taillé, C.; El-Benna, J.; Lanone, S.; Boczkowski, J.; Motterlini, R. Mitochondrial respiratory chain and NAD(P)H oxidase are targets for the antiproliferative effect of carbon monoxide in human airway smooth muscle. J. Biol. Chem. 2005, 280, 25350–25360. [Google Scholar] [CrossRef] [Green Version]
- Lanone, S.; Bloc, S.; Foresti, R.; Almolki, A.; Taillé, C.; Callebert, J.; Conti, M.; Goven, D.; Aubier, M.; Dureuil, B.; et al. Bilirubin decreases nos2 expression via inhibition of NAD(P)H oxidase: Implications for protection against endotoxic shock in rats. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 1890–1892. [Google Scholar] [CrossRef] [Green Version]
- Nakahira, K.; Kim, H.P.; Geng, X.H.; Nakao, A.; Wang, X.; Murase, N.; Drain, P.F.; Wang, X.; Sasidhar, M.; Nabel, E.G.; et al. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J. Exp. Med. 2006, 203, 2377–2389. [Google Scholar] [CrossRef]
- Taillé, C.; El-Benna, J.; Lanone, S.; Dang, M.-C.; Ogier-Denis, E.; Aubier, M.; Boczkowski, J. Induction of heme oxygenase-1 inhibits NAD(P)H oxidase activity by down-regulating cytochrome b558 expression via the reduction of heme availability. J. Biol. Chem. 2004, 279, 28681–28688. [Google Scholar] [CrossRef] [Green Version]
- Castro-Sánchez, S.; García-Yagüe, Á.J.; Kügler, S.; Lastres-Becker, I. CX3CR1-deficient microglia shows impaired signalling of the transcription factor NRF2: Implications in tauopathies. Redox Biol. 2019, 22, 101118. [Google Scholar] [CrossRef]
- Velagapudi, R.; El-Bakoush, A.; Olajide, O.A. Activation of Nrf2 Pathway Contributes to Neuroprotection by the Dietary Flavonoid Tiliroside. Mol. Neurobiol. 2018, 55, 8103–8123. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Choi, M.H.; Li, M.; Li, K.; Park, G.; Choi, Y.-W. AMPK/Nrf2 signaling is involved in the anti-neuroinflammatory action of Petatewalide B from Petasites japonicus against lipopolysaccharides in microglia. Immunopharmacol. Immunotoxicol. 2018, 40, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Jin, M.L.; Ko, M.J.; Park, G.; Choi, Y.-W. Anti-neuroinflammatory Effect of Emodin in LPS-Stimulated Microglia: Involvement of AMPK/Nrf2 Activation. Neurochem. Res. 2016, 41, 2981–2992. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.-J.; Ko, H.-M.; Jeong, Y.-H.; Park, E.-M.; Kim, H.-S. β-Lapachone suppresses neuroinflammation by modulating the expression of cytokines and matrix metalloproteinases in activated microglia. J. Neuroinflamm. 2015, 12, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henn, A.; Lund, S.; Hedtjärn, M.; Schrattenholz, A.; Pörzgen, P.; Leist, M. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX 2009, 26, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendiola, A.S.; Ryu, J.K.; Bardehle, S.; Meyer-Franke, A.; Ang, K.K.-H.; Wilson, C.; Baeten, K.M.; Hanspers, K.; Merlini, M.; Thomas, S.; et al. Transcriptional profiling and therapeutic targeting of oxidative stress in neuroinflammation. Nat. Immunol. 2020, 21, 513–524. [Google Scholar] [CrossRef]
- Finelli, M.J.; Sanchez-Pulido, L.; Liu, K.X.; Davies, K.E.; Oliver, P.L. The Evolutionarily Conserved Tre2/Bub2/Cdc16 (TBC), Lysin Motif (LysM), Domain Catalytic (TLDc) Domain Is Neuroprotective against Oxidative Stress. J. Biol. Chem. 2016, 291, 2751–2763. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Rousseau, J.; Kim, E.; Ehresmann, S.; Cheng, Y.-T.; Duraine, L.; Zuo, Z.; Park, Y.-J.; Li-Kroeger, D.; Bi, W.; et al. Loss of Oxidation Resistance 1, OXR1, Is Associated with an Autosomal-Recessive Neurological Disease with Cerebellar Atrophy and Lysosomal Dysfunction. Am. J. Hum. Genet. 2019, 105, 1237–1253. [Google Scholar] [CrossRef]
- Liu, K.X.; Edwards, B.; Lee, S.; Finelli, M.J.; Davies, B.; Davies, K.E.; Oliver, P.L. Neuron-specific antioxidant OXR1 extends survival of a mouse model of amyotrophic lateral sclerosis. Brain 2015, 138, 1167–1181. [Google Scholar] [CrossRef] [Green Version]
- Doyle, T.; Moncorgé, O.; Bonaventure, B.; Pollpeter, D.; Lussignol, M.; Tauziet, M.; Apolonia, L.; Catanese, M.-T.; Goujon, C.; Malim, M.H. The interferon-inducible isoform of NCOA7 inhibits endosome-mediated viral entry. Nat. Microbiol. 2018, 3, 1369–1376. [Google Scholar] [CrossRef]
- Baines, K.J.; Hsu, A.C.-Y.; Tooze, M.; Gunawardhana, L.P.; Gibson, P.G.; Wark, P.A.B. Novel immune genes associated with excessive inflammatory and antiviral responses to rhinovirus in COPD. Respir. Res. 2013, 14, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alasoo, K.; Martinez, F.O.; Hale, C.; Gordon, S.; Powrie, F.; Dougan, G.; Mukhopadhyay, S.; Gaffney, D.J. Transcriptional profiling of macrophages derived from monocytes and iPS cells identifies a conserved response to LPS and novel alternative transcription. Sci. Rep. 2015, 5, 12524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmerer, J.M.; Lesinski, G.B.; Ruppert, A.S.; Radmacher, M.D.; Noble, C.; Kendra, K.; Walker, M.J.; Carson, W.E. Gene expression profiling reveals similarities between the in vitro and in vivo responses of immune effector cells to IFN-alpha. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 5900–5906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, M.; Nakamura, M.; Tateno, M.; Sakai, A.; Shimakami, T.; Shirasaki, T.; Yamashita, T.; Arai, K.; Yamashita, T.; Sakai, Y.; et al. Differential interferon signaling in liver lobule and portal area cells under treatment for chronic hepatitis C. J. Hepatol. 2010, 53, 817–826. [Google Scholar] [CrossRef]
- Yu, L.; Croze, E.; Yamaguchi, K.D.; Tran, T.; Reder, A.T.; Litvak, V.; Volkert, M.R. Induction of a Unique Isoform of the NCOA7 Oxidation Resistance Gene by Interferon β-1b. J. Interferon Cytokine Res. 2015, 35, 186–199. [Google Scholar] [CrossRef] [Green Version]
- Shkolnik, K.; Ben-Dor, S.; Galiani, D.; Hourvitz, A.; Dekel, N. Molecular characterization and bioinformatics analysis of Ncoa7B, a novel ovulation-associated and reproduction system-specific Ncoa7 isoform. Reproduction 2008, 135, 321–333. [Google Scholar] [CrossRef] [Green Version]
- de Cabo, R.; Siendones, E.; Minor, R.; Navas, P. CYB5R3: A key player in aerobic metabolism and aging? Aging 2009, 2, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [Green Version]
- Wes, P.D.; Holtman, I.R.; Boddeke, E.W.G.M.; Möller, T.; Eggen, B.J.L. Next generation transcriptomics and genomics elucidate biological complexity of microglia in health and disease. Glia 2016, 64, 197–213. [Google Scholar] [CrossRef]
- Patel, M. Targeting Oxidative Stress in Central Nervous System Disorders. Trends Pharmacol. Sci. 2016, 37, 768–778. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. https://doi.org/10.3390/antiox9080743
Simpson DSA, Oliver PL. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants. 2020; 9(8):743. https://doi.org/10.3390/antiox9080743
Chicago/Turabian StyleSimpson, Dominic S. A., and Peter L. Oliver. 2020. "ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease" Antioxidants 9, no. 8: 743. https://doi.org/10.3390/antiox9080743
APA StyleSimpson, D. S. A., & Oliver, P. L. (2020). ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants, 9(8), 743. https://doi.org/10.3390/antiox9080743