Syk Kinase Inhibitors Synergize with Artemisinins by Enhancing Oxidative Stress in Plasmodium falciparum-Parasitized Erythrocytes

 ,
,  ,
,  ,
,
Abstract
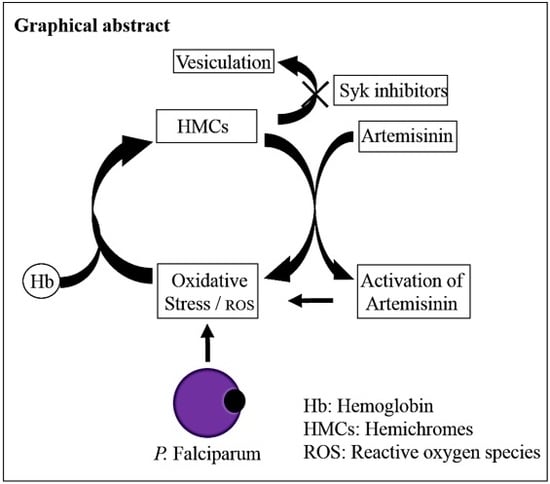
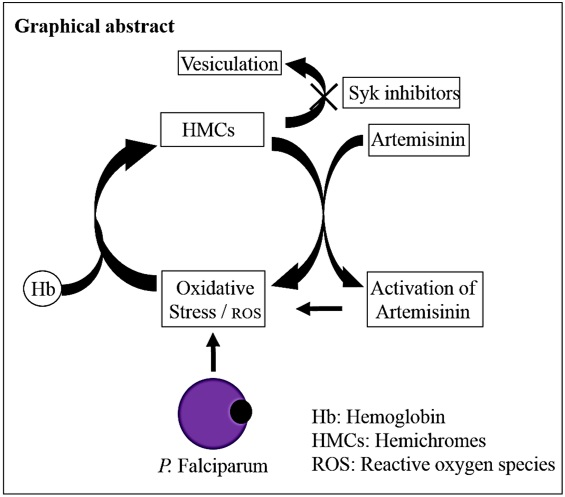
:
1. Introduction
2. Material and Methods
2.1. Cultivation of Plasmodium falciparum-Infected RBCs (pRBCs)
2.2. Ethics Statement
2.3. Drug Susceptibility Assays of Cultured Parasites
2.4. Preparation of Iron Chelator Deferasirox
2.5. Isobologram Preparation and Combination Index (CI) Measurement
2.6. Assessment of Parasitemia by Light Microscopy
2.7. IC50 Measurement
2.8. Preparation of Cells for Confocal Microscopy
2.9. Hemichrome Analysis
2.10. Hemoglobin Release Quantification
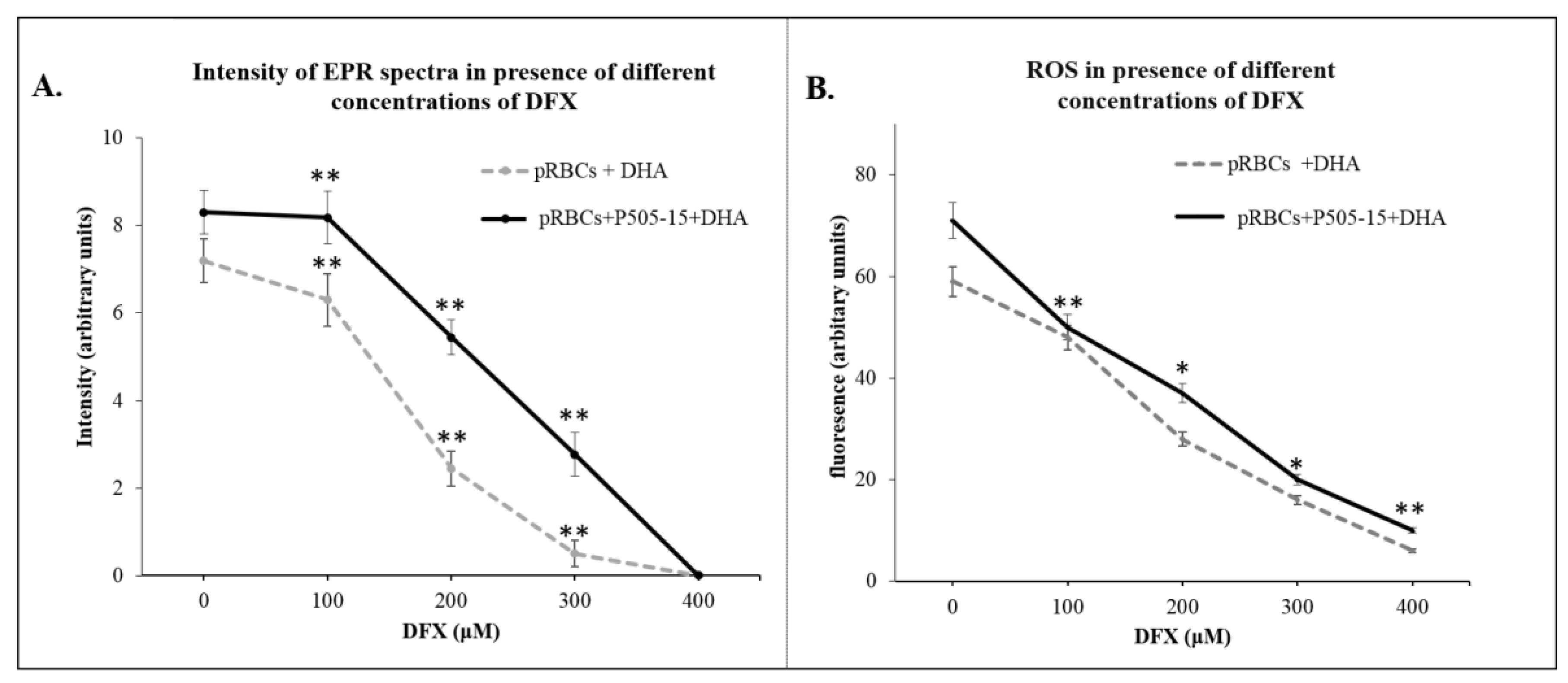
2.11. Electron Paramagnetic Resonance (EPR) Measurements
2.12. ROS Analysis
2.13. Isolation and Characterization of Microparticles (MPs)
2.14. Statistical Analysis
3. Results
3.1. Effect of Syk Inhibitors on Accumulation of Hemichromes in Parasitized RBCs
3.2. Effect of Syk Inhibitors on MP Production
3.3. Hemichromes Accumulation and ROS Production
3.4. Evaluation of Synergy between Syk Inhibitors and Artemisinins in Suppression of Parasitemia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rosenthal, P.J.; Meshnick, S.R. Hemoglobin catabolism and iron utilization by malaria parasites. Mol. Biochem. Parasitol. 1996, 83, 131–139. [Google Scholar] [CrossRef]
- Pandey, A.V.; Tekwani, B.L. Depolymerization of malarial hemozoin: A novel reaction initiated by blood schizontocidal antimalarials. FEBS Lett. 1997. [Google Scholar] [CrossRef] [Green Version]
- Lew, V.L.; Tiffert, T.; Ginsburg, H. Excess hemoglobin digestion and the osmotic stability of Plasmodium falciparum—Infected red blood cells. Blood 2003. [Google Scholar] [CrossRef] [PubMed]
- Mauritz, J.M.A.; Esposito, A.; Ginsburg, H.; Kaminski, C.F.; Tiffert, T.; Lew, V.L. The Homeostasis of Plasmodium falciparum-Infected Red Blood Cells. PLoS Comput. Biol. 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coronado, L.M.; Nadovich, C.T.; Spadafora, C. Malarial hemozoin: From target to tool. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 2032–2041. [Google Scholar] [CrossRef] [Green Version]
- Olafson, K.N.; Nguyen, T.Q.; Rimer, J.D.; Vekilov, P.G. Antimalarials inhibit hematin crystallization by unique drug–surface site interactions. Proc. Natl. Acad. Sci. USA 2017. [Google Scholar] [CrossRef] [Green Version]
- WHO. WHO Malaria Report. 2018; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Hommel, M. The future of artemisinins: Natural, synthetic or recombinant? J. Biol. 2008, 7, 38. [Google Scholar] [CrossRef]
- O’Neill, P.M.; Barton, V.E.; Ward, S.A. The molecular mechanism of action of artemisinin—The debate continues. Molecules 2010, 15, 1705–1721. [Google Scholar] [CrossRef]
- Cui, L.; Su, X.Z. Discovery, mechanisms of action and combination therapy of artemisinin. Expert Rev. Anti. Infect. Ther. 2009, 7, 999–1013. [Google Scholar] [CrossRef]
- Rudrapal, M.; Chetia, D. Endoperoxide antimalarials: Development, structural diversity and pharmacodynamic aspects with reference to 1,2,4-trioxane-based structural scaffold. Drug Des. Dev. Ther. 2016, 10, 3575. [Google Scholar] [CrossRef] [Green Version]
- Klonis, N.; Crespo-Ortiz, M.P.; Bottova, I.; Abu-Bakar, N.; Kenny, S.; Rosenthal, P.J.; Tilley, L. Artemisinin activity against Plasmodium falciparum requires hemoglobin uptake and digestion. Proc. Natl. Acad. Sci. USA 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayi, K.; Turrini, F.; Piga, A.; Arese, P. Enhanced phagocytosis of ring-parasitized mutant erythrocytes: A common mechanism that may explain protection against falciparum malaria in sickle trait and beta-thalassemia trait. Blood 2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantaleo, A.; Ferru, E.; Carta, F.; Valente, E.; Pippia, P.; Turrini, F. Effect of heterozygous beta thalassemia on the phosphorylative response to Plasmodium falciparum infection. J. Proteom. 2012. [Google Scholar] [CrossRef] [PubMed]
- Eridani, S. Sickle cell protection from malaria: A review. Hematol. Rep. Former. Hematol. Rev. 2011. [Google Scholar] [CrossRef]
- Mbanefo, E.C.; Ahmed, A.M.; Titouna, A.; Elmaraezy, A.; Trang, N.T.H.; Phuoc Long, N.; Hoang Anh, N.; Diem Nghi, T.; The Hung, B.; Van Hieu, M.; et al. Association of glucose-6-phosphate dehydrogenase deficiency and malaria: A systematic review and meta-analysis. Sci. Rep. 2017. [Google Scholar] [CrossRef] [Green Version]
- Roberts, D.J.; Williams, T.N. Haemoglobinopathies and resistance to malaria. Redox Rep. 2003. [Google Scholar] [CrossRef] [Green Version]
- López, C.; Saravia, C.; Gomez, A.; Hoebeke, J.; Patarroyo, M.A. Mechanisms of genetically-based resistance to malaria. Gene 2010, 467, 1–12. [Google Scholar] [CrossRef]
- Mohandas, N.; An, X. Malaria and human red blood cells. Med. Microbiol. Immunol. 2012, 201, 593–598. [Google Scholar] [CrossRef]
- Pantaleo, A.; Ferru, E.; Vono, R.; Giribaldi, G.; Lobina, O.; Nepveu, F.; Ibrahim, H.; Nallet, J.P.; Carta, F.; Mannu, F.; et al. New antimalarial indolone-N-oxides, generating radical species, destabilize the host cell membrane at early stages of Plasmodium falciparum growth: Role of band 3 tyrosine phosphorylation. Free Radic. Biol. Med. 2012. [Google Scholar] [CrossRef] [Green Version]
- Ehrhardt, K.; Deregnaucourt, C.; Goetz, A.A.; Tzanova, T.; Gallo, V.; Arese, P.; Pradines, B.; Adjalley, S.H.; Bagrel, D.; Blandin, S.; et al. The redox cycler plasmodione is a fast-acting antimalarial lead compound with pronounced activity against sexual and early asexual blood-stage parasites. Antimicrob. Agents Chemother. 2016. [Google Scholar] [CrossRef] [Green Version]
- Giribaldi, G.; Ulliers, D.; Mannu, F.; Arese, P.; Turrini, F. Growth of Plasmodium falciparum induces stage-dependent haemichrome formation, oxidative aggregation of band 3, membrane deposition of complement and antibodies, and phagocytosis of parasitized erythrocytes. Br. J. Haematol. 2001. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, B.; Jäger, J.; Lansche, C.; Sanchez, C.P.; Cyrklaff, M.; Buchholz, B.; Soubeiga, S.T.; Simpore, J.; Ito, H.; Schwarz, U.S.; et al. Hemoglobin S and C affect biomechanical membrane properties of P. falciparum-infected erythrocytes. Commun. Biol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Méndez, D.; Linares, M.; Diez, A.; Puyet, A.; Bautista, J.M. Stress response and cytoskeletal proteins involved in erythrocyte membrane remodeling upon Plasmodium falciparum invasion are differentially carbonylated in G6PD A- deficiency. Free Radic. Biol. Med. 2011. [Google Scholar] [CrossRef] [PubMed]
- Gallo, V.; Schwarzer, E.; Rahlfs, S.; Schirmer, R.H.; van Zwieten, R.; Roos, D.; Arese, P.; Becker, K. Inherited glutathione reductase deficiency and Plasmodium falciparum malaria—A case study. PLoS ONE 2009. [Google Scholar] [CrossRef]
- Tokumasu, F.; Nardone, G.A.; Ostera, G.R.; Fairhurst, R.M.; Beaudry, S.D.; Hayakawa, E.; Dvorak, J.A. Altered membrane structure and surface potential in homozygous hemoglobin C erythrocytes. PLoS ONE 2009. [Google Scholar] [CrossRef]
- Sherman, I.W.; Eda, S.; Winograd, E. Erythrocyte aging and malaria. Cell. Mol. Biol. (Noisy-le-grand) 2004, 50, 159–169. [Google Scholar]
- Welbourn, E.M.; Wilson, M.T.; Yusof, A.; Metodiev, M.V.; Cooper, C.E. The mechanism of formation, structure and physiological relevance of covalent hemoglobin attachment to the erythrocyte membrane. Free Radic. Biol. Med. 2017. [Google Scholar] [CrossRef] [Green Version]
- Turrini, F.; Arese, P.; Yuan, J.; Low, P.S. Clustering of integral membrane proteins of the human erythrocyte membrane stimulates autologous IgG binding, complement deposition, and phagocytosis. J. Biol. Chem. 1991, 266, 23611–23617. [Google Scholar]
- Tokumasu, F.; Fairhurst, R.M.; Ostera, G.R.; Brittain, N.J.; Hwang, J.; Wellems, T.E.; Dvorak, J.A. Band 3 modifications in Plasmodium falciparum-infected AA and CC erythrocytes assayed by autocorrelation analysis using quantum dots. J. Cell Sci. 2005. [Google Scholar] [CrossRef] [Green Version]
- Low, P.S.; Waugh, S.M.; Zinke, K.; Drenckhahn, D. The Role of Hemoglobin Denaturation and Band 3 Clustering in Red Blood Cell Aging. Science 1985. [Google Scholar] [CrossRef]
- Pantaleo, A.; Ferru, E.; Giribaldi, G.; Mannu, F.; Carta, F.; Matte, A.; de Franceschi, L.; Turrini, F. Oxidized and poorly glycosylated band 3 is selectively phosphorylated by Syk kinase to form large membrane clusters in normal and G6PD-deficient red blood cells. Biochem. J. 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferru, E.; Giger, K.; Pantaleo, A.; Campanella, E.; Grey, J.; Ritchie, K.; Vono, R.; Turrini, F.; Low, P.S. Regulation of membrane-cytoskeletal interactions by tyrosine phosphorylation of erythrocyte band 3. Blood 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappadoro, M.; Giribaldi, G.; O’Brien, E.; Turrini, F.; Mannu, F.; Ulliers, D.; Simula, G.; Luzzatto, L.; Arese, P. Early phagocytosis of glucose-6-phosphate dehydrogenase (G6PD)-deficient erythrocytes parasitized by Plasmodium falciparum may explain malaria protection in G6PD deficiency. Blood 1998. [Google Scholar] [CrossRef]
- Mohandas, N.; Gallagher, P.G. Red cell membrane: Past, present, and future. Blood 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thevenin, B.J.M.; Willardson, B.M.; Low, P.S. The redox state of cysteines 201 and 317 of the erythrocyte anion exchanger is critical for ankyrin binding. J. Biol. Chem. 1989, 264, 15886–15892. [Google Scholar]
- Pantaleo, A.; Ferru, E.; Pau, M.C.; Khadjavi, A.; Mandili, G.; Mattè, A.; Spano, A.; De Franceschi, L.; Pippia, P.; Turrini, F. Band 3 erythrocyte membrane protein acts as redox stress sensor leading to its phosphorylation by p72 Syk. Oxid. Med. Cell. Longev. 2016. [Google Scholar] [CrossRef] [Green Version]
- Turrini, F.; Giribaldi, G.; Carta, F.; Mannu, F.; Arese, P. Mechanisms of band 3 oxidation and clustering in the phagocytosis of Plasmodium falciparum -infected erythrocytes. Redox Rep. 2003. [Google Scholar] [CrossRef]
- Ferru, E.; Pantaleo, A.; Carta, F.; Mannu, F.; Khadjavi, A.; Gallo, V.; Ronzoni, L.; Graziadei, G.; Cappellini, M.D.; Turrini, F. Thalassemic erythrocytes release microparticles loaded with hemichromes by redox activation of p72Syk kinase. Haematologica 2014. [Google Scholar] [CrossRef]
- Leal, J.K.F.; Adjobo-Hermans, M.J.W.; Bosman, G.J.C.G.M. Red blood cell homeostasis: Mechanisms and effects of microvesicle generation in health and disease. Front. Physiol. 2018, 9, 703. [Google Scholar] [CrossRef]
- Aharon, A.; Rebibo-Sabbah, A.; Tzoran, I.; Levin, C. Extracellular Vesicles in Hematological Disorders. Rambam Maimonides Med. J. 2014. [Google Scholar] [CrossRef]
- Westerman, M.; Pizzey, A.; Hirschman, J.; Cerino, M.; Weil-Weiner, Y.; Ramotar, P.; Eze, A.; Lawrie, A.; Purdy, G.; Mackie, I.; et al. Microvesicles in haemoglobinopathies offer insights into mechanisms of hypercoagulability, haemolysis and the effects of therapy. Br. J. Haematol. 2008. [Google Scholar] [CrossRef] [PubMed]
- Kesely, K.R.; Pantaleo, A.; Turrini, F.M.; Olupot-Olupot, P.; Low, P.S. Inhibition of an erythrocyte tyrosine kinase with imatinib prevents Plasmodium falciparum egress and terminates parasitemia. PLoS ONE 2016. [Google Scholar] [CrossRef] [PubMed]
- Pantaleo, A.; Kesely, K.R.; Pau, M.C.; Tsamesidis, I.; Schwarzer, E.; Skorokhod, O.A.; Chien, H.D.; Ponzi, M.; Bertuccini, L.; Low, P.S.; et al. Syk inhibitors interfere with erythrocyte membrane modification during P falciparum growth and suppress parasite egress. Blood 2017, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantaleo, A.; Ferru, E.; Carta, F.; Mannu, F.; Giribaldi, G.; Vono, R.; Lepedda, A.J.; Pippia, P.; Turrini, F. Analysis of changes in tyrosine and serine phosphorylation of red cell membrane proteins induced by P. falciparum growth. Proteomics 2010. [Google Scholar] [CrossRef]
- Amato, R.; Pearson, R.D.; Almagro-Garcia, J.; Amaratunga, C.; Lim, P.; Suon, S.; Sreng, S.; Drury, E.; Stalker, J.; Miotto, O.; et al. Origins of the current outbreak of multidrug-resistant malaria in southeast Asia: A retrospective genetic study. Lancet Infect. Dis. 2018. [Google Scholar] [CrossRef] [Green Version]
- Dondorp, A.M.; Nosten, F.; Yi, P.; Das, D.; Phyo, A.P.; Tarning, J.; Lwin, K.M.; Ariey, F.; Hanpithakpong, W.; Lee, S.J.; et al. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009. [Google Scholar] [CrossRef] [Green Version]
- Hanboonkunupakarn, B.; White, N.J. Travel Medicine and Infectious Disease; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Pantaleo, A.; Pau, M.C.; Chien, H.D.; Turrini, F. Artemisinin resistance, some facts and opinions. J. Infect. Dev. Ctries. 2015. [Google Scholar] [CrossRef] [Green Version]
- Blasco, B.; Leroy, D.; Fidock, D.A. Antimalarial drug resistance: Linking Plasmodium falciparum parasite biology to the clinic. Nat. Med. 2017, 23, 917. [Google Scholar] [CrossRef]
- Cui, L.; Mharakurwa, S.; Ndiaye, D.; Rathod, P.K.; Rosenthal, P.J. Antimalarial drug resistance: Literature review and activities and findings of the ICEMR network. Am. J. Trop. Med. Hyg. 2015, 93, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Trager, W.; Jensen, J.B. Human malaria parasites in continuous culture. Science 1976. [Google Scholar] [CrossRef]
- Lambros, C.; Vanderberg, J.P. Synchronization of Plasmodium falciparum Erythrocytic Stages in Culture. J. Parasitol. 1979. [Google Scholar] [CrossRef]
- Rivadeneira, E.M.; Wasserman, M.; Espinal, C.T. Separation and Concentration of Schizonts of Plasmodium falciparum by Percoll Gradients. J. Protozool. 1983. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the chou-talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.-C. Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacol. Rev. 2006. [Google Scholar] [CrossRef] [PubMed]
- Mannu, F.; Arese, P.; Cappellini, M.D.; Fiorelli, G.; Cappadoro, M.; Giribaldi, G.; Turrini, F. Role of hemichrome binding to erythrocyte membrane in the generation of band-3 alterations in beta-thalassemia intermedia erythrocytes. Blood 1995, 86, 2014–2020. [Google Scholar] [CrossRef] [Green Version]
- Creek, D.J.; Ryan, E.; Charman, W.N.; Chiu, F.C.K.; Prankerd, R.J.; Vennerstrom, J.L.; Charman, S.A. Stability of peroxide antimalarials in the presence of human hemoglobin. Antimicrob. Agents Chemother. 2009. [Google Scholar] [CrossRef] [Green Version]
- Winterbourn, C.C. Free-radical production and oxidative reactions of hemoglobin. Environ. Health Perspect. 1985. [Google Scholar] [CrossRef]
- Antonini, E.; Brunori, M. “Hemoglobin and Methemoglobin” in the Red Blood; Surgenor, D.M.E., Ed.; NY Acad. Press: New York, NY, USA, 1975; Volume 2, pp. 753–797. [Google Scholar]
- Pantaleo, A.; Ferru, E.; Carta, F.; Mannu, F.; Simula, L.F.; Khadjavi, A.; Pippia, P.; Turrini, F. Irreversible AE1 tyrosine phosphorylation leads to membrane vesiculation in G6PD deficient red cells. PLoS ONE 2011. [Google Scholar] [CrossRef] [Green Version]
- Arese, P.; Gallo, V.; Pantaleo, A.; Turrini, F. Life and death of glucose-6-phosphate dehydrogenase (G6PD) deficient erythrocytes-role of redox stress and band 3 modifications. Transfus. Med. Hemotherapy 2012, 39, 328–334. [Google Scholar] [CrossRef] [Green Version]
- Luzzatto, L.; Arese, P. Favism and glucose-6-phosphate dehydrogenase deficiency. N. Engl. J. Med. 2018, 378, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Becker, K.; Tilley, L.; Vennerstrom, J.L.; Roberts, D.; Rogerson, S.; Ginsburg, H. Oxidative stress in malaria parasite-infected erythrocytes: Host-parasite interactions. Int. J. Parasitol. 2004. [Google Scholar] [CrossRef] [PubMed]
- Kavishe, R.A.; Koenderink, J.B.; Alifrangis, M. Oxidative stress in malaria and artemisinin combination therapy: Pros and Cons. FEBS J. 2017, 284, 2579–2591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, M.D.; Eaton, J.W. Thalassaemic erythrocytes: Cellular suicide arising from iron and glutathione-dependent oxidation reactions? Br. J. Haematol. 1995. [Google Scholar] [CrossRef] [PubMed]
- Bayer, S.B.; Low, F.M.; Hampton, M.B.; Winterbourn, C.C. Interactions between peroxiredoxin 2, hemichrome and the erythrocyte membrane. Free Radic. Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Voskou, S.; Aslan, M.; Fanis, P.; Phylactides, M.; Kleanthous, M. Oxidative stress in β-thalassaemia and sickle cell disease. Redox Biol. 2015, 6, 226–239. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Gerhard, G.S. Heme activates artemisinin more efficiently than hemin, inorganic iron, or hemoglobin. Bioorganic Med. Chem. 2008. [Google Scholar] [CrossRef]
- De Franceschi, L.; Bertoldi, M.; Matte, A.; Santos Franco, S.; Pantaleo, A.; Ferru, E.; Turrini, F. Oxidative stress and β -thalassemic erythroid cells behind the molecular defect. Oxid. Med. Cell. Longev. 2013. [Google Scholar] [CrossRef] [Green Version]
- Tsamesidis, I.; Perio, P.; Pantaleo, A.; Reybier, K. Oxidation of erythrocytes enhance the production of Reactive species in the presence of Artemisinins. Int. J. Mol. Sci. 2020, 21, 4799. [Google Scholar] [CrossRef]
- Baluom, M.; Grossbard, E.B.; Mant, T.; Lau, D.T.W. Pharmacokinetics of fostamatinib, a spleen tyrosine kinase (SYK) inhibitor, in healthy human subjects following single and multiple oral dosing in three phase I studies. Br. J. Clin. Pharmacol. 2013. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, E.M.; Stewart, E.; Zhu, F.; Bradley, C.; Griffiths, L.; Yang, L.; Suryadevara, P.K.; Zhang, J.; Freeman, B.B.; Guy, R.K.; et al. Pharmacokinetics and efficacy of the spleen tyrosine kinase inhibitor R406 after ocular delivery for retinoblastoma. Pharm. Res. 2014. [Google Scholar] [CrossRef] [Green Version]
- Doerig, C.; Abdi, A.; Bland, N.; Eschenlauer, S.; Dorin-Semblat, D.; Fennell, C.; Halbert, J.; Holland, Z.; Nivez, M.P.; Semblat, J.P.; et al. Malaria: Targeting parasite and host cell kinomes. Biochim. Biophys. Acta Proteins Proteom. 2010, 1804, 604–612. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Syk Inhibitors (nM) | Dihydroartemisinin | Artesunate | Artemether | |||
|---|---|---|---|---|---|---|
| 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | |
| P505-15 1 (IC50 = 1 nM) | ||||||
| 500 | 0.54 ± 0.03 | 0.75 ± 0.06 | 0.97 ± 0.03 | 0.55 ± 0.07 | 0.87 ± 0.03 | 0.83 ± 0.01 |
| 250 | 0.51 ± 0.01 | 0.71 ± 0.03 | 0.76 ± 0.08 | 0.51 ± 0.01 | 0.89 ± 0.01 | 0.65 ± 0.03 |
| 100 | 0.42 ± 0.04 | 0.55 ± 0.04 | 0.63 ± 0.03 | 0.61 ± 0.03 | 0.95 ± 0.01 | 0.63 ± 0.04 |
| 50 | 0.43 ± 0.05 | 0.49 ± 0.01 | 0.58 ± 0.07 | 0.49 ± 0.05 | 0.98 ± 0.04 | 0.58 ± 0.01 |
| Entospletinib 1 (IC50 = 7.7 nM) | ||||||
| 500 | 0.52 ± 0.01 | 0.85 ± 0.06 | 0.87 ± 0.03 | 0.74 ± 0.04 | 1.01 ± 0.03 | 0.83 ± 0.04 |
| 250 | 0.51 ± 0.07 | 0.84 ± 0.07 | 0.73 ± 0.02 | 0.62 ± 0.01 | 0.95 ± 0.05 | 0.68 ± 0.04 |
| 100 | 0.77 ± 0.04 | 0.78 ± 0.03 | 0.66 ± 0.01 | 0.60 ± 0.02 | 0.98 ± 0.03 | 0.63 ± 0.03 |
| 50 | 0.82 ± 0.03 | 0.69 ± 0.05 | 0.62 ± 0.02 | 0.55 ± 0.02 | 1.04 ± 0.07 | 0.72 ± 0.04 |
| R406 1 (IC50 = 41 nM) | ||||||
| 500 | 0.42 ± 0.04 | 0.95 ± 0.06 | 0.82 ± 0.04 | 0.75 ± 0.02 | 0.85 ± 0.03 | 0.93 ± 0.04 |
| 250 | 0.46 ± 0.02 | 0.81 ± 0.03 | 0.63 ± 0.01 | 0.72 ± 0.01 | 0.82 ± 0.01 | 0.88 ± 0.04 |
| 100 | 0.50 ± 0.03 | 0.60 ± 0.04 | 0.76 ± 0.03 | 0.50 ± 0.02 | 0.80 ± 0.04 | 0.73 ± 0.03 |
| 50 | 0.60 ± 0.05 | 0.58 ± 0.01 | 0.92 ± 0.02 | 0.44 ± 0.02 | 0.82 ± 0.04 | 0.72 ± 0.04 |
| SYK II 1 (IC50 = 41 nM) | ||||||
| 500 | 0.51 ± 0.01 | 0.92 ± 0.02 | 0.79 ± 0.04 | 0.89 ± 0.04 | 0.97 ± 0.01 | 0.73 ± 0.02 |
| 250 | 0.66 ± 0.01 | 0.93 ± 0.02 | 0.55 ± 0.01 | 0.99 ± 0.03 | 0.99 ± 0.03 | 0.58 ± 0.04 |
| 100 | 0.77 ± 0.06 | 0.52 ± 0.03 | 0.67 ± 0.04 | 0.96 ± 0.01 | 1.06 ± 0.05 | 0.53 ± 0.05 |
| 50 | 0.99 ± 0.04 | 0.46 ± 0.05 | 0.89 ± 0.02 | 0.90 ± 0.04 | 1.12 ± 0.01 | 0.62 ± 0.07 |
| Imatinib 1 (IC50 = 5 µM) | ||||||
| 500 | 0.79 ± 0.02 | 0.78 ± 0.01 | 0.67 ± 0.03 | 0.75 ± 0.02 | 1.01 ± 0.02 | 0.83 ± 0.01 |
| 250 | 0.73 ± 0.06 | 0.57 ± 0.07 | 0.61 ± 0.04 | 0.96 ± 0.04 | 0.89 ± 0.03 | 0.78 ± 0.06 |
| 100 | 0.83 ± 0.01 | 0.58 ± 0.02 | 0.70 ± 0.01 | 0.64 ± 0.01 | 0.91 ± 0.01 | 0.73 ± 0.04 |
| 50 | 0.92 ± 0.05 | 0.55 ± 0.05 | 0.90 ± 0.02 | 0.60 ± 0.02 | 0.85 ± 0.02 | 0.82 ± 0.04 |
| Piceatannol 1 (IC50 = 10 µM) | ||||||
| 500 | 0.57 ± 0.04 | 0.67 ± 0.02 | 0.77 ± 0.01 | 0.65 ± 0.03 | 0.95 ± 0.01 | 0.73 ± 0.02 |
| 250 | 0.56 ± 0.01 | 0.66 ± 0.04 | 0.71 ± 0.03 | 0.66 ± 0.07 | 0.78 ± 0.01 | 0.80 ± 0.01 |
| 100 | 0.68 ± 0.03 | 0.58 ± 0.05 | 0.80 ± 0.02 | 0.84 ± 0.02 | 0.85 ± 0.02 | 0.83 ± 0.04 |
| 50 | 0.79 ± 0.02 | 0.49 ± 0.03 | 085 ± 0.01 | 0.90 ± 0.04 | 0.78 ± 0.03 | 0.75 ± 0.01 |
| Syk Inhibitors (µM) | Deferasirox (50 µM) | |
|---|---|---|
| P505-15 | Dihydroartemisin | Artesunate |
| 500 | 1.08 ± 0.05 | 1.15 ± 0.07 |
| 250 | 1.05 ± 0.04 | 1.01 ± 0.09 |
| 100 | 1.08 ± 0.10 | 1.11 ± 0.03 |
| 50 | 1.29 ± 0.08 | 1.42 ± 0.05 |
| R406 | ||
| 500 | 1.03 ± 0.06 | 1.03 ± 0.02 |
| 250 | 0.96 ± 0.07 | 1.08 ± 0.04 |
| 100 | 1.17 ± 0.05 | 1.10 ± 0.06 |
| 50 | 1.29 ± 0.03 | 1.16 ± 0.08 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsamesidis, I.; Reybier, K.; Marchetti, G.; Pau, M.C.; Virdis, P.; Fozza, C.; Nepveu, F.; Low, P.S.; Turrini, F.M.; Pantaleo, A. Syk Kinase Inhibitors Synergize with Artemisinins by Enhancing Oxidative Stress in Plasmodium falciparum-Parasitized Erythrocytes. Antioxidants 2020, 9, 753. https://doi.org/10.3390/antiox9080753
Tsamesidis I, Reybier K, Marchetti G, Pau MC, Virdis P, Fozza C, Nepveu F, Low PS, Turrini FM, Pantaleo A. Syk Kinase Inhibitors Synergize with Artemisinins by Enhancing Oxidative Stress in Plasmodium falciparum-Parasitized Erythrocytes. Antioxidants. 2020; 9(8):753. https://doi.org/10.3390/antiox9080753
Chicago/Turabian StyleTsamesidis, Ioannis, Karine Reybier, Giuseppe Marchetti, Maria Carmina Pau, Patrizia Virdis, Claudio Fozza, Francoise Nepveu, Philip S. Low, Francesco Michelangelo Turrini, and Antonella Pantaleo. 2020. "Syk Kinase Inhibitors Synergize with Artemisinins by Enhancing Oxidative Stress in Plasmodium falciparum-Parasitized Erythrocytes" Antioxidants 9, no. 8: 753. https://doi.org/10.3390/antiox9080753
APA StyleTsamesidis, I., Reybier, K., Marchetti, G., Pau, M. C., Virdis, P., Fozza, C., Nepveu, F., Low, P. S., Turrini, F. M., & Pantaleo, A. (2020). Syk Kinase Inhibitors Synergize with Artemisinins by Enhancing Oxidative Stress in Plasmodium falciparum-Parasitized Erythrocytes. Antioxidants, 9(8), 753. https://doi.org/10.3390/antiox9080753