Systemic Oxidative Stress and Visceral Adipose Tissue Mediators of NLRP3 Inflammasome and Autophagy Are Reduced in Obese Type 2 Diabetic Patients Treated with Metformin


 , ,
, ,  and
and 
Abstract
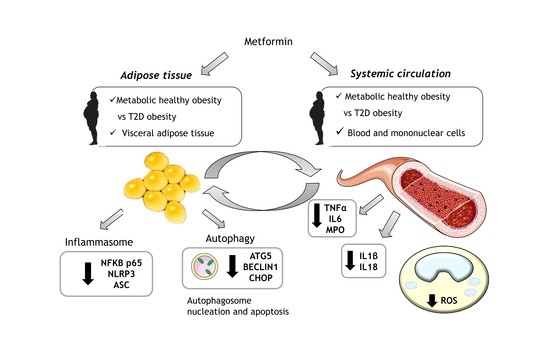
:
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Clinical and Biochemical Determinations
2.3. Cell Isolation
2.4. Analysis of Oxidative Stress Markers and Serum Cytokines
2.5. VAT Protein Analysis
2.6. Statistical Analysis
3. Results
3.1. Anthropometric and Biochemical Parameters
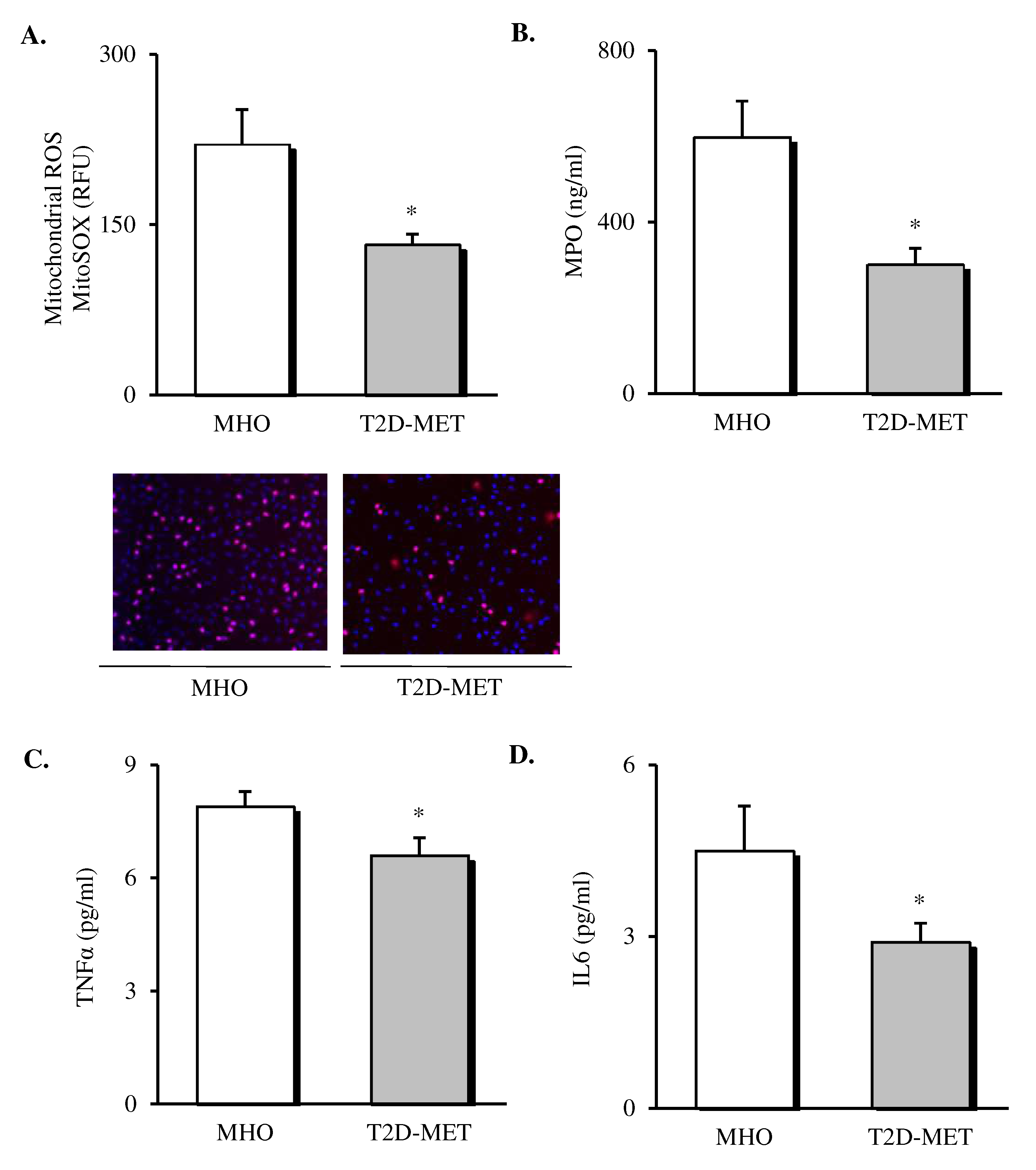
3.2. Systemic Oxidative Stress Markers and Pro-Inflammatory Cytokines
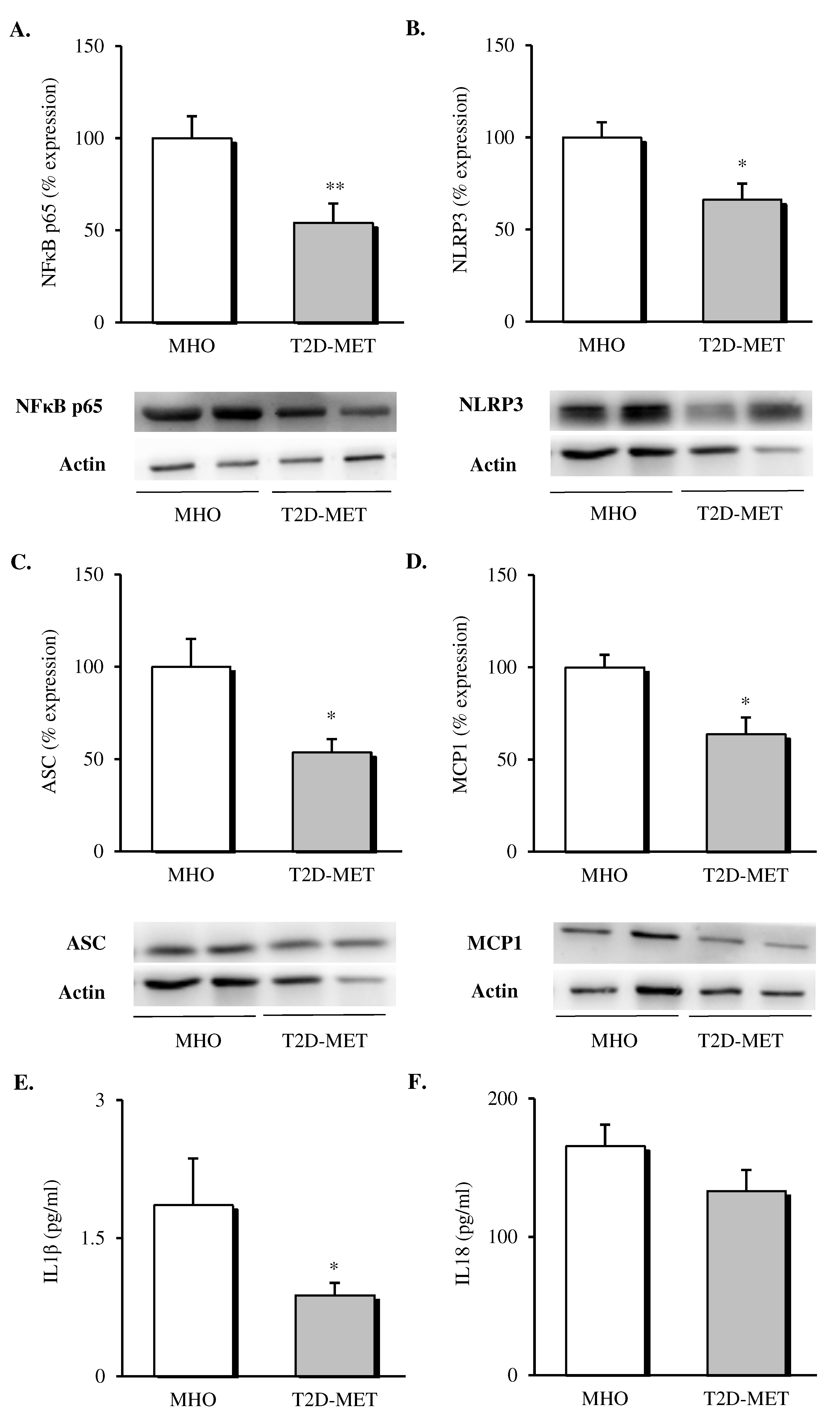
3.3. NLRP3 Inflammasome Complex Activation in VAT
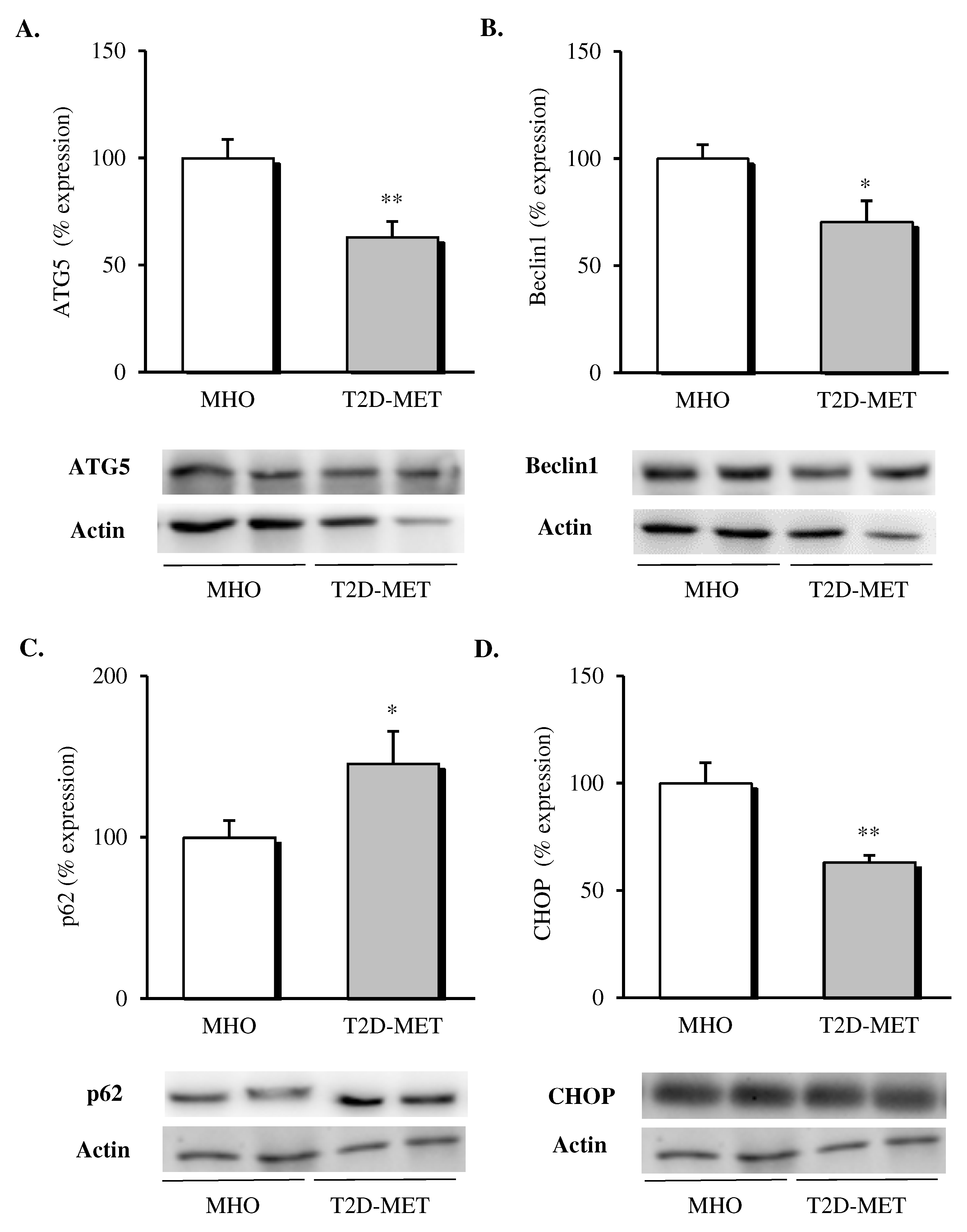
3.4. Protein Expression of Autophagy Mediators in VAT
3.5. Analysis of Correlations between Variables
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Després, J.-P. Body fat distribution and risk of cardiovascular disease: An update. Circulation 2012, 126, 1301–1313. [Google Scholar] [CrossRef] [Green Version]
- Frühbeck, G. Obesity: Screening for the evident in obesity. Nat. Rev. Endocrinol. 2012, 8, 570–572. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Eagon, J.C.; Trujillo, M.E.; Scherer, P.E.; Klein, S. Visceral Fat Adipokine Secretion Is Associated With Systemic Inflammation in Obese Humans. Diabetes 2007, 56, 1010–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernicova, I.; Korbonits, M. Metformin—mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2014, 10, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.R.; Morrison, V.L.; Levin, D.; Mohan, M.; Forteath, C.; Beall, C.; McNeilly, A.D.; Balfour, D.J.K.; Savinko, T.; Wong, A.K.F.; et al. Anti-Inflammatory Effects of Metformin Irrespective of Diabetes Status. Circ. Res. 2016, 119, 652–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, M.; Uchiba, M.; Komura, H.; Mizuochi, Y.; Harada, N.; Okajima, K. Metformin, an antidiabetic agent, suppresses the production of tumor necrosis factor and tissue factor by inhibiting early growth response factor-1 expression in human monocytes in vitro. J. Pharmacol. Exp. Ther. 2010, 334, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Russo, E.; Pellino, G.; D’Angelo, S.; Chiaravalloti, A.; De Sarro, G.; Manfredini, R.; De Giorgio, R. Metformin and Autoimmunity: A “New Deal” of an Old Drug. Front. Immunol. 2018, 9, 1236. [Google Scholar] [CrossRef] [Green Version]
- Foretz, M.; Guigas, B.; Viollet, B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 569–589. [Google Scholar] [CrossRef] [Green Version]
- Blüher, M. Metabolically Healthy Obesity. Endocr. Rev. 2020, 41, 405–420. [Google Scholar] [CrossRef] [Green Version]
- Klöting, N.; Fasshauer, M.; Dietrich, A.; Kovacs, P.; Schön, M.R.; Kern, M.; Stumvoll, M.; Blüher, M. Insulin-sensitive obesity. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E506–E515. [Google Scholar] [CrossRef]
- Iacobini, C.; Pugliese, G.; Blasetti Fantauzzi, C.; Federici, M.; Menini, S. Metabolically healthy versus metabolically unhealthy obesity. Metab. Clin. Exp. 2019, 92, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Esser, N.; L’homme, L.; De Roover, A.; Kohnen, L.; Scheen, A.J.; Moutschen, M.; Piette, J.; Legrand-Poels, S.; Paquot, N. Obesity phenotype is related to NLRP3 inflammasome activity and immunological profile of visceral adipose tissue. Diabetologia 2013, 56, 2487–2497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutterwala, F.S.; Haasken, S.; Cassel, S.L. Mechanism of NLRP3 inflammasome activation. Ann. N. Y. Acad. Sci. 2014, 1319, 82–95. [Google Scholar] [CrossRef]
- Vanaja, S.K.; Rathinam, V.A.K.; Fitzgerald, K.A. Mechanisms of inflammasome activation: Recent advances and novel insights. Trends Cell Biol. 2015, 25, 308–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abderrazak, A.; Syrovets, T.; Couchie, D.; El Hadri, K.; Friguet, B.; Simmet, T.; Rouis, M. NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015, 4, 296–307. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Lu, C.-H.; Hung, Y.-J.; Hsieh, P.-S. Additional effect of metformin and celecoxib against lipid dysregulation and adipose tissue inflammation in high-fat fed rats with insulin resistance and fatty liver. Eur. J. Pharmacol. 2016, 789, 60–67. [Google Scholar] [CrossRef]
- Li, A.; Zhang, S.; Li, J.; Liu, K.; Huang, F.; Liu, B. Metformin and resveratrol inhibit Drp1-mediated mitochondrial fission and prevent ER stress-associated NLRP3 inflammasome activation in the adipose tissue of diabetic mice. Mol. Cell. Endocrinol. 2016, 434, 36–47. [Google Scholar] [CrossRef]
- Ma, Y.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Autophagy and Cellular Immune Responses. Immunity 2013, 39, 211–227. [Google Scholar] [CrossRef] [Green Version]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesselborg, S.; Stork, B. Autophagy signal transduction by ATG proteins: From hierarchies to networks. Cell. Mol. Life Sci. 2015, 72, 4721–4757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovsan, J.; Blüher, M.; Tarnovscki, T.; Klöting, N.; Kirshtein, B.; Madar, L.; Shai, I.; Golan, R.; Harman-Boehm, I.; Schön, M.R.; et al. Altered autophagy in human adipose tissues in obesity. J. Clin. Endocrinol. Metab. 2011, 96, E268–E277. [Google Scholar] [CrossRef] [PubMed]
- Jansen, H.J.; van Essen, P.; Koenen, T.; Joosten, L.A.B.; Netea, M.G.; Tack, C.J.; Stienstra, R. Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology 2012, 153, 5866–5874. [Google Scholar] [CrossRef] [Green Version]
- Kosacka, J.; Kern, M.; Klöting, N.; Paeschke, S.; Rudich, A.; Haim, Y.; Gericke, M.; Serke, H.; Stumvoll, M.; Bechmann, I.; et al. Autophagy in adipose tissue of patients with obesity and type 2 diabetes. Mol. Cell. Endocrinol. 2015, 409, 21–32. [Google Scholar] [CrossRef]
- Ost, A.; Svensson, K.; Ruishalme, I.; Brännmark, C.; Franck, N.; Krook, H.; Sandström, P.; Kjolhede, P.; Strålfors, P. Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol. Med. 2010, 16, 235–246. [Google Scholar] [CrossRef]
- He, C.; Zhu, H.; Li, H.; Zou, M.-H.; Xie, Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes 2013, 62, 1270–1281. [Google Scholar] [CrossRef] [Green Version]
- Tomic, T.; Botton, T.; Cerezo, M.; Robert, G.; Luciano, F.; Puissant, A.; Gounon, P.; Allegra, M.; Bertolotto, C.; Bereder, J.-M.; et al. Metformin inhibits melanoma development through autophagy and apoptosis mechanisms. Cell Death Dis. 2011, 2, e199. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Sharma, A.; Yin, C.; Tan, X.; Xiao, Y. Metformin ameliorates hepatic steatosis and improves the induction of autophagy in HFD-induced obese mice. Mol. Med. Rep. 2017, 16, 680–686. [Google Scholar] [CrossRef] [Green Version]
- Lettieri Barbato, D.; Tatulli, G.; Aquilano, K.; Ciriolo, M.R. FoxO1 controls lysosomal acid lipase in adipocytes: Implication of lipophagy during nutrient restriction and metformin treatment. Cell Death Dis. 2013, 4, e861. [Google Scholar] [CrossRef]
- Berg, A.H.; Scherer, P.E. Adipose tissue, inflammation, and cardiovascular disease. Circ. Res. 2005, 96, 939–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M. Metabolic Syndrome Scientific Statement by the American Heart Association and the National Heart, Lung, and Blood Institute. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2243–2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Diabetes Association 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes—2018. Diabetes Care 2018, 41, S13–S27. [Google Scholar] [CrossRef] [Green Version]
- Bañuls, C.; Rovira-Llopis, S.; Lopez-Domenech, S.; Diaz-Morales, N.; Blas-Garcia, A.; Veses, S.; Morillas, C.; Victor, V.M.; Rocha, M.; Hernandez-Mijares, A. Oxidative and endoplasmic reticulum stress is impaired in leukocytes from metabolically unhealthy vs. healthy obese individuals. Int. J. Obes. 2017, 41, 1556–1563. [Google Scholar] [CrossRef]
- López-Domènech, S.; Bañuls, C.; Díaz-Morales, N.; Escribano-López, I.; Morillas, C.; Veses, S.; Orden, S.; Álvarez, Á.; Víctor, V.M.; Hernández-Mijares, A.; et al. Obesity impairs leukocyte-endothelium cell interactions and oxidative stress in humans. Eur. J. Clin. Investig. 2018, 48, e12985. [Google Scholar] [CrossRef]
- Diaz-Morales, N.; Rovira-Llopis, S.; Bañuls, C.; Lopez-Domenech, S.; Escribano-Lopez, I.; Veses, S.; Jover, A.; Rocha, M.; Hernandez-Mijares, A.; Victor, V.M. Does Metformin Protect Diabetic Patients from Oxidative Stress and Leukocyte-Endothelium Interactions? Antioxid. Redox Signal. 2017, 27, 1439–1445. [Google Scholar] [CrossRef]
- Bułdak, Ł.; Łabuzek, K.; Bułdak, R.J.; Kozłowski, M.; Machnik, G.; Liber, S.; Suchy, D.; Duława-Bułdak, A.; Okopień, B. Metformin affects macrophages’ phenotype and improves the activity of glutathione peroxidase, superoxide dismutase, catalase and decreases malondialdehyde concentration in a partially AMPK-independent manner in LPS-stimulated human monocytes/macrophages. Pharmacol. Rep. PR 2014, 66, 418–429. [Google Scholar] [CrossRef]
- Sena, C.M.; Leandro, A.; Azul, L.; Seiça, R.; Perry, G. Vascular Oxidative Stress: Impact and Therapeutic Approaches. Front. Physiol. 2018, 9, 1668. [Google Scholar] [CrossRef] [Green Version]
- Ghavimi, H.; Sheidaei, S.; Vaez, H.; Zolali, E.; Asgharian, P.; Hamishehkar, H. Metformin-attenuated sepsis-induced oxidative damages: A novel role for metformin. Iran. J. Basic Med. Sci. 2018, 21, 469–475. [Google Scholar]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef] [PubMed]
- Evia-Viscarra, M.L.; Rodea-Montero, E.R.; Apolinar-Jiménez, E.; Muñoz-Noriega, N.; García-Morales, L.M.; Leaños-Pérez, C.; Figueroa-Barrón, M.; Sánchez-Fierros, D.; Reyes-García, J.G. The effects of metformin on inflammatory mediators in obese adolescents with insulin resistance: Controlled randomized clinical trial. J. Pediatr. Endocrinol. Metab. JPEM 2012, 25, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Fidan, E.; Onder Ersoz, H.; Yilmaz, M.; Yilmaz, H.; Kocak, M.; Karahan, C.; Erem, C. The effects of rosiglitazone and metformin on inflammation and endothelial dysfunction in patients with type 2 diabetes mellitus. Acta Diabetol. 2011, 48, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Morita, N.; Hosaka, T.; Kitahara, A.; Murashima, T.; Onuma, H.; Sumitani, Y.; Takahashi, K.; Tanaka, T.; Kondo, T.; Ishida, H. Novel Mechanisms Modulating Palmitate-Induced Inflammatory Factors in Hypertrophied 3T3-L1 Adipocytes by AMPK. J. Diabetes Res. 2018, 2018, 9256482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Schuiveling, M.; Vazirpanah, N.; Radstake, T.R.D.J.; Zimmermann, M.; Broen, J.C.A. Metformin, A New Era for an Old Drug in the Treatment of Immune Mediated Disease? Curr. Drug Targets 2018, 19, 945–959. [Google Scholar] [CrossRef]
- Iannantuoni, F.; Diaz-Morales, N.; Escribano-Lopez, I.; Sola, E.; Roldan-Torres, I.; Apostolova, N.; Bañuls, C.; Rovira-Llopis, S.; Rocha, M.; Victor, V.M. Does Glycemic Control Modulate the Impairment of NLRP3 Inflammasome Activation in Type 2 Diabetes? Antioxid. Redox Signal. 2019, 30, 232–240. [Google Scholar] [CrossRef]
- Shi, C.-S.; Shenderov, K.; Huang, N.-N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [Green Version]
- Bjørkøy, G.; Lamark, T.; Pankiv, S.; Øvervatn, A.; Brech, A.; Johansen, T. Chapter 12 Monitoring Autophagic Degradation of p62/SQSTM1. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2009; Volume 452, pp. 181–197. ISBN 978-0-12-374547-7. [Google Scholar]
- Nuñez, C.E.; Rodrigues, V.S.; Gomes, F.S.; de Moura, R.F.; Victorio, S.C.; Bombassaro, B.; Chaim, E.A.; Pareja, J.C.; Geloneze, B.; Velloso, L.A.; et al. Defective regulation of adipose tissue autophagy in obesity. Int. J. Obes. 2013, 37, 1473–1480. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | MHO | T2D-Met | Corrected by Age |
|---|---|---|---|
| n (females %) | 34 (85.3) | 34 (70.6) | |
| Age (years) | 38.1 ± 9.1 | 51.8 ± 9.1 *** | |
| BMI (kg/m2) | 38.8 ± 4.3 | 39.6 ± 4.6 | p > 0.05 |
| Waist (cm) | 111 ± 11 | 120 ± 12 ** | p < 0.01 |
| Waist-to-hip ratio | 0.86 ± 0.06 | 0.93 ± 0.10 ** | p < 0.05 |
| SBP (mmHg) | 120 ± 9 | 138 ± 20 *** | p < 0.001 |
| DBP (mmHg) | 76 ± 7 | 84 ± 15 * | p < 0.05 |
| Glucose (mg/dL) | 90 ± 8 | 119 ± 37 *** | p < 0.001 |
| Insulin (μU/mL) | 12.90 ± 6.11 | 18.18 ± 9.76 * | p < 0.001 |
| HOMA-IR | 2.96 ± 1.60 | 5.81 ± 9.12 ** | p < 0.001 |
| HbA1c (%) | 5.24 ± 0.32 | 6.20 ± 0.88 *** | p < 0.001 |
| TC (mg/dL) | 182 ± 37 | 181 ± 42 | p > 0.05 |
| HDLc (mg/dL) | 46 ± 8 | 45 ± 9 | p > 0.05 |
| LDLc (mg/dL) | 116 ± 31 | 107 ± 36 | p > 0.05 |
| TG (mg/dL) | 85 (66, 109) | 122 (98, 163) ** | p < 0.05 |
| hsCRP (mg/L) | 4.79 (3.42, 11.05) | 4.00 (1.34, 7.98) | p > 0.05 |
| Leukocytes (cells × 103/μL) | 7.3 ± 1.8 | 7.8 ± 1.9 | p > 0.05 |
| Treatment n (%) | |||
| Hypertension | - | 21 (62%) | |
| Hyperlipidemia | - | 23 (67%) | |
| T2D | - | 34 (100%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abad-Jiménez, Z.; López-Domènech, S.; Díaz-Rúa, R.; Iannantuoni, F.; Gómez-Abril, S.Á.; Periañez-Gómez, D.; Morillas, C.; Víctor, V.M.; Rocha, M. Systemic Oxidative Stress and Visceral Adipose Tissue Mediators of NLRP3 Inflammasome and Autophagy Are Reduced in Obese Type 2 Diabetic Patients Treated with Metformin. Antioxidants 2020, 9, 892. https://doi.org/10.3390/antiox9090892
Abad-Jiménez Z, López-Domènech S, Díaz-Rúa R, Iannantuoni F, Gómez-Abril SÁ, Periañez-Gómez D, Morillas C, Víctor VM, Rocha M. Systemic Oxidative Stress and Visceral Adipose Tissue Mediators of NLRP3 Inflammasome and Autophagy Are Reduced in Obese Type 2 Diabetic Patients Treated with Metformin. Antioxidants. 2020; 9(9):892. https://doi.org/10.3390/antiox9090892
Chicago/Turabian StyleAbad-Jiménez, Zaida, Sandra López-Domènech, Rubén Díaz-Rúa, Francesca Iannantuoni, Segundo Ángel Gómez-Abril, Dolores Periañez-Gómez, Carlos Morillas, Víctor M. Víctor, and Milagros Rocha. 2020. "Systemic Oxidative Stress and Visceral Adipose Tissue Mediators of NLRP3 Inflammasome and Autophagy Are Reduced in Obese Type 2 Diabetic Patients Treated with Metformin" Antioxidants 9, no. 9: 892. https://doi.org/10.3390/antiox9090892
APA StyleAbad-Jiménez, Z., López-Domènech, S., Díaz-Rúa, R., Iannantuoni, F., Gómez-Abril, S. Á., Periañez-Gómez, D., Morillas, C., Víctor, V. M., & Rocha, M. (2020). Systemic Oxidative Stress and Visceral Adipose Tissue Mediators of NLRP3 Inflammasome and Autophagy Are Reduced in Obese Type 2 Diabetic Patients Treated with Metformin. Antioxidants, 9(9), 892. https://doi.org/10.3390/antiox9090892