
Chimeric Human Papillomavirus-16 Virus-like Particles Presenting P18I10 and T20 Peptides from HIV-1 Envelope Induce HPV16 and HIV-1-Specific Humoral and T Cell-Mediated Immunity in BALB/c Mice


Abstract
: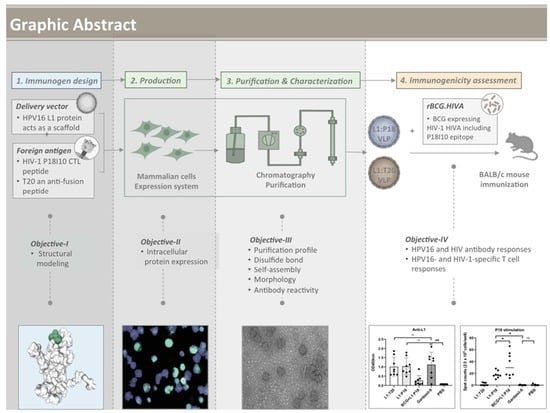
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Construction of the BCG.HIVA2auxo.int Vaccine Strain
2.2. Bacterial Cultures and Transformation
2.3. Cell Lines and Cell Culture
2.4. Production of L1:P18I10 and L1:T20 Proteins Using the 293F Expression System
2.5. Immunofluorescence Staining
2.6. Purification of HPV:HIV (L1:P18I10 and L1:T20) VLPs
2.7. Non-Reducing SDS-PAGE
2.8. Molecular Mass Analysis
2.9. Negative Staining and Transmission Electron Microscopy
2.10. Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis and Western Blotting Analysis
2.11. Immunization of Mice, Collection of Sera and Isolation of Splenocytes
2.12. Enzyme-Linked Immunosorbent Assay
2.13. IFN-γ ELISpot Assay
2.14. Statistical Analysis
2.15. Ethics Statements
3. Results
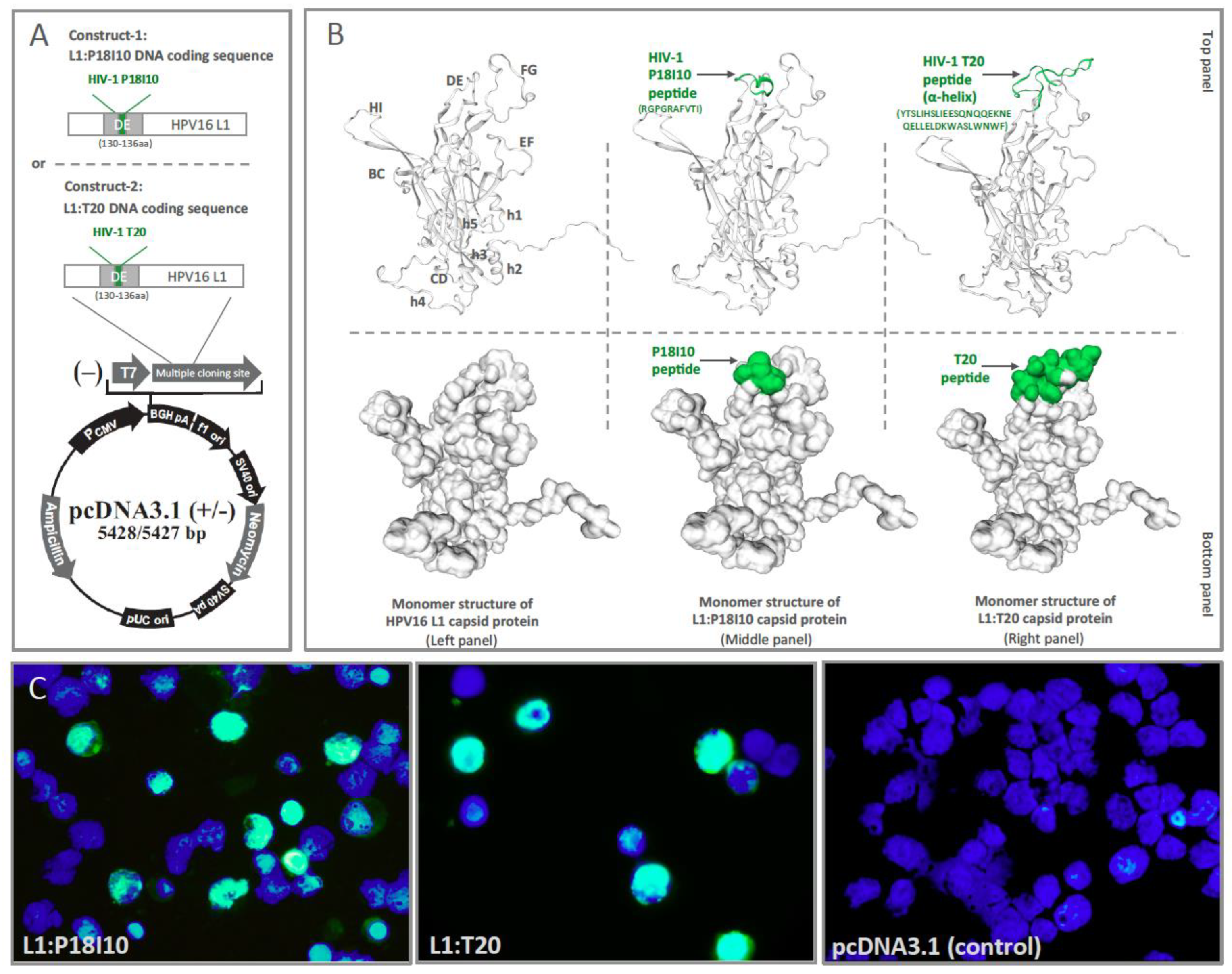
3.1. Design of L1:P18I10 and L1:T20 Immunogens and Evaluation of HPV:HIV Protein Expression by Using 293F Expression System
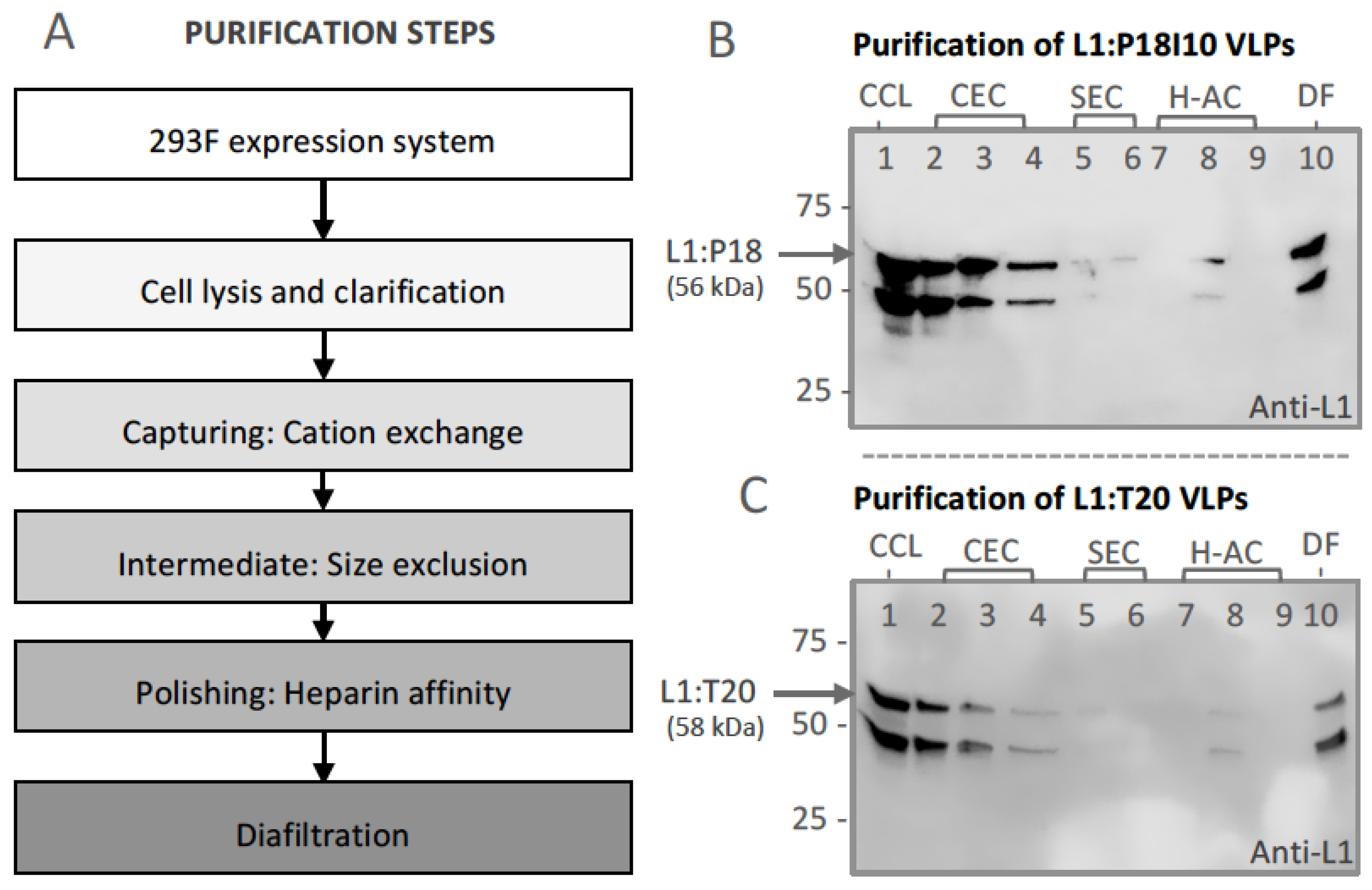
3.2. Purification of L1:P18I10 and L1:T20 VLPs by Using Chromatographic Methods
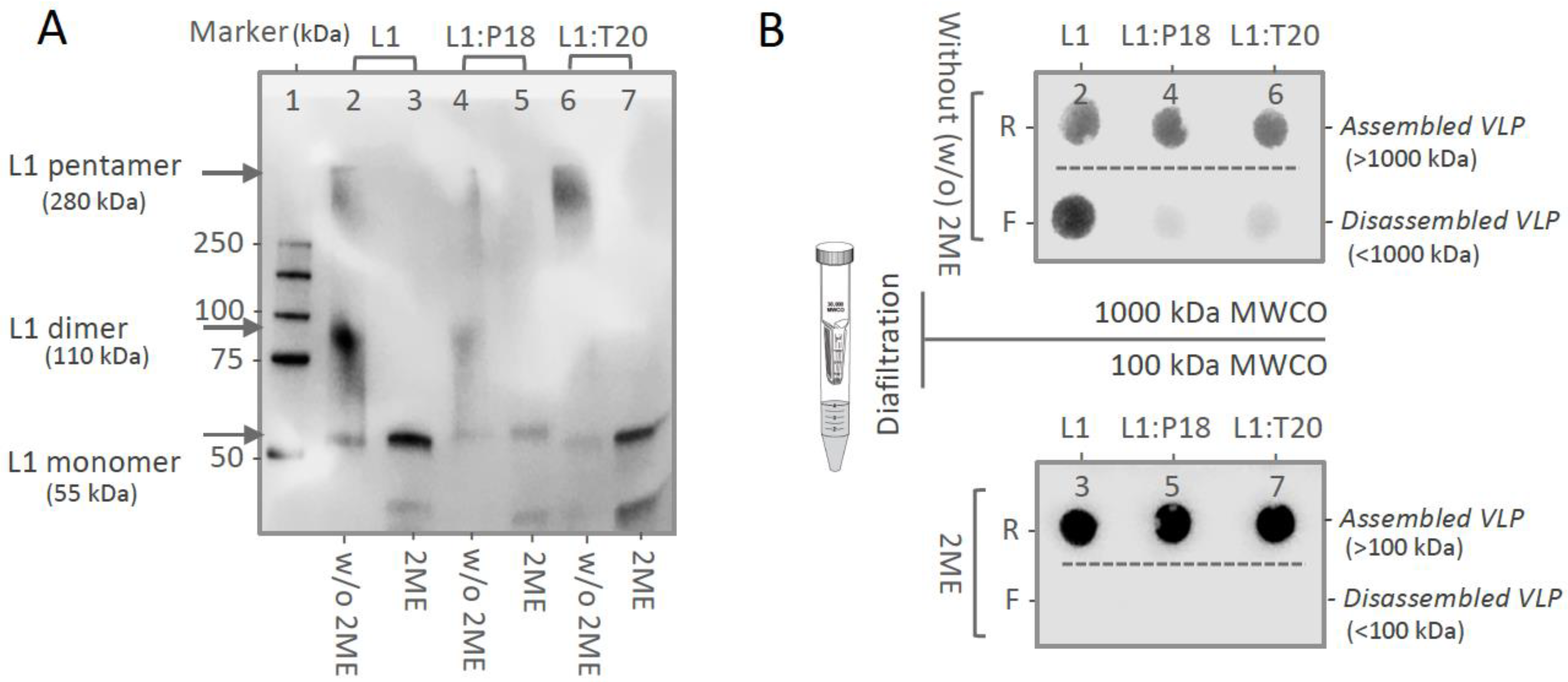
3.3. In Vitro Stability and Self-Assembly of L1:P18I10 and L1:T20 VLPs
3.4. Morphological Characterization of L1:P18I10 and L1:T20 VLPs
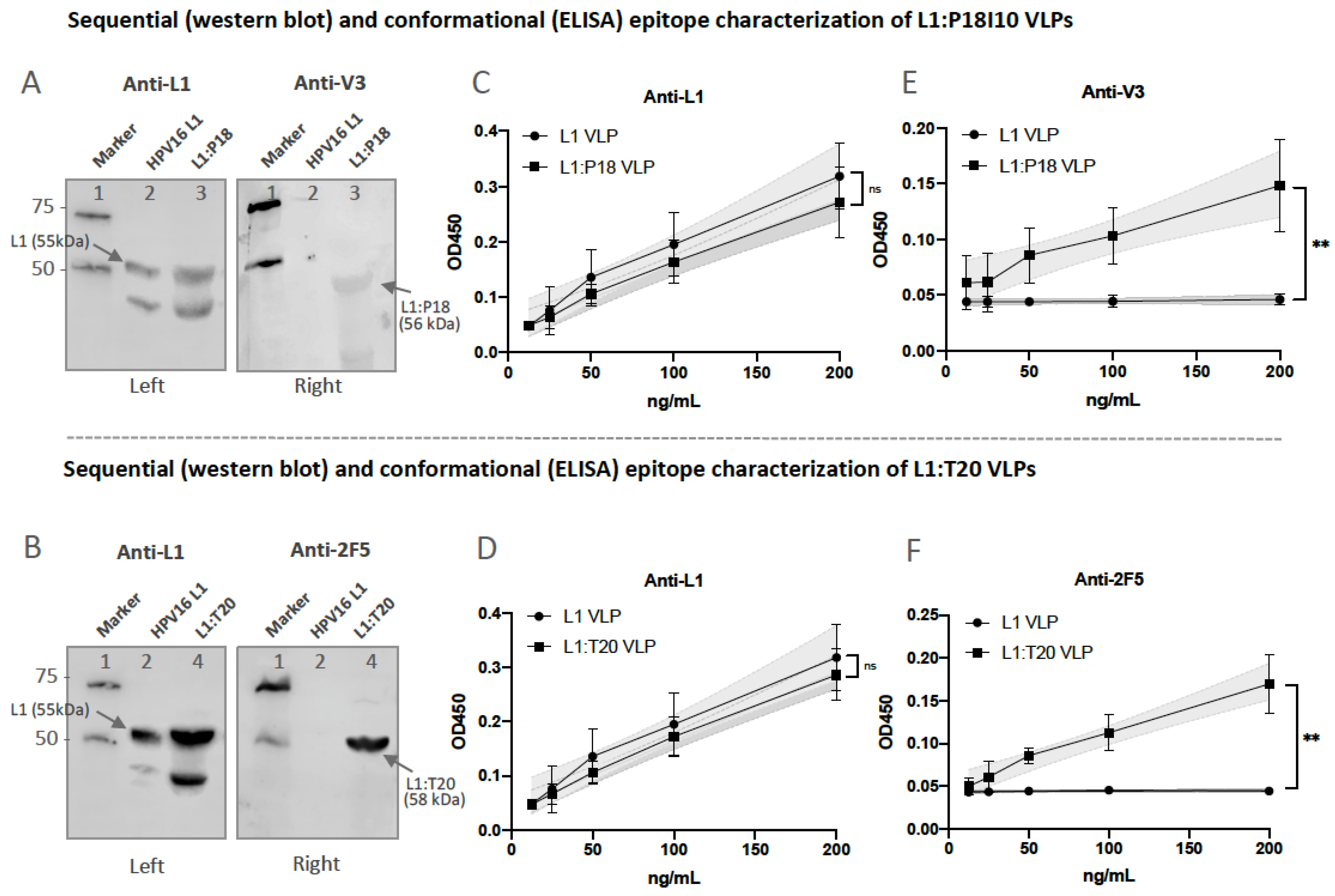
3.5. Presentation and Reactivity of the HPV-16 and HIV-1 Epitopes
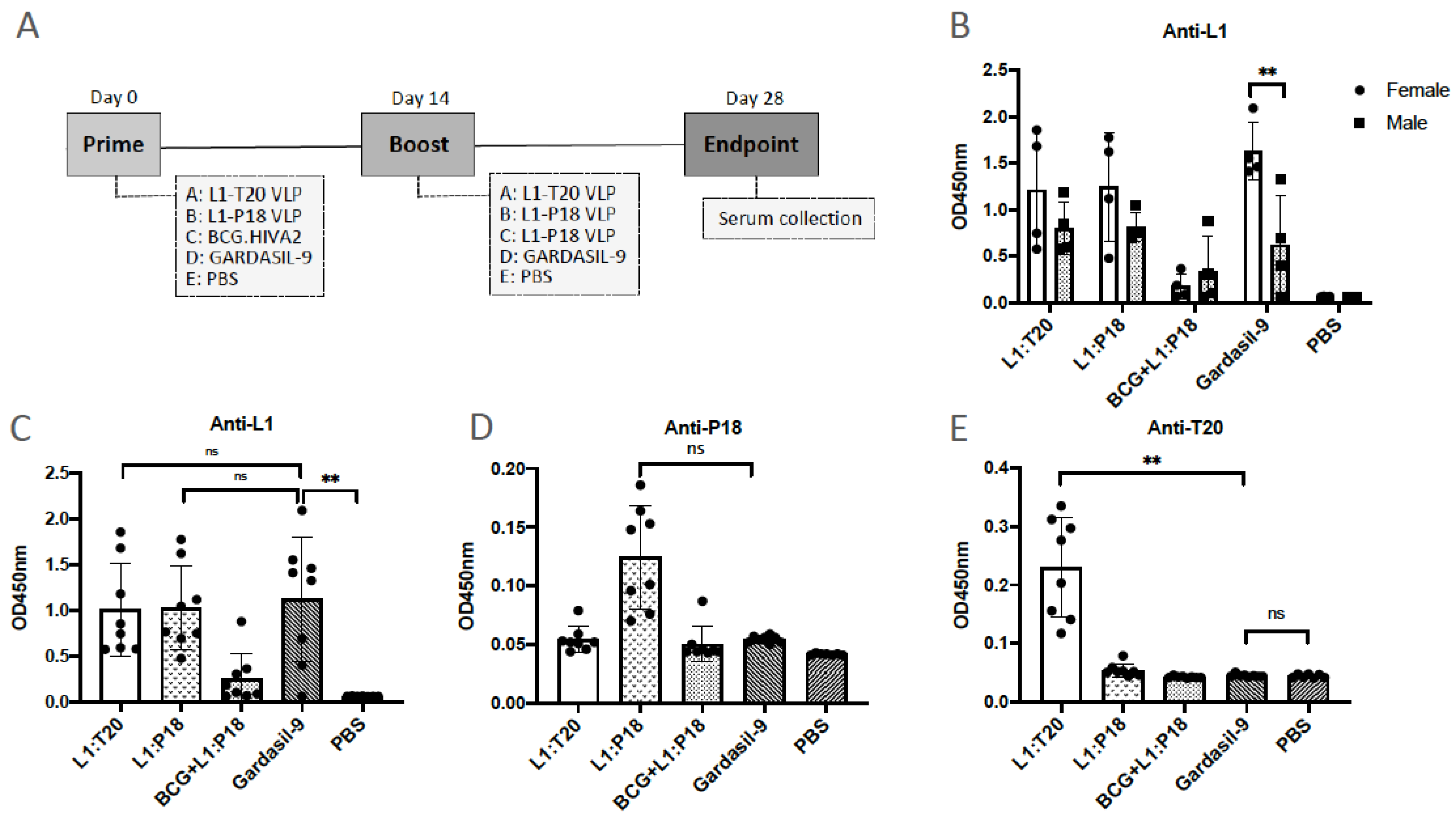
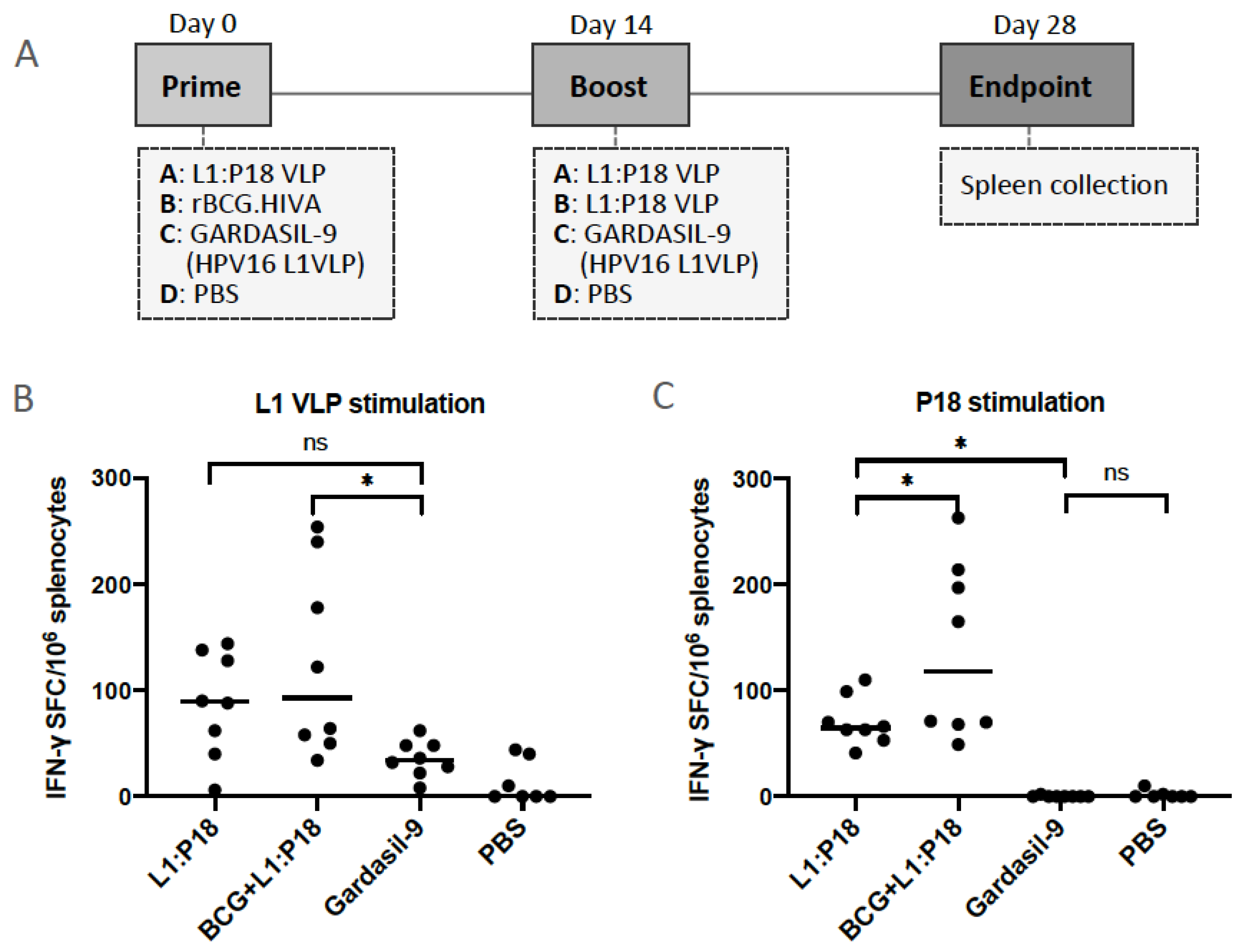
3.6. Immunogenicity of L1:P18I10 and L1:T20 VLPs after BALB/c Mice Immunization
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- UNAIDS Global HIV & AIDS Statistics—2018 Fact Sheet. 2019. Available online: http://www.unaids.org/en/resources/fact-sheet (accessed on 15 June 2020).
- Baeten, J.M. PrEP for HIV: Grade A for evidence but pending for impact. Nat. Rev. Urol. 2019, 16, 570–571. [Google Scholar] [CrossRef]
- Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayaphan, S.; Kaewkungwal, J.; Chiu, J.; Paris, R.; Premsri, N.; Namwat, C.; De Souza, M.; Adams, E.; et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 2009, 361, 2209–2220. [Google Scholar] [CrossRef]
- Ng’uni, T.; Chasara, C.; Ndhlovu, Z.M. Major scientific hurdles in HIV vaccine development: Historical perspective and future directions. Front. Immunol. 2020, 11, 590780. [Google Scholar] [CrossRef]
- Robinson, H.L. HIV/AIDS vaccines: 2018. Clin. Pharmacol. Ther. 2018, 104, 1062–1073. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhang, L. Broadly neutralizing antibodies and vaccine design against HIV-1 infection. Front. Med. 2020, 14, 30–42. [Google Scholar] [CrossRef] [Green Version]
- Collins, D.R.; Gaiha, G.D.; Walker, B.D. CD8+ T cells in HIV control, cure and prevention. Nat. Rev. Immunol. 2020, 20, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Pollard, A.J.; Bijker, E.M. A guide to vaccinology: From basic principles to new developments. Nat. Rev. Immunol. 2021, 21, 83–100. [Google Scholar] [CrossRef]
- Shapiro, S.Z. Lessons for general vaccinology research from attempts to develop an HIV vaccine. Vaccine 2019, 37, 3400–3408. [Google Scholar] [CrossRef]
- Ahmed, H.G.; Bensumaidea, S.H.; Alshammari, F.D.; Alenazi, F.S.H.; ALmutlaq, B.A.; Alturkstani, M.Z.; Aladani, I.A. Prevalence of human papillomavirus subtypes 16 and 18 among Yemeni patients with cervical cancer. Asian Pacific J. Cancer Prev. 2017, 18, 1543–1548. [Google Scholar] [CrossRef]
- Olcese, V.A.; Chen, Y.; Schlegel, R.; Yuan, H. Characterization of HPV16 L1 loop domains in the formation of a type-specific, conformational epitope. BMC Microbiol. 2004, 4, 29. [Google Scholar] [CrossRef]
- Dupuy, C.; Buzoni-Gate, D.; Touze, A.; Le Cann, P.; Bout, D.; Coursaget, P. Cell mediated immunity induced in mice by HPV 16 L1 virus-like particles. Microb. Pathog. 1997, 22, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.; Lowy, D. Explanations for the high potency of HPV prophylactic vaccines. Vaccine 2018, 36, 4768–4773. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, L.E.; Schiller, J.T. Human Papillomavirus Vaccines. J. Infect. Dis. 2021, 224, S367–S378. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.W.; Saubi, N.; Joseph-Munné, J. Design concepts of virus-like particle-based HIV-1 vaccines. Front. Immunol. 2020, 11, 573157. [Google Scholar] [CrossRef] [PubMed]
- Eto, Y.; Saubi, N.; Ferrer, P.; Joseph, J. Designing chimeric virus-like particle-based vaccines for human papillomavirus and HIV: Lessons learned. AIDS Rev. 2019, 21, 218–232. [Google Scholar] [CrossRef]
- Liu, W.J.; Liu, X.S.; Zhao, K.N.; Leggatt, G.R.; Frazer, I.H. Papillomavirus virus-like particles for the delivery of multiple cytotoxic T cell epitopes. Virology 2000, 273, 374–382. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.S.; Abdul-Jabbar, I.; Qi, Y.M.; Frazer, I.H.; Zhou, J. Mucosal immunisation with papillomavirus virus-like particles elicits systemic and mucosal immunity in mice. Virology 1998, 252, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.; Frazer, I.H.; Fernando, G.J.; Zhou, J. Papillomavirus virus-like particles can deliver defined CTL epitopes to the MHC class I pathway. Virology 1998, 240, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Zhai, Y.; Zhong, Z.; Zariffard, M.; Spear, G.T.; Qiao, L. Bovine papillomavirus-like particles presenting conserved epitopes from membrane-proximal external region of HIV-1 gp41 induced mucosal and systemic antibodies. Vaccine 2013, 31, 5422–5429. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Huang, Y.; Fayad, R.; Spear, G.T.; Qiao, L. Induction of mucosal and systemic neutralizing antibodies against human immunodeficiency virus type 1 (HIV-1) by oral immunization with bovine papillomavirus-HIV-1 gp41 chimeric virus-like particles. J. Virol. 2004, 78, 8342–8348. [Google Scholar] [CrossRef]
- Xiao, S.L.; Wen, J.L.; Kong, N.Z.; Yue, H.L.; Leggatt, G.; Frazer, I.H. Route of administration of chimeric BPV1 VLP determines the character of the induced immune responses. Immunol. Cell Biol. 2002, 80, 21–29. [Google Scholar] [CrossRef]
- Kirnbauer, R.; Taub, J.A.N.E.T.; Greenstone, H.E.A.T.H.E.R.; Roden, R.I.C.H.A.R.D.; Dürst, M.; Gissmann, L.; Lowy, D.R.; Schiller, J.T. Efficient Self-Assembly of Human Papillomavirus Type 16 LI and L1-L2 into Virus-Like Particles. J. Virol. 1993, 67, 6929–6936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Potter, C.S.; Carragher, B.; Lander, G.; Sworen, J.; Towne, V.; Abraham, D.; Duncan, P.; Washabaugh, M.W.; Sitrin, R.D. Characterization of virus-like particles in GARDASIL® by cryo transmission electron microscopy. Hum. Vaccines Immunother. 2014, 10, 734–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achour, A.; Lemhammedi, S.; Picard, O.; M’bika, J.P.; Zagury, J.F.; Moukrim, Z.; Willer, A.; Beix, F.; Burny, A.; Zagury, D. Cytotoxic T Lymphocytes Specific for HIV-1 gp160 Antigen and Synthetic P18IIIB Peptide in an HLA-A11-Immunized Individual. AIDS Res. Hum. Retroviruses 1994, 10, 19–25. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Kikuchi, H.; Takahashi, H. Molecular analysis of TCR and peptide/MHC interaction using P18-I10-derived peptides with a single D-amino acid substitution. Biophys. J. 2007, 92, 2570–2582. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.; Chong, H.; Yao, X.; Su, Y.; Cui, S.; He, Y. Identification and characterization of a subpocket on the N-trimer of HIV-1 Gp41: Implication for viral entry and drug target. Aids 2015, 29, 1015–1024. [Google Scholar] [CrossRef]
- Peters, B.S.; Cheingsong-Popov, R.; Callow, D.; Foxall, R.; Patou, G.; Hodgkin, K.; Weber, J.N. A pilot phase II study of the safety and immunogenicity of HIV p17/p24:VLP (p24-VLP) in asymptomatic HIV seropositive subjects. J. Infect. 1997, 35, 231–235. [Google Scholar] [CrossRef]
- Chege, G.K.; Burgers, W.A.; Stutz, H.; Meyers, A.E.; Chapman, R.; Kiravu, A.; Bunjun, R.; Shephard, E.G.; Jacobs, W.R.; Rybicki, E.P.; et al. Robust Immunity to an Auxotrophic Mycobacterium bovis BCG-VLP Prime-Boost HIV Vaccine Candidate in a Nonhuman Primate Model. J. Virol. 2013, 87, 5151–5160. [Google Scholar] [CrossRef] [Green Version]
- Chege, G.K.; Thomas, R.; Shephard, E.G.; Meyers, A.; Bourn, W.; Williamson, C.; Maclean, J.; Gray, C.M.; Rybicki, E.P.; Williamson, A.L. A prime-boost immunisation regimen using recombinant BCG and Pr55gag virus-like particle vaccines based on HIV type 1 subtype C successfully elicits Gag-specific responses in baboons. Vaccine 2009, 27, 4857–4866. [Google Scholar] [CrossRef]
- Joseph, J.; Saubi, N.; Im, E.J.; Fernández-Lloris, R.; Gil, O.; Cardona, P.J.; Gatell, J.M.; Hanke, T. Newborn mice vaccination with BCG.HIVA222 + MVA.HIVA enhances HIV-1-specific immune responses: Influence of age and immunization routes. Clin. Dev. Immunol. 2011, 2011, 516219. [Google Scholar] [CrossRef]
- Saubi, N.; Gea-Mallorquí, E.; Ferrer, P.; Hurtado, C.; Sánchez-Úbeda, S.; Eto, Y.; Gatell, J.M.; Hanke, T.; Joseph, J. Engineering new mycobacterial vaccine design for HIV-TB pediatric vaccine vectored by lysine auxotroph of BCG. Mol. Ther. Methods Clin. Dev. 2014, 1, 14017. [Google Scholar] [CrossRef]
- Mahant, A.; Saubi, N.; Eto, Y.; Guitart, N.; Gatell, J.M.; Hanke, T.; Joseph, J. Preclinical development of BCG.HIVA2auxo.int, harboring an integrative expression vector, for a HIV-TB Pediatric vaccine. Enhancement of stability and specific HIV-1 T-cell immunity. Hum. Vaccines Immunother. 2017, 13, 1798–1810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanke, T.; McMichael, A.J. Design and construction of an experimental HIV-1 vaccine for a year-2000 clinical trial in Kenya. Nat. Med. 2000, 6, 951–955. [Google Scholar] [CrossRef] [PubMed]
- Durocher, Y.; Perret, S.; Kamen, A. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 2002, 30, E9. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, S.Y.; Lim, S.J.; Kim, J.Y.; Lee, S.J.; Kim, H.J. One-step chromatographic purification of human papillomavirus type 16 L1 protein from Saccharomyces cerevisiae. Protein Expr. Purif. 2010, 70, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Eto, Y.; Saubi, N.; Ferrer, P.; Joseph-Munné, J. Expression of chimeric HPV-HIV protein L1P18 in pichia pastoris; purification and characterization of the virus-like particles. Pharmaceutics 2021, 13, 1967. [Google Scholar] [CrossRef]
- GE. The use of CaptoTM Core 700 and Capto Q ImpRes in the purification of human papilloma virus like particles. J. Asia’s Pharm. Biopharm. Ind. 2014, 1–4. [Google Scholar]
- McLean, C.S.; Churcher, M.J.; Meinke, J.; Smith, G.L.; Higgins, G.; Stanley, M.; Minson, A.C. Production and characterisation of a monoclonal antibody to human papillomavirus type 16 using recombinant vaccinia virus. J. Clin. Pathol. 1990, 43, 488–492. [Google Scholar] [CrossRef] [Green Version]
- Merck Canada Inc. Product Monograph Gardasil®. Prod. Monogr. Monopril Prod. Monogr. 2015, 1–71. [Google Scholar]
- Achour, A.; Persson, K.; Harris, R.A.; Sundbäck, J.; Sentman, C.L.; Lindqvist, Y.; Schneides, G.; Kärre, K. The crystal structure of H-2D(d) MHC class I complexed with the HIV-1- derived peptide P18-110 at 2.4 Å resolution: Implications for T cell and NK cell recognition. Immunity 1998, 9, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Pang, W.; Tom, S.C.; Zheng, Y.T. Current peptide HIV type-1 fusion inhibitors. Antivir. Chem. Chemother. 2009, 20. [Google Scholar] [CrossRef]
- Chen, X.S.; Garcea, R.L.; Goldberg, I.; Casini, G.; Harrison, S.C. Structure of Small Virus-like Particles Assembled from the L1 Protein of Human Papillomavirus 16. Mol. Cell 2000, 5, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Weisberg, A.S.; Thompson, C.D.; Hughes, M.M.; Pang, Y.Y.; Lowy, D.R.; Schiller, J.T. Human Papillomavirus 16 Capsids Mediate Nuclear Entry during Infection. J. Virol. 2019, 93, e00454-19. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Doorbar, J.; Sun, X.Y.; Crawford, L.V.; McLean, C.S.; Frazer, I.H. Identification of the nuclear localization signal of human papillomavirus type 16 L1 protein. Virology 1991, 185, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Vlps, A.; Middelberg, A.P.J.; Lua, L.H.L. Virus-like particle bioprocessing: Challenges and opportunities. Pharm. Bioprocess. 2013, 1, 407–409. [Google Scholar]
- Bousarghin, L.; Touzé, A.; Combita-Rojas, A.L.; Coursaget, P. Positively charged sequences of human papillomavirus type 16 capsid proteins are sufficient to mediate gene transfer into target cells via the heparan sulfate receptor. J. Gen. Virol. 2003, 84, 157–164. [Google Scholar] [CrossRef]
- Sasagawa, T.; Pushko, P.; Steers, G.; Gschmeissner, S.E.; Nasser Hajibagheri, M.A.; Finch, J.; Crawford, L.; Tommasino, M. Synthesis and assembly of virus-like particles of human papillomaviruses type 6and Type 16 in fission yeast Schizosaccharomyces pombe. Virology 1995, 206, 126–135. [Google Scholar] [CrossRef] [Green Version]
- Biemelt, S.; Sonnewald, U.; Galmbacher, P.; Willmitzer, L.; Müller, M. Production of Human Papillomavirus Type 16 Virus-Like Particles in Transgenic Plants. J. Virol. 2003, 77, 9211–9220. [Google Scholar] [CrossRef] [Green Version]
- Zahin, M.; Joh, J.; Khanal, S.; Husk, A.; Mason, H.; Warzecha, H.; Ghim, S.J.; Miller, D.M.; Matoba, N.; Jenson, A.B. Scalable production of HPV16 L1 protein and VLPs from tobacco leaves. PLoS ONE 2016, 11, e0160995. [Google Scholar] [CrossRef] [Green Version]
- Aires, K.A.; Cianciarullo, A.M.; Carneiro, S.M.; Villa, L.L.; Boccardo, E.; Pérez-Martinez, C.; Perez-Arellano, I.; Oliveira, M.L.S.; Ho, P.L. Production of human papillomavirus type 16 L1 virus-like particles by recombinant Lactobacillus casei cells. Appl. Environ. Microbiol. 2006, 72, 745–752. [Google Scholar] [CrossRef] [Green Version]
- Shank-Retzlaff, M.L.; Zhao, Q.; Anderson, C.; Hamm, M.; High, K.; Nguyen, M.; Wang, F.; Wang, N.; Wang, B.; Wang, Y.; et al. Evaluation of the thermal stability of Gardasil®. Hum. Vaccin. 2006, 2, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Thorsteinsson, M.V.; Johnston, L.B.; DePhillips, P.A.; Zlotnick, A. A Quantitative Description of In Vitro Assembly of Human Papillomavirus 16 Virus-Like Particles. J. Mol. Biol. 2008, 381, 229–237. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.P.; White, W.I.; Palmer-Hill, F.; Koenig, S.; Suzich, J.A. Quantitative Disassembly and Reassembly of Human Papillomavirus Type 11 Viruslike Particles In Vitro. J. Virol. 1998, 72, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Kirnbauer, R.; Booy, F.; Cheng, N.; Lowy, D.R.; Schiller, J.T. Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proc. Natl. Acad. Sci. USA 1992, 88, 12180–12184. [Google Scholar] [CrossRef] [Green Version]
- Patel, M.C.; Patkar, K.K.; Basu, A.; Mohandas, K.M.; Mukhopadhyaya, R. Production of immunogenic human papillomavirus-16 major capsid protein derived virus like particles. Indian J. Med. Res. 2009, 130, 213–218. [Google Scholar]
- Shi, L.; Sanyal, G.; Ni, A.; Luo, Z.; Doshna, S.; Wang, B.; Graham, T.L.; Wang, N.; Volkin, D.B. Stabilization of human papillomavirus virus-like particles by non-ionic surfactants. J. Pharm. Sci. 2005, 94, 1538–1551. [Google Scholar] [CrossRef]
- Carter, J.J.; Wipf, G.C.; Benki, S.F.; Christensen, N.D.; Galloway, D.A. Identification of a Human Papillomavirus Type 16-Specific Epitope on the C-Terminal Arm of the Major Capsid Protein L1. J. Virol. 2003, 77, 11625–11632. [Google Scholar] [CrossRef] [Green Version]
- Von Brunn, A.; Brand, M.; Reichhuber, C.; Morys-wortmann, C.; Deinhardt, F.; Schdelt, F. Principal neutralizing domain of HIV-I is highly immunogenic when expressed on the surface of hepatitis B core particles. Vaccine 1993, 11, 817–824. [Google Scholar] [CrossRef]
- Dennison, S.M.; Anasti, K.; Scearce, R.M.; Sutherland, L.; Parks, R.; Xia, S.-M.; Liao, H.-X.; Gorny, M.K.; Zolla-Pazner, S.; Haynes, B.F.; et al. Nonneutralizing HIV-1 gp41 Envelope Cluster II Human Monoclonal Antibodies Show Polyreactivity for Binding to Phospholipids and Protein Autoantigens. J. Virol. 2011, 85, 1340–1347. [Google Scholar] [CrossRef] [Green Version]
- Trkola, A.; Kuster, H.; Rusert, P.; Joos, B.; Fischer, M.; Leemann, C.; Manrique, A.; Huber, M.; Rehr, M.; Oxenius, A.; et al. Delay of HIV-1 rebound after cessation of antiretroviral therapy through passive transfer of human neutralizing antibodies. Nat. Med. 2005, 11, 615–622. [Google Scholar] [CrossRef]
- Shank-Retzlaff, M.; Wang, F.; Morley, T.; Anderson, C.; Hamm, M.; Brown, M.; Rowland, K.; Pancari, G.; Zorman, J.; Lowe, R.; et al. Correlation between mouse potency and in vitro relative potency for human papillomavirus Type 16 virus-like particles and Gardasil vaccine samples. Hum. Vaccin. 2005, 1, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Kilpeläinen, A.; Maya-Hoyos, M.; Saubí, N.; Soto, C.Y.; Joseph Munne, J. Advances and challenges in recombinant Mycobacterium bovis BCG-based HIV vaccine development: Lessons learned. Expert Rev. Vaccines 2018, 17, 1005–1020. [Google Scholar] [CrossRef] [PubMed]
- Kilpeläinen, A.; Saubi, N.; Guitart, N.; Moyo, N.; Wee, E.G.; Ravi, K.; Hanke, T.; Joseph, J. Priming with recombinant BCG expressing novel HIV-1 conserved mosaic immunogens and boosting with recombinant CHADOX1 is safe, stable, and elicits HIV-1specific T-cell responses in BALB/c mice. Front. Immunol. 2019, 10, 923. [Google Scholar] [CrossRef] [PubMed]
- Kilpeläinen, A.; Saubi, N.; Guitart, N.; Olvera, A.; Hanke, T.; Brander, C.; Joseph, J. Recombinant BCG expressing HTI prime and recombinant ChAdOx1 boost is safe and elicits HIV-1-specific T-cell responses in BALB/c mice. Vaccines 2019, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Saubi, N.; Kilpeläinen, A.; Eto, Y.; Chen, C.W.; Olvera, À.; Hanke, T.; Brander, C.; Joseph-Munné, J. Priming with recombinant bcg expressing hti enhances the magnitude and breadth of the t-cell immune responses elicited by mva.Hti in balb/c mice. Vaccines 2020, 8, 678. [Google Scholar] [CrossRef]
- Bissett, S.L.; Godi, A.; Beddows, S. The DE and FG loops of the HPV major capsid protein contribute to the epitopes of vaccine-induced cross-neutralising antibodies. Sci. Rep. 2016, 6, 39730. [Google Scholar] [CrossRef] [Green Version]
- Sapp, M.; Volpers, C.; Muller, M.; Streeck, R.E. Organization of the major and minor capsid proteins in human papillomavirus type 33 virus-like particles. J. Gen. Virol. 1995, 76, 2407–2412. [Google Scholar] [CrossRef]
- Volpers, C.; Schirmacher, P.; Streeck, R.E.; Sapp, M. Assembly of the Major and the Minor Capsid Protein of Human Papillomavirus Type 33 into Virus-like Particles and Tubular Structures in Insect Cells. Virology 1994, 200, 504–512. [Google Scholar] [CrossRef]
- Sapp, M.; Fligge, C.; Petzak, I.; Harris, J.R.; Streeck, R.E. Papillomavirus Assembly Requires Trimerization of the Major Capsid Protein by Disulfides between Two Highly Conserved Cysteines. J. Virol. 1998, 72, 6186–6189. [Google Scholar] [CrossRef] [Green Version]
- Sadeyen, J.R.; Tourne, S.; Shkreli, M.; Sizaret, P.Y.; Coursaget, P. Insertion of a foreign sequence on capsid surface loops of human papillomavirus type 16 virus-like particles reduces their capacity to induce neutralizing antibodies and delineates a conformational neutralizing epitope. Virology 2003, 309, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Yang, O.O.; Kalams, S.A.; Trocha, A.; Cao, H.; Luster, A.; Johnson, R.P.; Walker, B.D. Suppression of human immunodeficiency virus type 1 replication by CD8+ cells: Evidence for HLA class I-restricted triggering of cytolytic and noncytolytic mechanisms. J. Virol. 1997, 71, 3120–3128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, D.A.; Sewell, A.K.; Dong, T.; Tan, R.; Goulder, P.J.R.; Rowland-Jones, S.L.; Phillips, R.E. Antigen-specific release of β-chemokines by anti-HIV-1 cytotoxic T lymphocytes. Curr. Biol. 1998, 8, 355–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, L.; Yang, O.O.; Garcia-Zepeda, E.A.; Ge, Y.; Kalams, S.A.; Walker, B.D.; Pasternack, M.S.; Luster, A.D. β-Chemokines are released from HIV-1-specific cytolytic T-cell granules complexed to proteoglycans. Nature 1998, 391, 908–911. [Google Scholar] [CrossRef] [PubMed]
- Goulder, P.J.R.; Watkins, D.I. Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat. Rev. Immunol. 2008, 8, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Belyakov, I.M.; Moss, B.; Strober, W.; Berzofsky, J.A. Mucosal vaccination overcomes the barrier to recombinant vaccinia immunization caused by preexisting poxvirus immunity. Proc. Natl. Acad. Sci. USA 1999, 96, 4512–4517. [Google Scholar] [CrossRef] [Green Version]
- Belyakov, I.M.; Wyatt, L.S.; Ahlers, J.D.; Earl, P.; Pendleton, C.D.; Kelsall, B.L.; Strober, W.; Moss, B.; Berzofsky, J.A. Induction of a Mucosal Cytotoxic T-Lymphocyte Response by Intrarectal Immunization with a Replication-Deficient Recombinant Vaccinia Virus Expressing Human Immunodeficiency Virus 89.6 Envelope Protein. J. Virol. 1998, 72, 8264–8272. [Google Scholar] [CrossRef] [Green Version]
- Hanke, T.; Schneider, J.; Gilbert, S.C.; Hill, A.V.S.; McMichael, A. DNA multi-CTL epitope vaccines for HIV and Plasmodium falciparum: Immunogenicity in mice. Vaccine 1998, 16, 426–435. [Google Scholar] [CrossRef]
- Hanke, T.; Blanchard, T.J.; Schneider, J.; Hannan, C.M.; Becker, M.; Gilbert, S.C.; Hill, A.V.S.; Smith, G.L.; McMichael, A. Enhancement of MHC class I-restricted peptide-specific T cell induction by a DNA prime/MVA boost vaccination regime. Vaccine 1998, 16, 439–445. [Google Scholar] [CrossRef]
- Moore, M.W.; Carbone, F.R.; Bevan, M.J. Introduction of soluble protein into the class I pathway of antigen processing and presentation. Cell 1988, 54, 777–785. [Google Scholar] [CrossRef]
- Schulz, M.; Zinkernagel, R.M.; Hengartner, H. Peptide-induced antiviral protection by cytotoxic T cells. Proc. Natl. Acad. Sci. USA 1991, 88, 991–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahlers, J.D.; Takeshita, T.; Pendleton, C.D.; Berzofsky, J.A. Enhanced immunogenicity of HIV-1 vaccine construct by modification of the native peptide sequence. Proc. Natl. Acad. Sci. USA 1997, 94, 10856–10861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belyakov, I.M.; Derby, M.A.; Ahlers, J.D.; Kelsall, B.L.; Earl, P.; Moss, B.; Strober, W.; Berzofsky, J.A. Mucosal immunization with HIV-1 peptide vaccine induces mucosal and systemic cytotoxic T lymphocytes and protective immunity in mice against intrarectal recombinant HIV-vaccinia challenge. Proc. Natl. Acad. Sci. USA 1998, 95, 1709–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahlers, J.D.; Dunlop, N.; Pendleton, C.D.; Newman, M.; Nara, P.L.; Berzofsky, J.A. Candidate HIV type 1 multideterminant cluster peptide-P18MN vaccine constructs elicit type 1 helper T cells, cytotoxic T cells, and neutralizing antibody, all using the same adjuvant immunization. AIDS Res. Hum. Retrovir. 1996, 12, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Shirai, M.; Pendleton, C.D.; Ahlers, J.; Takeshita, T.; Newman, M.; Berzofsky, J.A. Helper-cytotoxic T lymphocyte (CTL) determinant linkage required for priming of anti-HIV CD8+ CTL in vivo with peptide vaccine constructs. J. Immunol. 1994, 152, 549–556. [Google Scholar] [PubMed]
- Griffiths, J.C.; Harris, S.J.; Layton, G.T.; Berrie, E.L.; French, T.J.; Burns, N.R.; Adams, S.E.; Kingsman, A.J. Hybrid human immunodeficiency virus Gag particles as an antigen carrier system: Induction of cytotoxic T-cell and humoral responses by a Gag:V3 fusion. J. Virol. 1993, 67, 3191–3198. [Google Scholar] [CrossRef] [Green Version]
- Schirmbeck, R.; Böhm, W.; Melber, K.; Reimann, J. Processing of exogenous heat-aggregated (denatured) and particulate (native) hepatitis B surface antigen for class I-restricted epitope presentation. J. Immunol. 1995, 155, 4676–4684. [Google Scholar]
- Sedlik, C.; Saron, M.F.; Sarraseca, J.; Casal, I.; Leclerc, C. Recombinant parvovirus-like particles as an antigen carrier: A novel nonreplicative exogenous antigen to elicit protective antiviral cytotoxic T cells. Proc. Natl. Acad. Sci. USA 1997, 94, 7503–7508. [Google Scholar] [CrossRef] [Green Version]
- Norbury, C.C.; Hewlett, L.J.; Prescott, A.R.; Shastri, N.; Watts, C. Class I MHC presentation of exogenous soluble antigen via macropinocytosis in bone marrow macrophages. Immunity 1995, 3, 783–791. [Google Scholar] [CrossRef] [Green Version]
- Lanzavecchia, A. Mechanisms of antigen uptake for presentation. Curr. Opin. Immunol. 1996, 8, 348–354. [Google Scholar] [CrossRef]
- Takeshita, T.; Takahashi, H.; Kozlowski, S.; Ahlers, J.D.; Pendleton, C.D.; Moore, R.L.; Nakagawa, Y.; Yokomuro, K.; Fox, B.S.; Margulies, D.H.; et al. Molecular analysis of the same HIV peptide functionally binding to both a class I and a class II MHC molecule. J. Immunol. 1995, 154, 1973–1986. [Google Scholar]
- Hosken, B.N.A.; Shibuya, K.; Heath, A.W.; Murphy, K.M.; Garra, A.O. An immunodominant class I-restricted cytotoxic T lymphocyte determinant of human immunodeficiency virus type 1 induces CD4 class II-restricted help for itself. Development 1995, 182, 20–22. [Google Scholar]
- Mann, J.K.; Ndung’u, T. HIV-1 vaccine immunogen design strategies. Virol. J. 2015, 12, 6–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korber, B.; Fischer, W. T cell-based strategies for HIV-1 vaccines. Hum. Vaccines Immunother. 2020, 16, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Koup, R.A.; Douek, D.C. Vaccine Design for CD8 T Lymphocyte Responses. Cold Spring Harb. Perspect. Med. 2011, 1, a007252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMichael, A. T cell responses and viral escape. Cell 1998, 93, 673–676. [Google Scholar] [CrossRef] [Green Version]
- Mothe, B.; Llano, A.; Ibarrondo, J.; Daniels, M.; Miranda, C.; Zamarreño, J.; Bach, V.; Zuniga, R.; Pérez-Álvarez, S.; Berger, C.T.; et al. Definition of the viral targets of protective HIV-1-specific T cell responses. J. Transl. Med. 2011, 9, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, S.A.; Khanna, R.; Gardner, J.; Burrows, S.R.; Coupar, B.; Moss, D.J.; Suhrbier, A. Minimal epitopes expressed in a recombinant polyepitope protein are processed and presented to CD8+ cytotoxic T cells: Implications for vaccine design. Proc. Natl. Acad. Sci. USA 1995, 92, 5845–5849. [Google Scholar] [CrossRef]
- Thomson, S.A.; Sherritt, M.A.; Medveczky, J.; Elliott, S.L.; Moss, D.J.; Fernando, G.J.; Brown, L.E.; Suhrbier, A. Delivery of multiple CD8 cytotoxic T cell epitopes by DNA vaccination. J. Immunol. 1998, 160, 1717–1723. [Google Scholar]
- Hanke, T.; Neumann, V.C.; Blanchard, T.J.; Sweeney, P.; Hill, A.V.S.; Smith, G.L.; McMichael, A. Effective induction of HIV-specific CTL by multi-epitope using gene gun in a combined vaccination regime. Vaccine 1999, 17, 589–596. [Google Scholar] [CrossRef]
- Sharan, R.; Kaushal, D. Vaccine strategies for the Mtb/HIV copandemic. Vaccines 2020, 5, 95. [Google Scholar] [CrossRef]
- Im, E.-J.; Saubi, N.; Virgili, G.; Sander, C.; Teoh, D.; Gatell, J.M.; McShane, H.; Joseph, J.; Hanke, T. Vaccine Platform for Prevention of Tuberculosis and Mother-to-Child Transmission of Human Immunodeficiency Virus Type 1 through Breastfeeding. J. Virol. 2007, 81, 9408–9418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chege, G.K.; Shephard, E.G.; Meyers, A.; van Harmelen, J.; Williamson, C.; Lynch, A.; Gray, C.M.; Rybicki, E.P.; Williamson, A.L. HIV-1 subtype C Pr55gag virus-like particle vaccine efficiently boosts baboons primed with a matched DNA vaccine. J. Gen. Virol. 2008, 89, 2214–2227. [Google Scholar] [CrossRef] [PubMed]
- Buonaguro, L.; Devito, C.; Tornesello, M.L.; Schröder, U.; Wahren, B.; Hinkula, J.; Buonaguro, F.M. DNA-VLP prime-boost intra-nasal immunization induces cellular and humoral anti-HIV-1 systemic and mucosal immunity with cross-clade neutralizing activity. Vaccine 2007, 25, 5968–5977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hovav, A.-H.; Cayabyab, M.J.; Panas, M.W.; Santra, S.; Greenland, J.; Geiben, R.; Haynes, B.F.; Jacobs, W.R.; Letvin, N.L. Rapid Memory CD8 + T-Lymphocyte Induction through Priming with Recombinant Mycobacterium smegmatis. J. Virol. 2007, 81, 74–83. [Google Scholar] [CrossRef] [Green Version]
- Muster, T.; Steindl, F.; Purtscher, M.; Trkola, A.; Klima, A.; Himmler, G.; Rüker, F.; Katinger, H. A conserved neutralizing epitope on gp41 of human immunodeficiency virus type 1. J. Virol. 1993, 67, 6642–6647. [Google Scholar] [CrossRef] [Green Version]
- Conley, A.J.; Kessler, J.A.; Boots, L.J.; Tung, J.S.; Arnold, B.A.; Keller, P.M.; Shaw, A.R.; Emini, E.A. Neutralization of divergent human immunodeficiency virus type 1 variants and primary isolates by IAM-41-2F5, an anti-gp41 human monoclonal antibody. Proc. Natl. Acad. Sci. USA 1994, 91, 3348–3352. [Google Scholar] [CrossRef] [Green Version]
- Montero, M.; van Houten, N.E.; Wang, X.; Scott, J.K. The Membrane-Proximal External Region of the Human Immunodeficiency Virus Type 1 Envelope: Dominant Site of Antibody Neutralization and Target for Vaccine Design. Microbiol. Mol. Biol. Rev. 2008, 72, 54–84. [Google Scholar] [CrossRef] [Green Version]
- Stenler, S.; Lundin, K.E.; Hansen, L.; Petkov, S.; Mozafari, N.; Isaguliants, M.; Blomberg, P.; Smith, C.I.E.; Goldenberg, D.M.; Chang, C.H.; et al. Immunization with HIV-1 envelope T20-encoding DNA vaccines elicits cross-clade neutralizing antibody responses. Hum. Vaccines Immunother. 2017, 13, 2849–2858. [Google Scholar] [CrossRef]
- Lai, R.P.J.; Hock, M.; Radzimanowski, J.; Tonks, P.; Hulsik, D.L.; Effantin, G.; Seilly, D.J.; Dreja, H.; Kliche, A.; Wagner, R.; et al. A fusion intermediate gp41 immunogen elicits neutralizing antibodies to HIV-1. J. Biol. Chem. 2014, 289, 29912–29926. [Google Scholar] [CrossRef] [Green Version]
- Hanson, M.C.; Abraham, W.; Crespo, M.P.; Chen, S.H.; Liu, H.; Szeto, G.L.; Kim, M.; Reinherz, E.L.; Irvine, D.J. Liposomal vaccines incorporating molecular adjuvants and intrastructural T-cell help promote the immunogenicity of HIV membrane-proximal external region peptides. Vaccine 2015, 33, 861–868. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, E.; Joyce, J.G.; Miller, M.D.; Finnefrock, A.C.; Liang, X.; Finotto, M.; Ingallinella, P.; McKenna, P.; Citron, M.; Ottinger, E.; et al. Vaccination with peptide mimetics of the gp41 prehairpin fusion intermediate yields neutralizing antisera against HIV-1 isolates. Proc. Natl. Acad. Sci. USA 2010, 107, 10655–10660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.; Munshi, S.; Shendure, J.; Mark, G.; Davies, M.E.; Freed, D.C.; Montefiori, D.C.; Shiver, J.W. Epitope insertion into variable loops of HIV-1 gp120 as a potential means to improve immunogenicity of viral envelope protein. Vaccine 1999, 17, 2862–2872. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, Z.; Cheng, X.; Chen, Y.H. The recombinant immunogen with high-density epitopes of ELDKWA and ELDEWA induced antibodies recognizing both epitopes on HIV-1 gp41. Microbiol. Immunol. 2005, 49, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; MacDonald, K.S.; Barber, B.H. Construction of recombinant targeting immunogens incorporating an HIV-1 neutralizing epitope into sites of differing conformational constraint. Vaccine 2002, 20, 1169–1180. [Google Scholar] [CrossRef]
- Coëffier, E.; Clément, J.M.; Cussac, V.; Khodaei-Boorane, N.; Jehanno, M.; Rojas, M.; Dridi, A.; Latour, M.; El Habib, R.; Barré-Sinoussi, F.; et al. Antigenicity and immunogenicity of the HIV-1 gp41 epitope ELDKWA inserted into permissive sites of the MalE protein. Vaccine 2000, 19, 684–693. [Google Scholar] [CrossRef]
- Eckhart, L.; Raffelsberger, W.; Ferko, B.; Klima, A.; Purtscher, M.; Katinger, H.; Rüker, F. Immunogenic presentation of a conserved gp41 epitope of human immunodeficiency virus type 1 an recombinant surface antigen of hepatitis B virus. J. Gen. Virol. 1996, 77, 2001–2008. [Google Scholar] [CrossRef]
- Phogat, S.; Svehla, K.; Tang, M.; Spadaccini, A.; Muller, J.; Mascola, J.; Berkower, I.; Wyatt, R. Analysis of the Human Immunodeficiency Virus Type 1 gp41 Membrane Proximal External Region Arrayed on Hepatitis B Surface Antigen Particles. Virology 2008, 373, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Schlienger, K.; Mancini, M.; Rivière, Y.; Dormont, D.; Tiollais, P.; Michel, M.L. Human immunodeficiency virus type 1 major neutralizing determinant exposed on hepatitis B surface antigen particles is highly immunogenic in primates. J. Virol. 1992, 66, 2570–2576. [Google Scholar] [CrossRef]
- McCoy, L.E. The expanding array of HIV broadly neutralizing antibodies. Retrovirology 2018, 15, 70. [Google Scholar] [CrossRef]
- Azoitei, M.L.; Correia, B.E.; Ban, Y.E.A.; Carrico, C.; Kalyuzhniy, O.; Chen, L.; Schroeter, A.; Huang, P.S.; McLellan, J.S.; Kwong, P.D.; et al. Computation-guided backbone grafting of a discontinuous motif onto a protein scaffold. Science 2011, 334, 373–376. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.; Ho, J.X.; Keeling, K.; Gilliland, G.L.; Ji, X.; Rüker, F.; Carter, D.C. Three-Dimensional structure of schistosoma japonicum glutathione s-transferase fused with a six-amino acid conserved neutralizing epitope of gp41 from hiv. Protein Sci. 1994, 3, 2233–2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dervillez, X.; Hüther, A.; Schuhmacher, J.; Griesinger, C.; Cohen, J.H.; Von Laer, D.; Dietrich, U. Stable expression of soluble therapeutic peptides in eukaryotic cells by multimerisation: Application to the HIV-1 fusion inhibitory peptide C46. ChemMedChem 2006, 1, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.R.; Mascola, J.R. Antibody responses to envelope glycoproteins in HIV-1 infection. Nat. Immunol. 2015, 16, 571–576. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-W.; Saubi, N.; Kilpeläinen, A.; Joseph-Munné, J. Chimeric Human Papillomavirus-16 Virus-like Particles Presenting P18I10 and T20 Peptides from HIV-1 Envelope Induce HPV16 and HIV-1-Specific Humoral and T Cell-Mediated Immunity in BALB/c Mice. Vaccines 2023, 11, 15. https://doi.org/10.3390/vaccines11010015
Chen C-W, Saubi N, Kilpeläinen A, Joseph-Munné J. Chimeric Human Papillomavirus-16 Virus-like Particles Presenting P18I10 and T20 Peptides from HIV-1 Envelope Induce HPV16 and HIV-1-Specific Humoral and T Cell-Mediated Immunity in BALB/c Mice. Vaccines. 2023; 11(1):15. https://doi.org/10.3390/vaccines11010015
Chicago/Turabian StyleChen, Chun-Wei, Narcís Saubi, Athina Kilpeläinen, and Joan Joseph-Munné. 2023. "Chimeric Human Papillomavirus-16 Virus-like Particles Presenting P18I10 and T20 Peptides from HIV-1 Envelope Induce HPV16 and HIV-1-Specific Humoral and T Cell-Mediated Immunity in BALB/c Mice" Vaccines 11, no. 1: 15. https://doi.org/10.3390/vaccines11010015
APA StyleChen, C. -W., Saubi, N., Kilpeläinen, A., & Joseph-Munné, J. (2023). Chimeric Human Papillomavirus-16 Virus-like Particles Presenting P18I10 and T20 Peptides from HIV-1 Envelope Induce HPV16 and HIV-1-Specific Humoral and T Cell-Mediated Immunity in BALB/c Mice. Vaccines, 11(1), 15. https://doi.org/10.3390/vaccines11010015