
mRNA Therapeutic Vaccine for Hepatitis B Demonstrates Immunogenicity and Efficacy in the AAV-HBV Mouse Model

 , , , and
, , , and 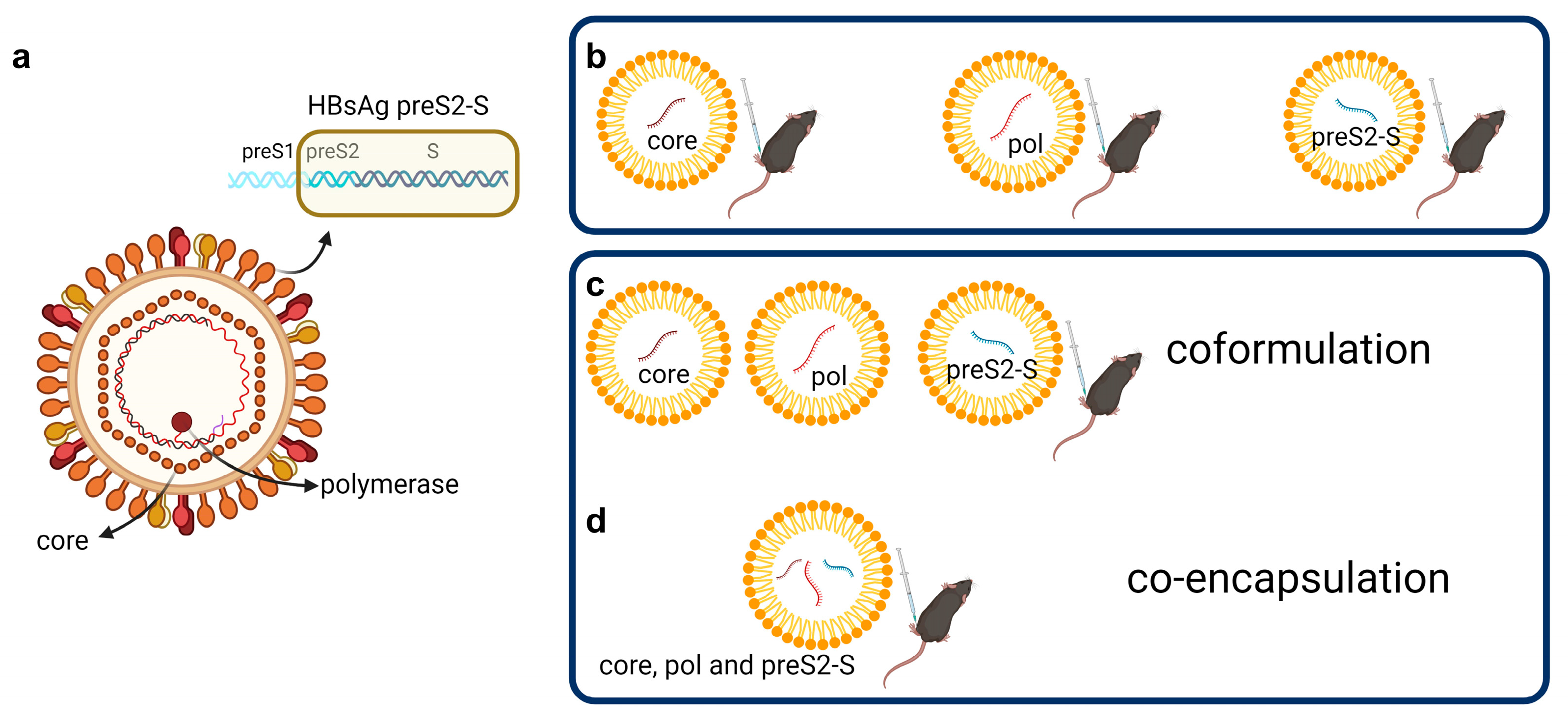{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation and LNP Formulation of mRNA
2.2. In Vivo
2.2.1. Immunization of Naïve Mice
2.2.2. Immunization of AAV-HBV-Transduced Mice
2.3. Ex Vivo
2.3.1. Viral Parameters, Antibodies, and Alanine Aminotransferase (ALT) Analyses
2.3.2. Isolation of Splenocytes
2.3.3. Isolation of Blood Leucocytes
2.3.4. Detection of HBV-Specific T Cells by IFN-γ ELISpot
2.3.5. Measurement of HBV-Specific Polyfunctional T Cells
2.3.6. Immunohistochemistry (IHC)
2.3.7. Statistical Analysis
3. Results
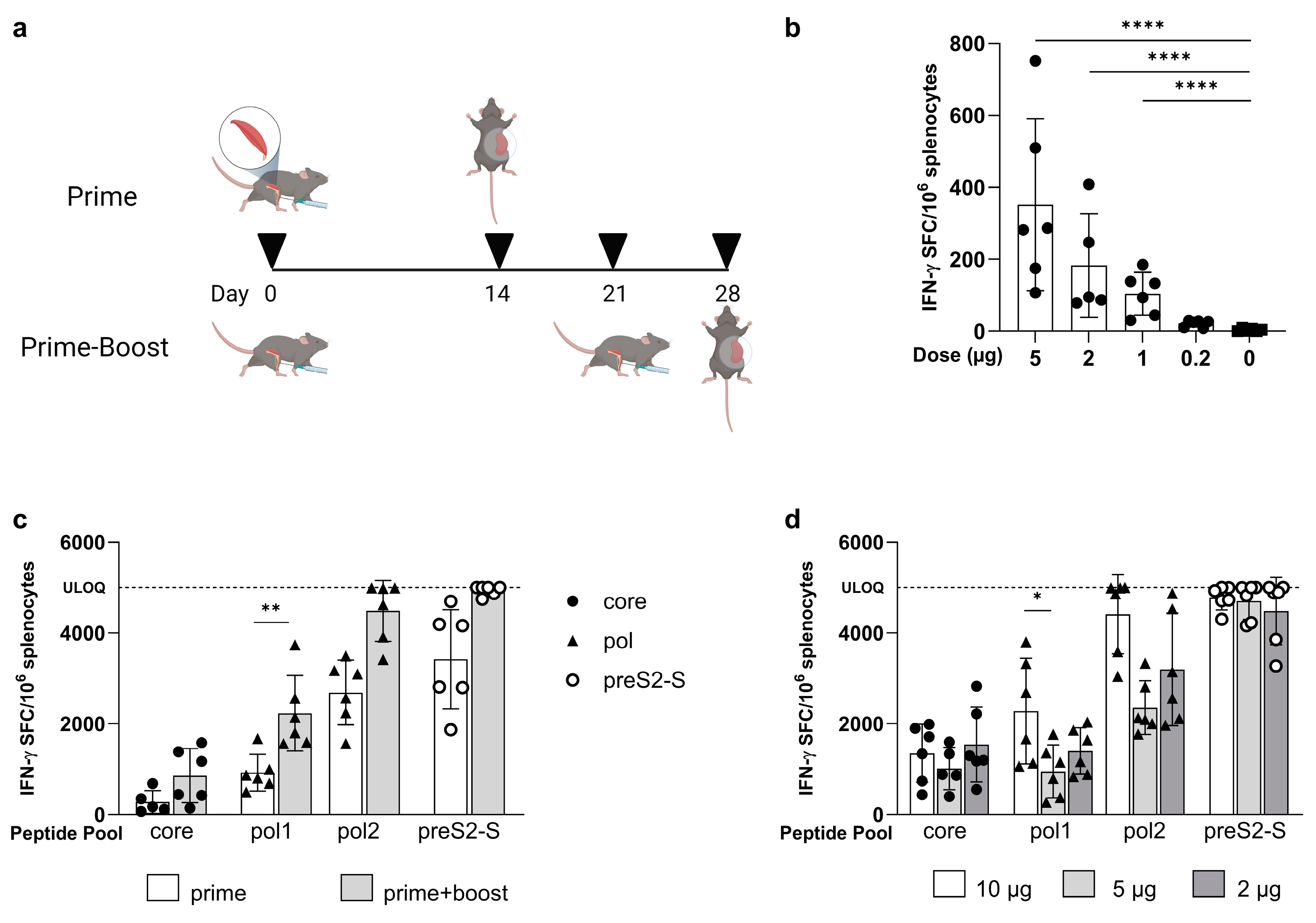
3.1. Low Doses of LNP-Formulated mRNA Encoding for Single HBV Antigen Induced Strong Immune Responses in Naive C57BL/6 Mice
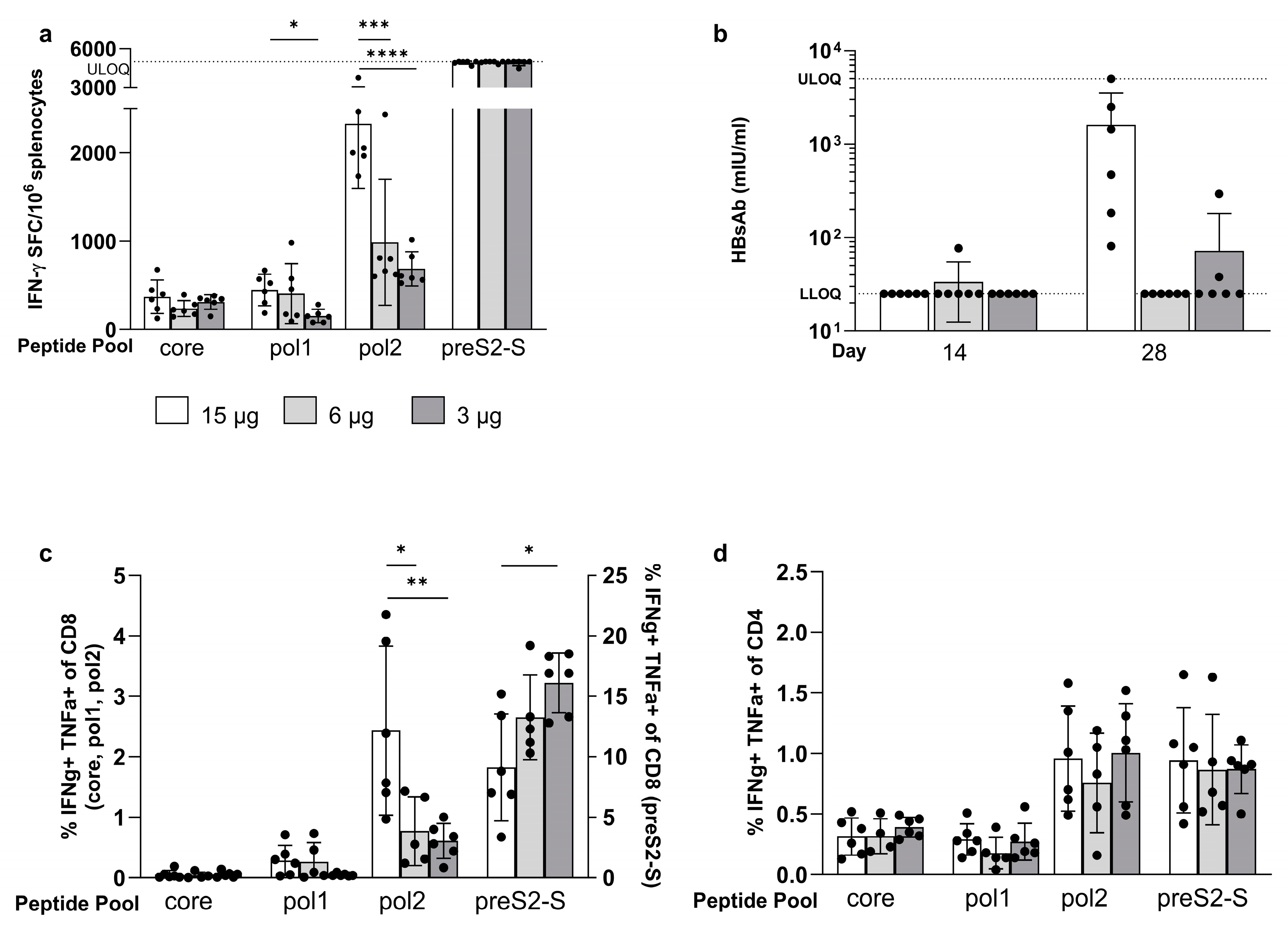
3.2. Coformulated mRNAs Encoding for Core, Pol, and preS2-S Induce Polyfunctional Responses and Antibody Production
3.3. Minor Differences in Response between Co-Encapsulation and Coformulation of Three mRNAs
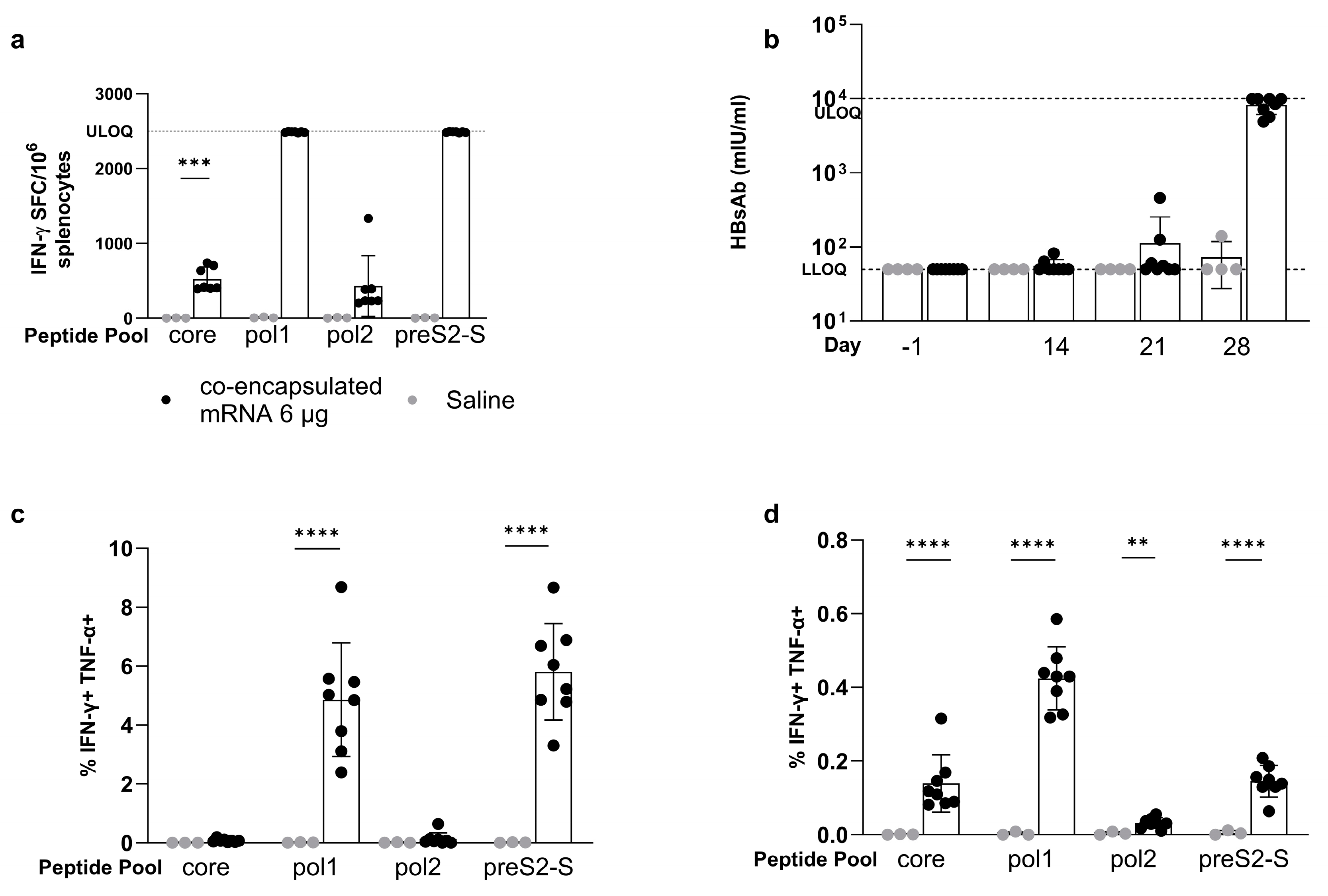
3.4. The Strong Vaccine-Induced Response Was Confirmed in the BALB/c Mouse Strain
3.5. Co-Encapsulated mRNA Vaccine Achieves up to 1.7 Log10 Reduction in HBsAg Levels in the AAV-HBV-Transduced Mouse Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Hepatitis B. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b (accessed on 18 July 2023).
- Noordeen, F. Hepatitis B virus infection: An insight into infection outcomes and recent treatment options. Virusdisease 2015, 26, 1–8. [Google Scholar] [CrossRef]
- Gerlich, W.H. Prophylactic vaccination against hepatitis B: Achievements, challenges and perspectives. Med. Microbiol. Immunol. 2015, 204, 39–55. [Google Scholar] [CrossRef]
- Grossi, G.; Vigano, M.; Loglio, A.; Lampertico, P. Hepatitis B virus long-term impact of antiviral therapy nucleot(s)ide analogues (NUCs). Liver Int. 2017, 37 (Suppl. S1), 45–51. [Google Scholar] [CrossRef] [PubMed]
- Boni, C.; Barili, V.; Acerbi, G.; Rossi, M.; Vecchi, A.; Laccabue, D.; Penna, A.; Missale, G.; Ferrari, C.; Fisicaro, P. HBV Immune-Therapy: From Molecular Mechanisms to Clinical Applications. Int. J. Mol. Sci. 2019, 20, 2754. [Google Scholar] [CrossRef]
- Xia, Y.; Guo, H. Hepatitis B virus cccDNA: Formation, regulation and therapeutic potential. Antivir. Res. 2020, 180, 104824. [Google Scholar] [CrossRef] [PubMed]
- Allweiss, L.; Dandri, M. The Role of cccDNA in HBV Maintenance. Viruses 2017, 9, 156. [Google Scholar] [CrossRef] [PubMed]
- Medas, R.; Liberal, R.; Macedo, G. Discontinuation of antiviral therapy in chronic hepatitis B patients. World J. Clin. Cases 2021, 9, 6979–6986. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef] [PubMed]
- Terrault, N.A.; Lok, A.S.F.; McMahon, B.J.; Chang, K.M.; Hwang, J.P.; Jonas, M.M.; Brown, R.S., Jr.; Bzowej, N.H.; Wong, J.B. Update on Prevention, Diagnosis, and Treatment of Chronic Hepatitis B: AASLD 2018 Hepatitis B Guidance. Clin. Liver Dis. 2018, 12, 33–34. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Chen, J. Interferon and Hepatitis B: Current and Future Perspectives. Front. Immunol. 2021, 12, 733364. [Google Scholar] [CrossRef]
- Wong, G.L.H.; Gane, E.; Lok, A.S.F. How to achieve functional cure of HBV: Stopping NUCs, adding interferon or new drug development? J. Hepatol. 2022, 76, 1249–1262. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.C.; Sung, P.S.; Park, S.H. Immune responses and immunopathology in acute and chronic viral hepatitis. Nat. Rev. Immunol. 2016, 16, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.; Liu, X.; Li, X.; Kong, H.; Tian, L.; Chen, Y. T-cell exhaustion in chronic hepatitis B infection: Current knowledge and clinical significance. Cell Death Dis. 2015, 6, e1694. [Google Scholar] [CrossRef] [PubMed]
- Gehring, A.J.; Protzer, U. Targeting Innate and Adaptive Immune Responses to Cure Chronic HBV Infection. Gastroenterology 2019, 156, 325–337. [Google Scholar] [CrossRef]
- De Pooter, D.; Van Gulck, E.; Chen, A.; Evans, C.F.; Neefs, J.M.; Horton, H.; Boden, D. A Therapeutic Hepatitis B Virus DNA Vaccine Induces Specific Immune Responses in Mice and Non-Human Primates. Vaccines 2021, 9, 969. [Google Scholar] [CrossRef] [PubMed]
- De Creus, A. Therapeutic Vaccine JNJ-0535 Induces a Strong HBV-Specific T-Cell Response in Healthy Adults and a Modest Response in Chronic HBV-Infected Patients. 2022. Available online: https://www.natap.org/2022/EASL/EASL_38.htm (accessed on 18 January 2024).
- Michler, T.; Kosinska, A.D.; Festag, J.; Bunse, T.; Su, J.; Ringelhan, M.; Imhof, H.; Grimm, D.; Steiger, K.; Mogler, C.; et al. Knockdown of Virus Antigen Expression Increases Therapeutic Vaccine Efficacy in High-Titer Hepatitis B Virus Carrier Mice. Gastroenterology 2020, 158, 1762–1775.e9. [Google Scholar] [CrossRef] [PubMed]
- Knolle, P.A.; Huang, L.R.; Kosinska, A.; Wohlleber, D.; Protzer, U. Improving Therapeutic Vaccination against Hepatitis B-Insights from Preclinical Models of Immune Therapy against Persistent Hepatitis B Virus Infection. Vaccines 2021, 9, 1333. [Google Scholar] [CrossRef] [PubMed]
- Schuch, A.; Salimi Alizei, E.; Heim, K.; Wieland, D.; Kiraithe, M.M.; Kemming, J.; Llewellyn-Lacey, S.; Sogukpinar, O.; Ni, Y.; Urban, S.; et al. Phenotypic and functional differences of HBV core-specific versus HBV polymerase-specific CD8+ T cells in chronically HBV-infected patients with low viral load. Gut 2019, 68, 905–915. [Google Scholar] [CrossRef]
- Hobernik, D.; Bros, M. DNA Vaccines-How Far From Clinical Use? Int. J. Mol. Sci. 2018, 19, 3605. [Google Scholar] [CrossRef]
- Anand, P.; Stahel, V.P. Review the safety of COVID-19 mRNA vaccines: A review. Patient Saf. Surg. 2021, 15, 20. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, T.Y.; Yuan, Q.; Xia, N.S. Antibody-mediated immunotherapy against chronic hepatitis B virus infection. Hum. Vaccines Immunother. 2017, 13, 1768–1773. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.A. A Comparison of Plasmid DNA and mRNA as Vaccine Technologies. Vaccines 2019, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Kiaie, S.H.; Majidi Zolbanin, N.; Ahmadi, A.; Bagherifar, R.; Valizadeh, H.; Kashanchi, F.; Jafari, R. Recent advances in mRNA-LNP therapeutics: Immunological and pharmacological aspects. J. Nanobiotechnol. 2022, 20, 276. [Google Scholar] [CrossRef]
- Rivino, L.; Le Bert, N.; Gill, U.S.; Kunasegaran, K.; Cheng, Y.; Tan, D.Z.; Becht, E.; Hansi, N.K.; Foster, G.R.; Su, T.H.; et al. Hepatitis B virus-specific T cells associate with viral control upon nucleos(t)ide-analogue therapy discontinuation. J. Clin. Investig. 2018, 128, 668–681. [Google Scholar] [CrossRef]
- Hassett, K.J.; Benenato, K.E.; Jacquinet, E.; Lee, A.; Woods, A.; Yuzhakov, O.; Himansu, S.; Deterling, J.; Geilich, B.M.; Ketova, T.; et al. Optimization of Lipid Nanoparticles for Intramuscular Administration of mRNA Vaccines. Mol. Ther. Nucleic Acids 2019, 15, 1–11. [Google Scholar] [CrossRef]
- Saadati, F.; Cammarone, S.; Ciufolini, M.A. A Route to Lipid ALC-0315: A Key Component of a COVID-19 mRNA Vaccine. Chemistry 2022, 28, e202200906. [Google Scholar] [CrossRef]
- Dong, X.Y.; Yu, C.J.; Wang, G.; Tian, W.H.; Lu, Y.; Zhang, F.W.; Wang, W.; Wang, Y.; Tan, W.J.; Wu, X.B. Establishment of hepatitis B virus (HBV) chronic infection mouse model by in vivo transduction with a recombinant adeno-associated virus 8 carrying 1. 3 copies of HBV genome (rAAN8-1. 3HBV). Chin. J. Virol. 2010, 26, 425–431. [Google Scholar]
- Conceição-Neto, N.P.W.; Vacca, M.; Beyens, M.; De Clerck, B.; Aerts, L.; Voeten, B.; De Pooter, D.; Verschueren, L.; Dockx, K.; Vandenberk, M.; et al. Sustained Liver HBsAg Loss and Clonal T- and B-Cell Expansion upon Therapeutic DNA Vaccination Require Low HBsAg Levels. Vaccines 2023, 11, 1825. [Google Scholar] [CrossRef]
- Jessen, B.; Faller, S.; Krempl, C.D.; Ehl, S. Major histocompatibility complex-dependent cytotoxic T lymphocyte repertoire and functional avidity contribute to strain-specific disease susceptibility after murine respiratory syncytial virus infection. J. Virol. 2011, 85, 10135–10143. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Lau, G.K.; Suri, D.; Liang, R.; Rigopoulou, E.I.; Thomas, M.G.; Mullerova, I.; Nanji, A.; Yuen, S.T.; Williams, R.; Naoumov, N.V. Resolution of chronic hepatitis B and anti-HBs seroconversion in humans by adoptive transfer of immunity to hepatitis B core antigen. Gastroenterology 2002, 122, 614–624. [Google Scholar] [CrossRef]
- Li, Y.; Wen, C.; Gu, S.; Wang, W.; Guo, L.; Li, C.K.; Yi, X.; Zhou, Y.; Dong, Z.; Fu, X.; et al. Differential response of HBV envelope-specific CD4+ T cells is related to HBsAg loss after stopping nucleos(t)ide analogue therapy. Hepatology 2023, 78, 592–606. [Google Scholar] [CrossRef]
- Suzuki, Y.; Ishihara, H. Difference in the lipid nanoparticle technology employed in three approved siRNA (Patisiran) and mRNA (COVID-19 vaccine) drugs. Drug Metab. Pharmacokinet. 2021, 41, 100424. [Google Scholar] [CrossRef]
- Zeng, C.; Zhang, C.; Walker, P.G.; Dong, Y. Formulation and Delivery Technologies for mRNA Vaccines. Curr. Top. Microbiol. Immunol. 2022, 440, 71–110. [Google Scholar] [CrossRef]
- Ferraresso, F.; Strilchuk, A.W.; Juang, L.J.; Poole, L.G.; Luyendyk, J.P.; Kastrup, C.J. Comparison of DLin-MC3-DMA and ALC-0315 for siRNA Delivery to Hepatocytes and Hepatic Stellate Cells. Mol. Pharm. 2022, 19, 2175–2182. [Google Scholar] [CrossRef]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Sasaki, K.; Sato, Y.; Okuda, K.; Iwakawa, K.; Harashima, H. mRNA-Loaded Lipid Nanoparticles Targeting Dendritic Cells for Cancer Immunotherapy. Pharmaceutics 2022, 14, 1572. [Google Scholar] [CrossRef]
- Okuda, K.; Sato, Y.; Iwakawa, K.; Sasaki, K.; Okabe, N.; Maeki, M.; Tokeshi, M.; Harashima, H. On the size-regulation of RNA-loaded lipid nanoparticles synthesized by microfluidic device. J. Control. Release 2022, 348, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.; Hatit, M.Z.C.; Zhao, K.; Loughrey, D.; Lokugamage, M.P.; Peck, H.E.; Cid, A.D.; Muralidharan, A.; Kim, Y.; Santangelo, P.J.; et al. Piperazine-derived lipid nanoparticles deliver mRNA to immune cells in vivo. Nat. Commun. 2022, 13, 4766. [Google Scholar] [CrossRef] [PubMed]
- Bowen, W.S.; Svrivastava, A.K.; Batra, L.; Barsoumian, H.; Shirwan, H. Current challenges for cancer vaccine adjuvant development. Expert Rev. Vaccines 2018, 17, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.R.; Wang, Z.L.; Feng, B. Hepatitis B functional cure and immune response. Front. Immunol. 2022, 13, 1075916. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J.; Lok, A.S.; Zoulim, F. New Perspectives on Development of Curative Strategies for Chronic Hepatitis B. Clin. Gastroenterol. Hepatol. 2023, 21, 2040–2050. [Google Scholar] [CrossRef]
- Yuen, M.F.; Lim, S.G.; Plesniak, R.; Tsuji, K.; Janssen, H.L.A.; Pojoga, C.; Gadano, A.; Popescu, C.P.; Stepanova, T.; Asselah, T.; et al. Efficacy and Safety of Bepirovirsen in Chronic Hepatitis B Infection. N. Engl. J. Med. 2022, 387, 1957–1968. [Google Scholar] [CrossRef] [PubMed]
- Gane, E.; Lim, Y.S.; Kim, J.B.; Jadhav, V.; Shen, L.; Bakardjiev, A.I.; Huang, S.A.; Cathcart, A.L.; Lempp, F.A.; Janas, M.M.; et al. Evaluation of RNAi therapeutics VIR-2218 and ALN-HBV for chronic hepatitis B: Results from randomized clinical trials. J. Hepatol. 2023, 79, 924–932. [Google Scholar] [CrossRef]
- Yuen, M.F.; Locarnini, S.; Lim, T.H.; Strasser, S.I.; Sievert, W.; Cheng, W.; Thompson, A.J.; Given, B.D.; Schluep, T.; Hamilton, J.; et al. Combination treatments including the small-interfering RNA JNJ-3989 induce rapid and sometimes prolonged viral responses in patients with CHB. J. Hepatol. 2022, 77, 1287–1298. [Google Scholar] [CrossRef]
- Kosinska, A.D.; Festag, J.; Muck-Hausl, M.; Festag, M.M.; Asen, T.; Protzer, U. Immunogenicity and Antiviral Response of Therapeutic Hepatitis B Vaccination in a Mouse Model of HBeAg-Negative, Persistent HBV Infection. Vaccines 2021, 9, 841. [Google Scholar] [CrossRef]
- Yuen, M.; Agarwal, K.; Roberts, S.; Lo, G.; Hsu, C.; Chuang, W.; Chen, C.; Su, P.; Galhenage, S.; Yang, S.; et al. Preliminary pharmacodynamics and safety of repeat dosing of imdusiran (AB-729) followed by VTP-300 or placebo in virally-suppressed, non-cirrhotic subjects with chronic hepatitis B (CHB). In Proceedings of the AASLD Liver Meeting, Boston, MA, USA, 10–14 November 2023. LB#5036-C. [Google Scholar]
- Kayesh, M.E.H.; Kohara, M.; Tsukiyama-Kohara, K. Toll-Like Receptor Response to Hepatitis B Virus Infection and Potential of TLR Agonists as Immunomodulators for Treating Chronic Hepatitis B: An Overview. Int. J. Mol. Sci. 2021, 22, 10462. [Google Scholar] [CrossRef]
- Tait, D. A Phase 2b, Open-label Study to Evaluate the Efficacy, Safety, Tolerability, Immunogenicity and Treatment Regimens of VTP-300 Combined with Low-dose Nivolumab in Chronic hepatitis B Infection. In Proceedings of the AASLD Liver Meeting, Boston, MA, USA, 10–14 November 2023. [Google Scholar]
- Geiben-Lynn, R.; Greenland, J.R.; Frimpong-Boateng, K.; van Rooijen, N.; Hovav, A.H.; Letvin, N.L. CD4+ T lymphocytes mediate in vivo clearance of plasmid DNA vaccine antigen expression and potentiate CD8+ T-cell immune responses. Blood 2008, 112, 4585–4590. [Google Scholar] [CrossRef]
- Hanson, H.L.; Kang, S.S.; Norian, L.A.; Matsui, K.; O’Mara, L.A.; Allen, P.M. CD4-directed peptide vaccination augments an antitumor response, but efficacy is limited by the number of CD8+ T cell precursors. J. Immunol. 2004, 172, 4215–4224. [Google Scholar] [CrossRef]
- Reinscheid, M.; Luxenburger, H.; Karl, V.; Graeser, A.; Giese, S.; Ciminski, K.; Reeg, D.B.; Oberhardt, V.; Roehlen, N.; Lang-Meli, J.; et al. COVID-19 mRNA booster vaccine induces transient CD8+ T effector cell responses while conserving the memory pool for subsequent reactivation. Nat. Commun. 2022, 13, 4631. [Google Scholar] [CrossRef]
- Vieillard, V.; Combadiere, B.; Tubiana, R.; Launay, O.; Pialoux, G.; Cotte, L.; Girard, P.M.; Simon, A.; Dudoit, Y.; Reynes, J.; et al. HIV therapeutic vaccine enhances non-exhausted CD4+ T cells in a randomised phase 2 trial. NPJ Vaccines 2019, 4, 25. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Brunner, L.; Ates Oz, E.; Sacherl, J.; Frank, G.; Kerth, H.A.; Thiele, F.; Wiegand, M.; Mogler, C.; Aguilar, J.C.; et al. Activation of CD4 T cells during prime immunization determines the success of a therapeutic hepatitis B vaccine in HBV-carrier mouse models. J. Hepatol. 2023, 78, 717–730. [Google Scholar] [CrossRef] [PubMed]
- American Chemical, S. Ethical guidelines to publication of chemical research. J. Med. Chem. 2004, 47, 17A–19A. [Google Scholar]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Pooter, D.; Pierson, W.; Pourshahian, S.; Dockx, K.; De Clerck, B.; Najera, I.; Davis, H.; Van Gulck, E.; Boden, D. mRNA Therapeutic Vaccine for Hepatitis B Demonstrates Immunogenicity and Efficacy in the AAV-HBV Mouse Model. Vaccines 2024, 12, 237. https://doi.org/10.3390/vaccines12030237
De Pooter D, Pierson W, Pourshahian S, Dockx K, De Clerck B, Najera I, Davis H, Van Gulck E, Boden D. mRNA Therapeutic Vaccine for Hepatitis B Demonstrates Immunogenicity and Efficacy in the AAV-HBV Mouse Model. Vaccines. 2024; 12(3):237. https://doi.org/10.3390/vaccines12030237
Chicago/Turabian StyleDe Pooter, Dorien, Wim Pierson, Soheil Pourshahian, Koen Dockx, Ben De Clerck, Isabel Najera, Heather Davis, Ellen Van Gulck, and Daniel Boden. 2024. "mRNA Therapeutic Vaccine for Hepatitis B Demonstrates Immunogenicity and Efficacy in the AAV-HBV Mouse Model" Vaccines 12, no. 3: 237. https://doi.org/10.3390/vaccines12030237
APA StyleDe Pooter, D., Pierson, W., Pourshahian, S., Dockx, K., De Clerck, B., Najera, I., Davis, H., Van Gulck, E., & Boden, D. (2024). mRNA Therapeutic Vaccine for Hepatitis B Demonstrates Immunogenicity and Efficacy in the AAV-HBV Mouse Model. Vaccines, 12(3), 237. https://doi.org/10.3390/vaccines12030237