
The Use of Synthetic Carriers in Malaria Vaccine Design

Abstract
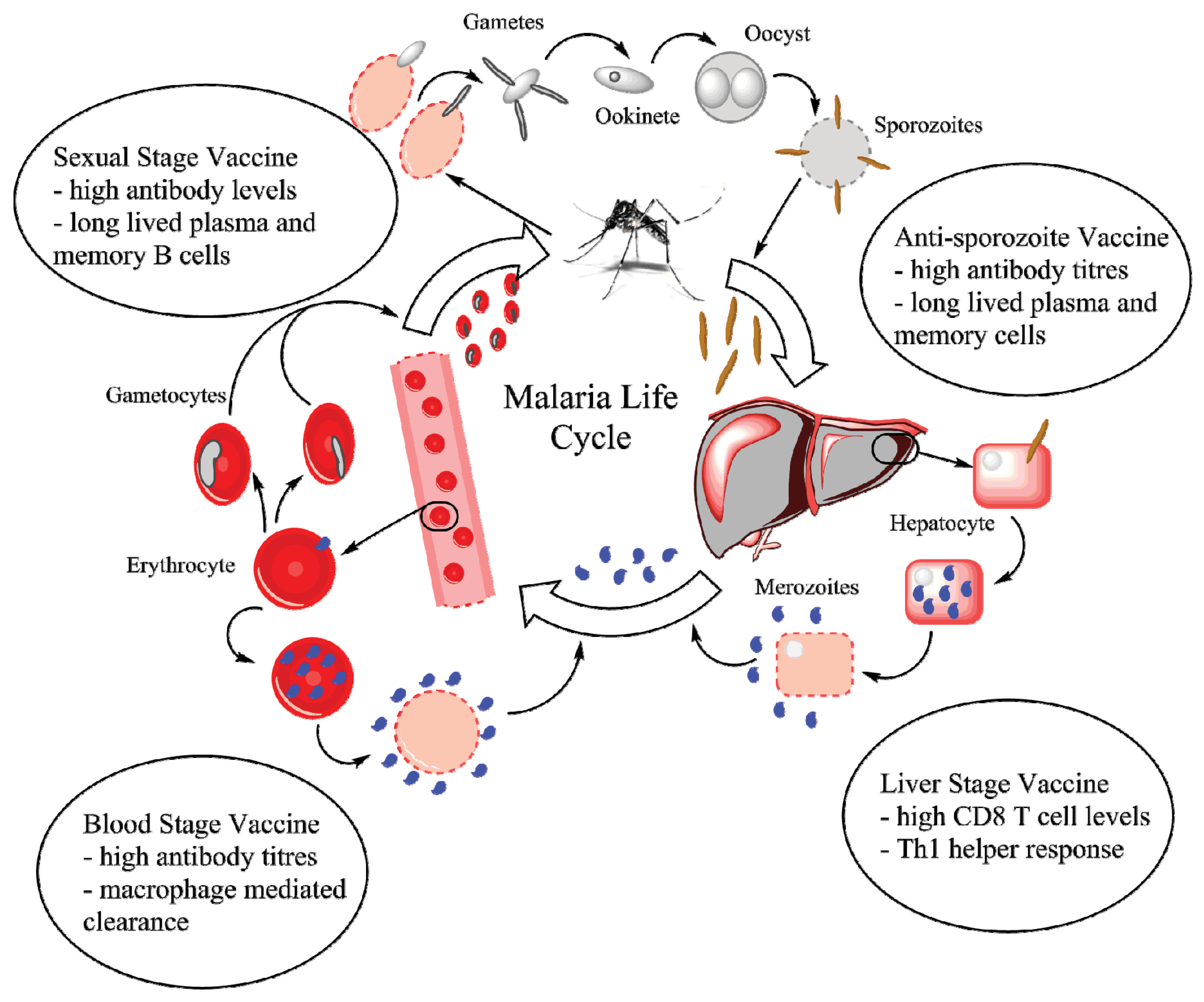
:1. Introduction
2. Vaccine Carrier/Vector Requirements
3. Malaria Vaccine Requirements

4. Particle Use in Malaria Vaccine Development


{kind=link}
{kind=link}
{kind=link}
| Vector | Size | Vaccine | Antibodies | Cytotoxic Response | Ref. |
|---|---|---|---|---|---|
| Pre-Erythrocytic Candidates | |||||
| Lipid Based Particles | |||||
| PC 1 liposomes | Not reported | PyCSP 2 (tetrapeptide B cell, other B, T cell epitopes, RTS,S) | Yes | Some antigens | [12,62,63,64,65,66,67,68,69] |
| Lipid core peptides | Not reported | PyCSP (CD4 and CD8 epitopes) | Minor IgE | ND 3 | [70] |
| ICMV 4 | 180 nm (DLS 5) | PvCSP 6 (VMP001) | Yes | ND | [71] |
| Polymeric Particles | |||||
| PCL 7/PLA 8 | 23–45 µm | PfCSP 9 (tetrapeptide, universal CD4) | Yes | ND | [72] |
| PLA/PLGA 10 | 1–100 µm | PfCSP (tetrapeptide, universal CD4), Pb911 | Yes | Against Pb9 | [13,73,74] |
| PLGA | 0.45–32.1 µm | Pb9 | ND | Yes | [75] |
| Lipid enveloped PLGA | 290 nm (DLS) | PvCSP (VMP001) | Yes | ND | [76] |
| Polystyrene Nanoparticles | 48 nm (DLS) | Pb9 | ND | Yes | [77] |
| Other Particles | |||||
| SAPN 12 | 40 nm (TEM 13, DLS) | PfCSP, PvCSP, CD8 and B epitopes, universal CD4 | Yes | Some antigens | [78,79,80] |
| Polymer coated calcium carbonate | Not reported | PfCSP (B and T cell epitopes) | Yes | Yes | [81] |
| Blood Stage Candidates | |||||
| Lipid Based Particles | |||||
| PC Liposome | Not reported | PfMSP1–19 14 | Yes | ND | [82] |
| PC Liposome | Not reported | PyIMP-66 15 | Yes | ND | [83] |
| E. coli 16 Liposome | Not reported | Py soluble | Yes | Yes | [84] |
| S. cer. 17 Liposome | Not reported | Py soluble | Yes | Yes | [85] |
| pH sensitive Liposome | 325–390 nm | PfMSP1–19 | Minor | ND | [70] |
| Other Organic Particles | |||||
| PEI 18, γ-PGA 19 | 68 nm (DLS) | PyMSP1-C-terminus, PyTAM 20 (DNA) | Yes | With PyTAM | [86,87,88] |
| ISCOM 21 | Not Reported | PfRESA 22 | Yes | ND | [89,90] |
| ISCOM | Not Reported | PfRESA peptides | Yes | ND | [91] |
| Inorganic Particles | |||||
| SPIONs 23 | 20 nm (unknown) | PfMSP1–42 | Yes | ND | [14] |
| PEI coated SPIONs | 147 nm (DLS) | PyMSP1–19 (DNA) | Yes | ND | [92] |
| Gold nanoparticle | 17 nm (TEM) | Pf/PvMSP1–19 | Yes | ND | [93] |
| Quantum Dots | 15 nm (unknown) | PfMSP1–42 | Yes | ND | [94] |
| Carbon Nanotubes | 20–30/500–2000 nm | PvAMA1 4peptides | Yes | ND | [95] |
| Hydroxyapatite | 784 nm (DLS) | MSP1–19 | Yes | ND | [15] |
| Mixed Stage Candidates | |||||
| PLGA | 0.5–2 µm | SPf66, PfMSP2 25 peptides, PfS3 | Yes | ND | [96,97,98,99,100,101,102] |
| PLGA-alginate-RGD | 0.8–1 µm | SPf66, PfS3 | Yes | ND | [103] |
| PLGA | 1–2 µm | PvCSP, MSP1, AMA1, Pvs24 (all with B and T epitopes) | Yes | ND | [104,105] |
| Sexual Stage Candidate | |||||
| Gel core liposomes | 1–1.2 µm | Pfs25 | Yes | ND | [106] |
4.1. Pre-Erythrocytic Vaccines
| Particle Type | Advantages | Disadvantages | Ref. |
|---|---|---|---|
| Lipid-Based | Wide size range Antigen encapsulated or on surface Hydrophobic or hydrophilic antigen FDA approved/Non-toxic Biodegradable | Expensive materials Reproducibility issues Oxidative Degradation | [107,108,109,110] |
| PLGA | Antigen encapsulated or on surface Biodegradable FDA approved/Non-toxic Prolonged release of antigen | Antigen degradation Scale-up Antigen burst releases | [110,111,112,113,114] |
| Polystyrene | Biocompatible Non-toxic Wide size range Readily available | Non-biodegradable | [115] |
| SAPN | Repetitive presentation Biodegradable | Complex design Limited Data | [116] |
| PEI/γ-PGA | Good for DNA vaccine Small size | Limited Data | [117] |
| ISCOM | Natural adjuvant Readily available Biodegradable Scalable Well-tolerated | Encapsulation limited Single size | [118,119,120] |
| SPION | Biodegradable Magnetic FDA approved Size control | Coating required Stability issues | [121,122] |
| Quantum Dot | Fluorescent Stable | Toxic materials Non-biodegradable | [123,124,125] |
| Calcium Based | Low cytotoxicity Surface modification | Limited degradability Limited study | [126,127] |
| Gold | Size control Low cytotoxicity | Non-biodegradable Coating required | [128,129] |
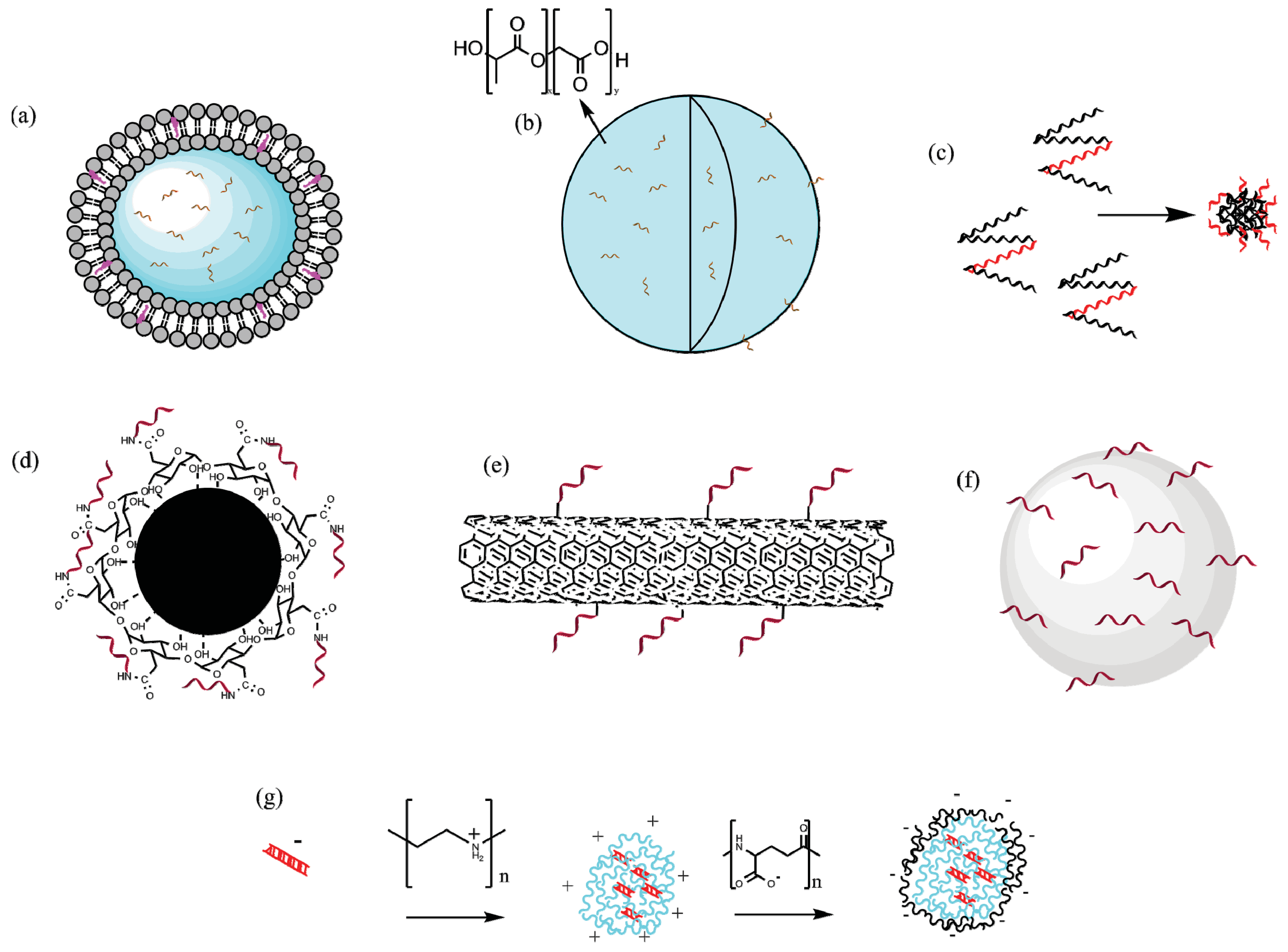
4.1.1. Lipid-Based Particles
4.1.2. Polymeric Particles
4.1.3. Self-Assembling Protein Nanoparticles (SAPNs)
4.1.4. Inorganic Particles
4.2. Blood Stage Vaccines
4.2.1. Lipid Based Particles
4.2.2. Immune Stimulating Complexes (ISCOMs)
4.2.3. Inorganic Particles
4.2.4. DNA Vaccine Carriers
4.3. Multi-Stage Vaccines
4.4. Sexual Stage Vaccines
5. Connecting Nanotechnology and Malaria Vaccines

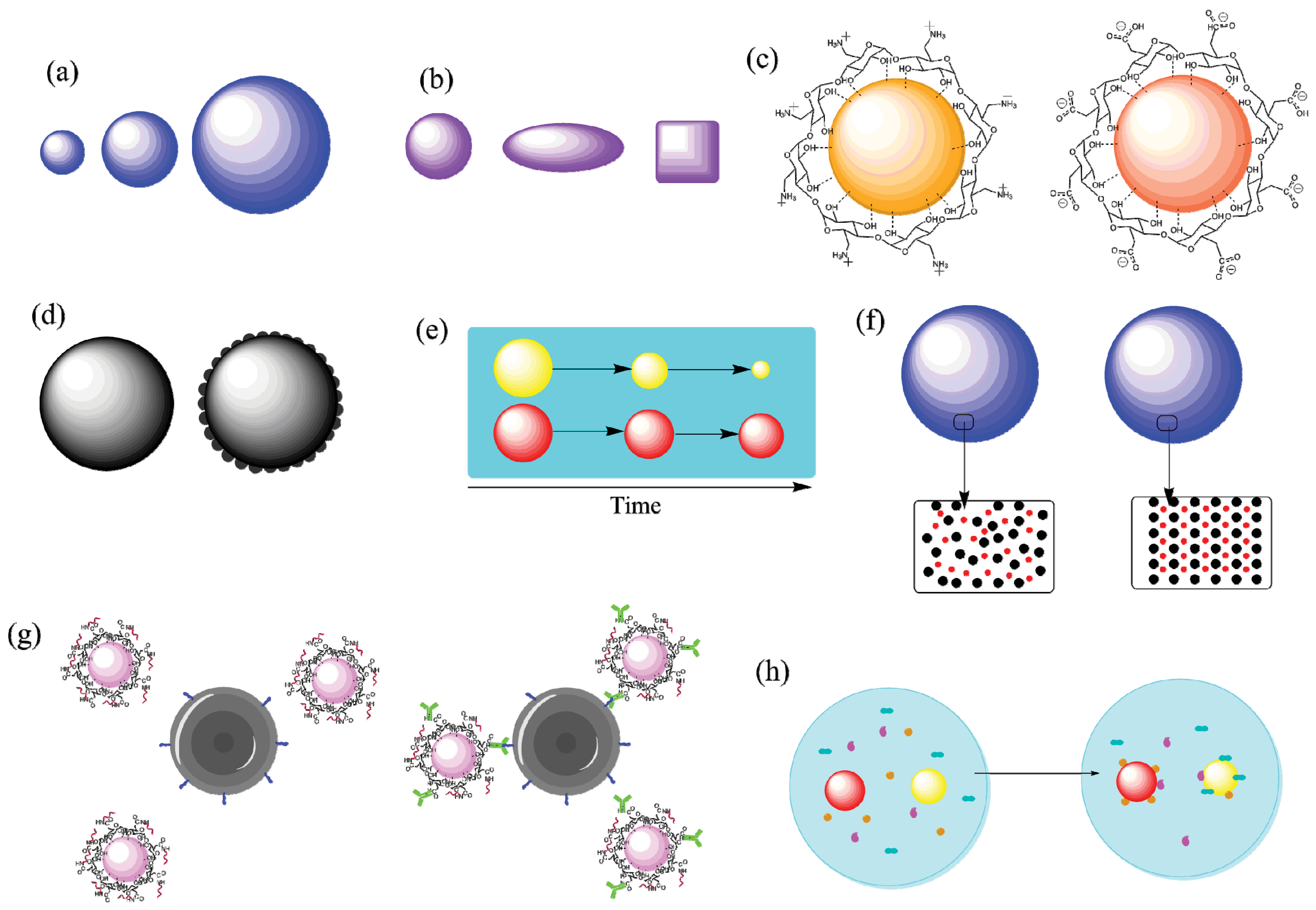
5.1. Size
5.2. Surface Charge
5.3. Targeting Ligands
5.4. Protein Corona and Material Characteristics
5.5. Method of Antigen Incorporation
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- WHO. World Malaria Report 2014; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Murray, C.J.L.; Rosenfeld, L.C.; Lim, S.S.; Andrews, K.G.; Foreman, K.J.; Haring, D.; Fullman, N.; Naghavi, M.; Lozano, R.; Lopez, A.D. Global malaria mortality between 1980 and 2010: A systematic analysis. Lancet 2012, 379, 413–431. [Google Scholar] [CrossRef]
- Playfair, J.H.L.; Taverne, J.; Bate, C.A.W.; Brian de Souza, J. The malaria vaccine: Anti-parasite or anti-disease? Immunol. Today 1990, 11, 25–27. [Google Scholar] [CrossRef]
- Conway, D.J. Natural selection on polymorphic malaria antigens and the search for a vaccine. Parasitol. Today 1997, 13, 26–29. [Google Scholar] [CrossRef]
- Schwartz, L.; Brown, G.V.; Genton, B.; Moorthy, V.S. A review of malaria vaccine clinical projects based on the WHO rainbow table. Malar. J. 2012. [Google Scholar] [CrossRef] [PubMed]
- Seder, R.A.; Chang, L.-J.; Enama, M.E.; Zephir, K.L.; Sarwar, U.N.; Gordon, I.J.; Holman, L.A.; James, E.R.; Billingsley, P.F.; Gunasekera, A.; et al. Protection against malaria by intravenous immunization with a nonreplicating sporozoite vaccine. Science 2013, 341, 1359–1365. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.E.; Williamson, D.; Titball, R. Vaccine delivery using nanoparticles. Front. Cell. Infect. Microbiol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Doolan, D.L. Plasmodium immunomics. Int. J. Parasitol. 2011, 41, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Vogel, F.R. Immunologic adjuvants for modern vaccine formulations. Ann. N.Y. Acad. Sci. 1995, 754, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Schijns, V.E.J.C. Immunological concepts of vaccine adjuvant activity: Commentary. Curr. Opin. Immunol. 2000, 12, 456–463. [Google Scholar] [CrossRef]
- Beeson, J.G.; Osier, F.H.A.; Engwerda, C.R. Recent insights into humoral and cellular immune responses against malaria. Trends Parasitol. 2008, 24, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Fries, L.F.; Gordon, D.M.; Richards, R.L.; Egan, J.E.; Hollingdale, M.R.; Gross, M.; Silverman, C.; Alving, C.R. Liposomal malaria vaccine in humans: A safe and potent adjuvant strategy. Proc. Natl. Acad. Sci. USA 1992, 89, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Thomasin, C.; Corradin, G.; Men, Y.; Merkle, H.P.; Gander, B. Tetanus toxoid and synthetic malaria antigen containing poly (lactide)/poly (lactide-co-glycolide) microspheres: Importance of polymer degradation and antigen release for immune response. J. Controll. Release 1996, 41, 131–145. [Google Scholar] [CrossRef]
- Pusic, K.; Aguilar, Z.; McLoughlin, J.; Kobuch, S.; Xu, H.; Tsang, M.; Wang, A.; Hui, G. Iron oxide nanoparticles as a clinically acceptable delivery platform for a recombinant blood-stage human malaria vaccine. FASEB J. 2013, 27, 1153–1166. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Goyal, A.K.; Khatri, K.; Mishra, N.; Mehta, A.; Vaidya, B.; Tiwari, S.; Paliwal, R.; Paliwal, S. Development of self-assembled nanoceramic carrier construct(s) for vaccine delivery. J. Biomater. Appl. 2009, 24, 65–84. [Google Scholar]
- Ruigrok, R.W.H.; Gerlier, D. Structure of the measles virus H glycoprotein sheds light on an efficient vaccine. Proc. Natl. Acad. Sci. USA 2007, 104, 20639–20640. [Google Scholar] [CrossRef] [PubMed]
- Hilleman, M.R.; Buynak, E.B.; Weibel, R.E.; Stokes, J. Live, attenuated mumps-virus vaccine. New Engl. J. Med. 1968, 278, 227–232. [Google Scholar] [CrossRef] [PubMed]
- American Academy of Pediatrics Committee on Infectious Diseases. Poliomyelitis prevention: Recommendations for use of inactivated poliovirus vaccine and live oral poliovirus vaccine. Pediatrics 1997, 99, 300–305. [Google Scholar]
- Schild, G.; Corbel, M.; Corran, P.; Minor, P. Vaccines: Past, Present and Future. In Vaccines; Perlmann, P., Wigzell, H., Eds.; Springer: Berlin, Germany; Heidelberg, Germany, 1999; Volume 133, pp. 1–19. [Google Scholar]
- Ulmer, J.B.; Donnelly, J.J.; Liu, M.A. DNA Vaccines: Immunogenicity and Preclinical Efficacy. In Vaccines; Perlmann, P., Wigzell, H., Eds.; Springer: Berlin, Germany; heidelberg, Germany, 1999; Volume 133, pp. 43–55. [Google Scholar]
- Robinson, H.L.; Pertmer, T.M. DNA vaccines for viral infections: Basic studies and applications. In Advances in Virus Research; Academic Press: Waltham, MA, USA, 2000; Volume 55, pp. 1–74. [Google Scholar]
- Male, D.; Brostoff, J.; Roth, D.; Roitt, I. Immunology, 8 ed.; Saunders Elsevier: Philadelphia, PA, USA, 2012. [Google Scholar]
- Ferraro, B.; Morrow, M.P.; Hutnick, N.A.; Shin, T.H.; Lucke, C.E.; Weiner, D.B. Clinical applications of DNA vaccines: Current progress. Clin. Infect. Dis. 2011, 53, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Friede, M.; Aguado, M.T. Need for new vaccine formulations and potential of particulate antigen and DNA delivery systems. Adv. Drug Deliv. Rev. 2005, 57, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, J.B.; Valley, U.; Rappuoli, R. Vaccine manufacturing: Challenges and solutions. Nat. Biotech. 2006, 24, 1377–1383. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.; Xia, T.; Mädler, L.; Li, N. Toxic Potential of Materials at the Nanolevel. Science 2006, 311, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Crompton, P.D.; Pierce, S.K.; Miller, L.H. Advances and challenges in malaria vaccine development. J. Clin. Investig. 2010, 120, 4168–4178. [Google Scholar] [CrossRef] [PubMed]
- Riley, E.M.; Stewart, V.A. Immune mechanisms in malaria: New insights in vaccine development. Nat. Med. 2013, 19, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Finney, O.C.; Keitany, G.J.; Smithers, H.; Kaushansky, A.; Kappe, S.; Wang, R. Immunization with genetically attenuated P. falciparum parasites induces long-lived antibodies that efficiently block hepatocyte invasion by sporozoites. Vaccine 2014, 32, 2135–2138. [Google Scholar] [CrossRef] [PubMed]
- Coppi, A.; Natarajan, R.; Pradel, G.; Bennett, B.L.; James, E.R.; Roggero, M.A.; Corradin, G.; Persson, C.; Tewari, R.; Sinnis, P. The malaria circumsporozoite protein has two functional domains, each with distinct roles as sporozoites journey from mosquito to mammalian host. J. Exp. Med. 2011, 208, 341–356. [Google Scholar] [CrossRef] [PubMed]
- Tewari, R.; Spaccapelo, R.; Bistoni, F.; Holder, A.A.; Crisanti, A. Function of region I and II adhesive motifs of Plasmodium falciparum circumsporozoite protein in sporozoite motility and infectivity. J. Biol. Chem. 2002, 277, 47613–47618. [Google Scholar] [CrossRef] [PubMed]
- Sultan, A.A.; Thathy, V.; Frevert, U.; Robson, K.J.H.; Crisanti, A.; Nussenzweig, V.; Nussenzweig, R.S.; Ménard, R. TRAP Is necessary for gliding motility and infectivity of Plasmodium sporozoites. Cell 1997, 90, 511–522. [Google Scholar] [CrossRef]
- Baldacci, P.; Ménard, R. The elusive malaria sporozoite in the mammalian host. Mol. Microbiol. 2004, 54, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Silvie, O.; Franetich, J.-F.; Charrin, S.; Mueller, M.S.; Siau, A.; Bodescot, M.; Rubinstein, E.; Hannoun, L.; Charoenvit, Y.; Kocken, C.H.; et al. A role for apical membrane antigen 1 during invasion of hepatocytes by Plasmodium falciparum sporozoites. J. Biol. Chem. 2004, 279, 9490–9496. [Google Scholar] [CrossRef] [PubMed]
- Kariu, T.; Ishino, T.; Yano, K.; Chinzei, Y.; Yuda, M. CelTOS, a novel malarial protein that mediates transmission to mosquito and vertebrate hosts. Mol. Microbiol. 2006, 59, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Ishino, T.; Yano, K.; Chinzei, Y.; Yuda, M. Cell-passage activity is required for the malarial parasite to cross the liver sinusoidal cell layer. PLoS Biol. 2004, 2, e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cockburn, I.A.; Tse, S.-W.; Radtke, A.J.; Srinivasan, P.; Chen, Y.-C.; Sinnis, P.; Zavala, F. Dendritic cells and hepatocytes use distinct pathways to process protective antigen from plasmodium in vivo. PLoS Pathog. 2011, 7, e1001318. [Google Scholar] [CrossRef] [PubMed]
- Butler, N.S.; Schmidt, N.W.; Harty, J.T. Differential effector pathways regulate memory CD8 T cell immunity against Plasmodium berghei versus P. yoelii sporozoites. J. Immunol. 2010, 184, 2528–2538. [Google Scholar] [CrossRef] [PubMed]
- Günther, K.; Tümmler, M.; Arnold, H.-H.; Ridley, R.; Goman, M.; Scaife, J.G.; Lingelbach, K. An exported protein of Plasmodium falciparum is synthesized as an integral membrane protein. Mol. Biochem. Parasitol. 1991, 46, 149–157. [Google Scholar] [CrossRef]
- Mikolajczak, S.A.; Sacci, J.J.B.; de la Vega, P.; Camargo, N.; VanBuskirk, K.; Krzych, U.; Cao, J.; Jacobs-Lorena, M.; Cowman, A.F.; Kappe, S.H.I. Disruption of the Plasmodium falciparum liver-stage antigen-1 locus causes a differentiation defect in late liver-stage parasites. Cell. Microbiol. 2011, 13, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Prieur, E.; Druilhe, P. The malaria candidate vaccine liver stage antigen-3 is highly conserved in Plasmodium falciparum isolates from diverse geographical areas. Malar. J. 2009. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.W.; Butler, N.S.; Badovinac, V.P.; Harty, J.T. Extreme CD8 T cell requirements for anti-malarial liver-stage immunity following immunization with radiation attenuated sporozoites. PLoS Pathog. 2010, 6, e1000998. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.W.; Podyminogin, R.L.; Butler, N.S.; Badovinac, V.P.; Tucker, B.J.; Bahjat, K.S.; Lauer, P.; Reyes-Sandoval, A.; Hutchings, C.L.; Moore, A.C.; et al. Memory CD8 T cell responses exceeding a large but definable threshold provide long-term immunity to malaria. Proc. Natl. Acad. Sci. USA 2008, 105, 14017–14022. [Google Scholar] [CrossRef] [PubMed]
- Osier, F.H.A.; Fegan, G.; Polley, S.D.; Murungi, L.; Verra, F.; Tetteh, K.K.A.; Lowe, B.; Mwangi, T.; Bull, P.C.; Thomas, A.W.; et al. Breadth and magnitude of antibody responses to multiple Plasmodium falciparum merozoite antigens are associated with protection from clinical malaria. Infect. Immun. 2008, 76, 2240–2248. [Google Scholar] [CrossRef] [PubMed]
- McCall, M.B.; Roestenberg, M.; Ploemen, I.; Teirlinck, A.; Hopman, J.; de Mast, Q.; Dolo, A.; Doumbo, O.K.; Luty, A.; van der Ven, A.J. Memory-like IFN-γ response by NK cells following malaria infection reveals the crucial role of T cells in NK cell activation by P. falciparum. Eur. J. Immunol. 2010, 40, 3472–3477. [Google Scholar] [CrossRef] [PubMed]
- McCall, M.B.B.; Sauerwein, R.W. Interferon-γ—Central mediator of protective immune responses against the pre-erythrocytic and blood stage of malaria. J. Leukocyte Biol. 2010, 88, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.C.; Corran, P.H.; Mangia, E.; Gaunt, M.W.; Li, Q.; Tetteh, K.K.A.; Polley, S.D.; Conway, D.J.; Holder, A.A.; Bacarese-Hamilton, T.; et al. Profiling the antibody immune response against blood stage malaria vaccine candidates. Clin. Chem. 2007, 53, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Volkman, S.K.; Hartl, D.L.; Wirth, D.F.; Nielsen, K.M.; Choi, M.; Batalov, S.; Zhou, Y.; Plouffe, D.; le Roch, K.G.; Abagyan, R.; et al. Excess polymorphisms in genes for membrane proteins in Plasmodium falciparum. Science 2002, 298, 216–218. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.H.; Roberts, T.; Shahabuddin, M.; McCutchan, T.F. Analysis of sequence diversity in the Plasmodium falciparum merozoite surface protein-1 (MSP-1). Mol. Biochem. Parasitol. 1993, 59, 1–14. [Google Scholar] [CrossRef]
- Goel, V.K.; Li, X.; Chen, H.; Liu, S.-C.; Chishti, A.H.; Oh, S.S. Band 3 is a host receptor binding merozoite surface protein 1 during the Plasmodium falciparum invasion of erythrocytes. Proc. Natl. Acad. Sci. USA 2003, 100, 5164–5169. [Google Scholar] [CrossRef] [PubMed]
- Cowman, A.F.; Crabb, B.S. Invasion of red blood cells by malaria parasites. Cell 2006, 124, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Remarque, E.J.; Faber, B.W.; Kocken, C.H.M.; Thomas, A.W. Apical membrane antigen 1: A malaria vaccine candidate in review. Trends Parasitol. 2008, 24, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Lee Sim, B.K.; Toyoshima, T.; David Haynes, J.; Aikawa, M. Localization of the 175-kilodalton erythrocyte binding antigen in micronemes of Plasmodium falciparum merozoites. Mol. Biochem. Parasitol. 1992, 51, 157–159. [Google Scholar] [CrossRef]
- Douglas, A.D.; Baldeviano, G.C.; Miura, K.; Wright, G.J.; Draper, S.J. PfRH5 vaccine efficacy against heterologous strain blood-stage Plasmodium falciparum. Lancet 2014. [Google Scholar] [CrossRef]
- Bustamante, L.Y.; Bartholdson, S.J.; Crosnier, C.; Campos, M.G.; Wanaguru, M.; Nguon, C.; Kwiatkowski, D.P.; Wright, G.J.; Rayner, J.C. A full-length recombinant Plasmodium falciparum PfRH5 protein induces inhibitory antibodies that are effective across common PfRH5 genetic variants. Vaccine 2013, 31, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Healer, J.; McGuinness, D.; Hopcroft, P.; Haley, S.; Carter, R.; Riley, E. Complement-mediated lysis of Plasmodium falciparum gametes by malaria-immune human sera is associated with antibodies to the gamete surface antigen Pfs230. Infect. Immun. 1997, 65, 3017–3023. [Google Scholar] [PubMed]
- Vaughan, A.M.; Kappe, S.H.I. Malaria vaccine development: Persistent challenges. Curr. Opin. Immunol. 2012, 24, 324–331. [Google Scholar] [PubMed]
- Barr, P.J.; Green, K.M.; Gibson, H.L.; Bathurst, I.C.; Quakyi, I.A.; Kaslow, D.C. Recombinant Pfs25 protein of Plasmodium falciparum elicits malaria transmission-blocking immunity in experimental animals. J. Exp. Med. 1991, 174, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Sato, C.; Otsuki, H.; Sattabongkot, J.; Kaneko, O.; Torii, M.; Tsuboi, T. Plasmodium vivax gametocyte protein Pvs230 is a transmission-blocking vaccine candidate. Vaccine 2012, 30, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Eksi, S.; Czesny, B.; van Gemert, G.-J.; Sauerwein, R.W.; Eling, W.; Williamson, K.C. Malaria transmission-blocking antigen, Pfs230, mediates human red blood cell binding to exflagellating male parasites and oocyst production. Mol. Microbiol. 2006, 61, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, D.R.; Angov, E.; Kariuki, T.; Kumar, N. A potent malaria transmission blocking vaccine based on codon harmonized full length Pfs48/45 expressed in Escherichia coli. PLoS ONE 2009, 4, e6352. [Google Scholar] [PubMed]
- Richards, R.; Hayre, M.; Hockmeyer, W.; Alving, C. Liposomes, lipid A, and aluminum hydroxide enhance the immune response to a synthetic malaria sporozoite antigen. Infect. Immun. 1988, 56, 682–686. [Google Scholar] [PubMed]
- Alving, C.R.; Richards, R.L.; Moss, J.; Alving, L.I.; Clements, J.D.; Shiba, T.; Kotani, S.; Wirtz, R.A.; Hockmeyer, W.T. Effectiveness of liposomes as potential carriers of vaccines: Applications to cholera toxin and human malaria sporozoite antigen. Vaccine 1986, 4, 166–172. [Google Scholar] [CrossRef]
- Richards, R.L.; Swartz, G.M., Jr.; Schultz, C.; Hayre, M.D.; Ward, G.S.; Ballou, W.R.; Chulay, J.D.; Hockmeyer, W.T.; Berman, S.L.; Alving, C.R. Immunogenicity of liposomal malaria sporozoite antigen in monkeys: Adjuvant effects of aluminium hydroxide and non-pyrogenic liposomal lipid A. Vaccine 1989, 7, 506–512. [Google Scholar] [CrossRef]
- White, K.; Krzych, U.; Gordon, D.M.; Porter, T.G.; Richards, R.L.; Alving, C.R.; Deal, C.D.; Hollingdale, M.; Silverman, C.; Sylvester, D.R. Induction of cytolytic and antibody responses using Plasmodium falciparum repeatless circumsporozoite protein encapsulated in liposomes. Vaccine 1993, 11, 1341–1346. [Google Scholar] [CrossRef]
- Verma, J.; Rao, M.; Amselem, S.; Krzych, U.; Alving, C.; Green, S.; Wassef, N. Adjuvant effects of liposomes containing lipid A: Enhancement of liposomal antigen presentation and recruitment of macrophages. Infect. Immun. 1992, 60, 2438–2444. [Google Scholar] [PubMed]
- Wang, R.; Charoenvit, Y.; Corradin, G.; Porrozzi, R.; Hunter, R.L.; Glenn, G.; Alving, C.R.; Church, P.; Hoffman, S.L. Induction of protective polyclonal antibodies by immunization with a Plasmodium yoelii circumsporozoite protein multiple antigen peptide vaccine. J. Immunol. 1995, 154, 2784–2793. [Google Scholar] [PubMed]
- Richards, R.L.; Rao, M.; Wassef, N.M.; Glenn, G.M.; Rothwell, S.W.; Alving, C.R. Liposomes containing lipid A serve as an adjuvant for induction of antibody and cytotoxic T-cell responses against RTS,S malaria antigen. Infect. Immun. 1998, 66, 2859–2865. [Google Scholar] [PubMed]
- Heppner, D.; Gordon, D.; Gross, M.; Wellde, B.; Leitner, W.; Krzych, U.; Schneider, I.; Wirtz, R.; Richards, R.; Trofa, A. Safety, immunogenicity, and efficacy of Plasmodium falciparum repeatless circumsporozoite protein vaccine encapsulated in liposomes. J. Infect. Dis. 1996, 174, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.P.; Jadon, R.S.; Goyal, A.K.; Mishra, N.; Gupta, P.N.; Khatri, K.; Tyagi, R. pH sensitive liposomes enhances immunogenicity of 19 kDa carboxyl-terminal fragment of Plasmodium falciparum. Int. J. Pharm. Sci. Nanotechnol. 2008, 1, 78–86. [Google Scholar]
- Moon, J.J.; Suh, H.; Li, A.V.; Ockenhouse, C.F.; Yadava, A.; Irvine, D.J. Enhancing humoral responses to a malaria antigen with nanoparticle vaccines that expand Tfh cells and promote germinal center induction. Proc. Natl. Acad. Sci. USA 2012, 109, 1080–1085. [Google Scholar] [CrossRef] [PubMed]
- Amselem, S.; Alving, C.R.; Domb, A.J. Polymeric biodegradable lipospheres™ as vaccine delivery systems. Polymers Adv. Technol. 1992, 3, 351–357. [Google Scholar] [CrossRef]
- Men, Y.; Gander, B.; Merkle, H.P.; Corradin, G. Induction of sustained and elevated immune responses to weakly immunogenic synthetic malarial peptides by encapsulation in biodegradable polymer microspheres. Vaccine 1996, 14, 1442–1450. [Google Scholar] [PubMed]
- Men, Y.; Tamber, H.; Audran, R.; Gander, B.; Corradin, G. Induction of a cytotoxic T lymphocyte response by immunization with a malaria specific CTL peptide entrapped in biodegradable polymer microspheres. Vaccine 1997, 15, 1405–1412. [Google Scholar] [CrossRef]
- Nixon, D.F.; Hioe, C.; Chen, P.-D.; Bian, Z.; Kuebler, P.; Li, M.-L.; Qiu, H.; Li, X.-M.; Singh, M.; Richardson, J.; et al. Synthetic peptides entrapped in microparticles can elicit cytotoxic T cell activity. Vaccine 1996, 14, 1523–1530. [Google Scholar] [CrossRef]
- Moon, J.J.; Suh, H.; Polhemus, M.E.; Ockenhouse, C.F.; Yadava, A.; Irvine, D.J. Antigen-displaying lipid-enveloped PLGA nanoparticles as delivery agents for a Plasmodium vivax malaria vaccine. PLoS ONE 2012, 7, e31472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, K.L.; Xiang, S.D.; Plebanski, M. Montanide, Poly I: C and nanoparticle based vaccines promote differential suppressor and effector cell expansion: A study of induction of CD8 T cells to a minimal Plasmodium berghei epitope. Front. Microbiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kaba, S.A.; Brando, C.; Guo, Q.; Mittelholzer, C.; Raman, S.; Tropel, D.; Aebi, U.; Burkhard, P.; Lanar, D.E. A nonadjuvanted polypeptide nanoparticle vaccine confers long-lasting protection against rodent malaria. J. Immunol. 2009, 183, 7268–7277. [Google Scholar] [CrossRef] [PubMed]
- Kaba, S.A.; McCoy, M.E.; Doll, T.A.; Brando, C.; Guo, Q.; Dasgupta, D.; Yang, Y.; Mittelholzer, C.; Spaccapelo, R.; Crisanti, A. Protective antibody and CD8+ T-cell responses to the Plasmodium falciparum circumsporozoite protein induced by a nanoparticle vaccine. PLoS ONE 2012, 7, e48304. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.E.; Golden, H.E.; Doll, T.A.; Yang, Y.; Kaba, S.A.; Burkhard, P.; Lanar, D.E. Mechanisms of protective immune responses induced by the Plasmodium falciparum circumsporozoite protein-based, self-assembling protein nanoparticle vaccine. Malar. J. 2013. [Google Scholar] [CrossRef]
- Powell, T.J.; Tang, J.; DeRome, M.E.; Mitchell, R.A.; Jacobs, A.; Deng, Y.; Palath, N.; Cardenas, E.; Boyd, J.G.; Nardin, E. Plasmodium falciparum synthetic LbL microparticle vaccine elicits protective neutralizing antibody and parasite-specific cellular immune responses. Vaccine 2013, 31, 1898–1904. [Google Scholar] [CrossRef] [PubMed]
- Burghaus, P.A.; Wellde, B.T.; Hall, T.; Richards, R.L.; Egan, A.F.; Riley, E.M.; Ballou, W.R.; Holder, A.A. Immunization of Aotus nancymai with recombinant C terminus of Plasmodium falciparum merozoite surface protein 1 in liposomes and alum adjuvant does not induce protection against a challenge infection. Infect. Immun. 1996, 64, 3614–3619. [Google Scholar] [PubMed]
- Sharma, S.K.; Gupta, C.; Dwivedi, V.; Misra-Bhattacharya, S.; Mohammad, O. Prophylactic potential of liposomized integral membrane protein of Plasmodium yoelii nigeriensis against blood stage infection in BALB/c mice. Vaccine 2007, 25, 2103–2111. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Farah, D.; Misra-Bhattacharya, S.; Bajpai, P.; Agarwal, A.; Mohammad, O. Escheriosome entrapped soluble blood stage antigens impart protective immunity against a multi-drug resistant isolate of Plasmodium yoelii nigeriensis in BALB/c mice. Vaccine 2006, 24, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, V.; Vasco, A.; Vedi, S.; Dangi, A.; Arif, K.; Bhattacharya, S.M.; Owais, M. Adjuvanticity and protective immunity of Plasmodium yoelii nigeriensis blood-stage soluble antigens encapsulated in fusogenic liposome. Vaccine 2009, 27, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Shuaibu, M.; Cherif, M.; Kurosaki, T.; Helegbe, G.; Kikuchi, M.; Yanagi, T.; Sasaki, H.; Hirayama, K. Effect of nanoparticle coating on the immunogenicity of plasmid DNA vaccine encoding P. yoelii MSP-1 C-terminal. Vaccine 2011, 29, 3239–3247. [Google Scholar] [CrossRef] [PubMed]
- Cherif, M.S.; Shuaibu, M.N.; Kurosaki, T.; Helegbe, G.K.; Kikuchi, M.; Yanagi, T.; Tsuboi, T.; Sasaki, H.; Hirayama, K. Immunogenicity of novel nanoparticle-coated MSP-1 C-terminus malaria DNA vaccine using different routes of administration. Vaccine 2011, 29, 9038–9050. [Google Scholar] [CrossRef] [PubMed]
- Cherif, M.S.; Shuaibu, M.N.; Kodama, Y.; Kurosaki, T.; Helegbe, G.K.; Kikuchi, M.; Ichinose, A.; Yanagi, T.; Sasaki, H.; Yui, K. Nanoparticle formulation enhanced protective immunity provoked by PYGPI8p-transamidase related protein (PyTAM) DNA vaccine in Plasmodium yoelii malaria model. Vaccine 2014, 32, 1998–2006. [Google Scholar] [CrossRef] [PubMed]
- Sjölander, A.; Lövgren, K.; Ståhl, S.; Åslund, L.; Hansson, M.; Nygren, P.-Å.; Larsson, M.; Hagstedt, M.; Wåhlin, B.; Berzins, K. High antibody responses in rabbits immunized with influenza virus ISCOMs containing a repeated sequence of the Plasmodium falciparum antigen Pf155/RESA. Vaccine 1991, 9, 443–450. [Google Scholar]
- Sjölander, A.; Hansson, M.; Lövgren, K.; Wåhlin, B.; Berzins, K.; Perlmann, P. Immunogenicity in rabbits and monkeys of influenza ISCOMs conjugated with repeated sequences of the Plasmodium falciparum antigen Pf155/RESA. Parasite Immunol. 1993, 15, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Chopra, N.; Biswas, S.; Thomas, B.; Sabhnani, L.; Rao, D. Inducing protective antibodies against ring-infected erythrocyte surface peptide antigen of Plasmodium falciparum using immunostimulating complex (ISCOMs) delivery. Med. Microbiol. Immunol. 2000, 189, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Nawwab Al-Deen, F.; Ma, C.; Xiang, S.D.; Selomulya, C.; Plebanski, M.; Coppel, R.L. On the efficacy of malaria DNA vaccination with magnetic gene vectors. J. Controll. Release 2013, 168, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Parween, S.; Gupta, P.K.; Chauhan, V.S. Induction of humoral immune response against PfMSP-1(19) and PvMSP-1(19) using gold nanoparticles along with alum. Vaccine 2011, 29, 2451–2460. [Google Scholar] [CrossRef] [PubMed]
- Pusic, K.; Xu, H.; Stridiron, A.; Aguilar, Z.; Wang, A.; Hui, G. Blood stage merozoite surface protein conjugated to nanoparticles induce potent parasite inhibitory antibodies. Vaccine 2011, 29, 8898–8908. [Google Scholar] [CrossRef] [PubMed]
- Yandar, N.; Pastorin, G.; Prato, M.; Bianco, A.; Patarroyo, M.E.; Manuel Lozano, J. Immunological profile of a Plasmodium vivax AMA-1 N-terminus peptide-carbon nanotube conjugate in an infected Plasmodium berghei mouse model. Vaccine 2008, 26, 5864–5873. [Google Scholar] [CrossRef] [PubMed]
- Rosas, J.; Hernández, R.; Gascon, A.; Igartua, M.; Guzman, F.; Patarroyo, M.; Pedraz, J. Biodegradable PLGA microspheres as a delivery system for malaria synthetic peptide SPf66. Vaccine 2001, 19, 4445–4451. [Google Scholar] [CrossRef]
- Carcaboso, A.; Hernandez, R.; Igartua, M.; Rosas, J.; Patarroyo, M.; Pedraz, J. Potent, long lasting systemic antibody levels and mixed Th1/Th2 immune response after nasal immunization with malaria antigen loaded PLGA microparticles. Vaccine 2004, 22, 1423–1432. [Google Scholar] [CrossRef] [PubMed]
- Rosas, J.; Pedraz, J.; Hernandez, R.; Gascon, A.; Igartua, M.; Guzman, F.; Rodrı́guez, R.; Cortes, J.; Patarroyo, M. Remarkably high antibody levels and protection against P. falciparum malaria in Aotus monkeys after a single immunisation of SPf66 encapsulated in PLGA microspheres. Vaccine 2002, 20, 1707–1710. [Google Scholar] [PubMed]
- Carcaboso, A.; Hernández, R.; Igartua, M.; Gascón, A.; Rosas, J.; Patarroyo, M.; Pedraz, J. Immune response after oral administration of the encapsulated malaria synthetic peptide SPf66. Int. J. Pharm. 2003, 260, 273–282. [Google Scholar] [CrossRef]
- Carcaboso, Á.M.; Hernández, R.M.; Igartua, M.; Rosas, J.E.; Patarroyo, M.E.; Pedraz, J.L. Enhancing immunogenicity and reducing dose of microparticulated synthetic vaccines: Single intradermal administration. Pharm. Res. 2004, 21, 121–126. [Google Scholar]
- Mata, E.; Igartua, M.; Hernández, R.M.; Rosas, J.E.; Patarroyo, M.E.; Pedraz, J.L. Comparison of the adjuvanticity of two different delivery systems on the induction of humoral and cellular responses to synthetic peptides. Drug Deliv. 2010, 17, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Mata, E.; Carcaboso, A.; Hernandez, R.; Igartua, M.; Corradin, G.; Pedraz, J. Adjuvant activity of polymer microparticles and montanide ISA 720 on immune responses to Plasmodium falciparum MSP2 long synthetic peptides in mice. Vaccine 2007, 25, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Mata, E.; Igartua, M.; Patarroyo, M.E.; Pedraz, J.L.; Hernández, R.M. Enhancing immunogenicity to PLGA microparticulate systems by incorporation of alginate and RGD-modified alginate. Eur. J. Pharm. Sci. 2011, 44, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.A.; Seth, R.K.; Kumar, S.; Ali, R.; Mohan, T.; Biswas, S.; Rao, D. Induction of cell-mediated immune responses to peptide antigens of P. vivax in microparticles using intranasal immunization. Immunol. Investig. 2010, 39, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.A.; Seth, R.K.; Babu, J.; Biswas, S.; Rao, D. Induction of mucosal and systemic humoral immune responses in murine system by intranasal immunization with peptide antigens of P. vivax and CpG oligodeoxynucleotide (ODN) in microparticle delivery. Int. Immunopharmacol. 2009, 9, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Goyal, A.K.; Mishra, N.; Khatri, K.; Vaidya, B.; Mehta, A.; Wu, Y.; Vyas, S.P. Development and characterization of novel carrier gel core liposomes based transmission blocking malaria vaccine. J. Controll. Release 2009, 140, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Van Rooijen, N.; van Nieuwmegen, R. Liposomes in immunology: Multilamellar phosphatidylcholine liposomes as a simple, biodegradable and harmless adjuvant without any immunogenic activity of its own. Immunol. Investig. 1980, 9, 243–256. [Google Scholar] [CrossRef]
- Toh, M.-R.; Chiu, G.N.C. Liposomes as sterile preparations and limitations of sterilisation techniques in liposomal manufacturing. Asian J. Pharm. Sci. 2013, 8, 88–95. [Google Scholar] [CrossRef]
- Barenholz, Y. Liposome application: Problems and prospects. Curr. Opin. Colloid Interface Sci. 2001, 6, 66–77. [Google Scholar] [CrossRef]
- Patravale, V.; Prabhu, P. Potential of nanocarriers in antigen delivery: The path to successful vaccine delivery. Nanocarriers 2014. [Google Scholar] [CrossRef]
- Anderson, J.M.; Shive, M.S. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 2012, 64, 72–82. [Google Scholar] [CrossRef]
- Slütter, B.; Bal, S.; Keijzer, C.; Mallants, R.; Hagenaars, N.; Que, I.; Kaijzel, E.; van Eden, W.; Augustijns, P.; Löwik, C.; et al. Nasal vaccination with N-trimethyl chitosan and PLGA based nanoparticles: Nanoparticle characteristics determine quality and strength of the antibody response in mice against the encapsulated antigen. Vaccine 2010, 28, 6282–6291. [Google Scholar] [CrossRef] [PubMed]
- Elamanchili, P.; Lutsiak, C.M.; Hamdy, S.; Diwan, M.; Samuel, J. “Pathogen-mimicking” nanoparticles for vaccine delivery to dendritic cells. J. Immunother. 2007, 30, 378–395. [Google Scholar] [CrossRef] [PubMed]
- Mallapragada, S.K.; Narasimhan, B. Immunomodulatory biomaterials. Int. J. Pharm. 2008, 364, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Loos, C.; Syrovets, T.; Musyanovych, A.; Mailänder, V.; Landfester, K.; Nienhaus, G.U.; Simmet, T. Functionalized polystyrene nanoparticles as a platform for studying bio-nano interactions. Beilstein J. Nanotechnol. 2014, 5, 2403–2412. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.; Machaidze, G.; Lustig, A.; Aebi, U.; Burkhard, P. Structure-based design of peptides that self-assemble into regular polyhedral nanoparticles. Nanomedicine 2006, 2, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, T.; Kitahara, T.; Fumoto, S.; Nishida, K.; Nakamura, J.; Niidome, T.; Kodama, Y.; Nakagawa, H.; To, H.; Sasaki, H. Ternary complexes of pDNA, polyethylenimine, and γ-polyglutamic acid for gene delivery systems. Biomaterials 2009, 30, 2846–2853. [Google Scholar] [CrossRef] [PubMed]
- Sanders, M.T.; Brown, L.E.; Deliyannis, G.; Pearse, M.J. ISCOMTM-based vaccines: The second decade. Immunol. Cell Biol. 2005, 83, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.J.; Pedersen, G.; Madhun, A.S.; Svindland, S.; Sævik, M.; Breakwell, L.; Hoschler, K.; Willemsen, M.; Campitelli, L.; Nøstbakken, J.K.; et al. Evaluation of a virosomal H5N1 vaccine formulated with Matrix M™ adjuvant in a phase I clinical trial. Vaccine 2011, 29, 8049–8059. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.-X.; Xie, Y.; Ye, Y.-P. ISCOMs and ISCOMATRIX™. Vaccine 2009, 27, 4388–4401. [Google Scholar] [CrossRef] [PubMed]
- Amiji, M.M. Nanotechnology for Cancer Therapy; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2006. [Google Scholar]
- Kunzmann, A.; Andersson, B.; Thurnherr, T.; Krug, H.; Scheynius, A.; Fadeel, B. Toxicology of engineered nanomaterials: Focus on biocompatibility, biodistribution and biodegradation. Biochim. Biophys. Acta 2011, 1810, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Biju, V.; Mundayoor, S.; Omkumar, R.V.; Anas, A.; Ishikawa, M. Bioconjugated quantum dots for cancer research: Present status, prospects and remaining issues. Biotechnol. Adv. 2010, 28, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Ron, H. A toxicologic review of quantum dots: Toxicity depends on physicochemical and environmental factors. Environ. Health Perspect. 2006, 114, 165–172. [Google Scholar]
- Chan, W.C.W.; Nie, S. Quantum dot bioconjugates for ultrasensitive nonisotopic detection. Science 1998, 281, 2016–2018. [Google Scholar] [CrossRef] [PubMed]
- Loo, S.; Moore, T.; Banik, B.; Alexis, F. Biomedical applications of hydroxyapatite nanoparticles. Curr. Pharm. Biotechnol. 2010, 11, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Epple, M.; Ganesan, K.; Heumann, R.; Klesing, J.; Kovtun, A.; Neumann, S.; Sokolova, V. Application of calcium phosphate nanoparticles in biomedicine. J. Mater. Chem. 2010, 20, 18–23. [Google Scholar] [CrossRef]
- Boisselier, E.; Astruc, D. Gold nanoparticles in nanomedicine: Preparations, imaging, diagnostics, therapies and toxicity. Chem. Soc. Rev. 2009, 38, 1759–1782. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ralston, J.; Sedev, R.; Beattie, D.A. Functionalized gold nanoparticles: Synthesis, structure and colloid stability. J. Colloid Interface Sci. 2009, 331, 251–262. [Google Scholar] [CrossRef] [PubMed]
- De Araújo Lopes, S.C.; dos Santos Giuberti, C.; Rocha, T.G.R.; dos Santos Ferreira, D.; Leite, E.A.; Oliveira, M.C. Liposomes as Carriers of Anticancer Drugs. In Cancer Treatment—Conventional and Innovative Approaches; Rangel, L., Ed.; InTech: Rijeka, Croatia, 2013. [Google Scholar]
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 1965, 13, 238–252. [Google Scholar] [CrossRef]
- Koo, O.M.; Rubinstein, I.; Onyuksel, H. Role of nanotechnology in targeted drug delivery and imaging: A concise review. Nanomedicine 2005, 1, 193–212. [Google Scholar] [CrossRef] [PubMed]
- Papahadjopoulos, D.; Allen, T.M.; Gabizon, A.; Mayhew, E.; Matthay, K.; Huang, S.K.; Lee, K.D.; Woodle, M.C.; Lasic, D.D.; Redemann, C. Sterically stabilized liposomes: Improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc. Natl. Acad. Sci. USA 1991, 88, 11460–11464. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Nishida, M.; Maekawa, N.; Yamamura, H.; Tanaka, Y.; Kasai, M.; Taneichi, M.; Uchida, T. An increased adjuvanticity of liposomes by the inclusion of phosphatidylserine in immunization with surface-coupled liposomal antigen. Int. Arch. Allergy Immunol. 2005, 136, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.J.; Suh, H.; Bershteyn, A.; Stephan, M.T.; Liu, H.; Huang, B.; Sohail, M.; Luo, S.; Ho Um, S.; Khant, H.; et al. Interbilayer-crosslinked multilamellar vesicles as synthetic vaccines for potent humoral and cellular immune responses. Nat. Mater. 2011, 10, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Kowalczuk, A.; Trzcinska, R.; Trzebicka, B.; Müller, A.H.; Dworak, A.; Tsvetanov, C.B. Loading of polymer nanocarriers: Factors, mechanisms and applications. Prog. Polymer Sci. 2014, 39, 43–86. [Google Scholar] [CrossRef]
- Yilgor, P.; Hasirci, N.; Hasirci, V. Sequential BMP-2/BMP-7 delivery from polyester nanocapsules. J. Biomed. Mater. Res. 2010, 93, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; le Breton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Controll. Release 2012, 161, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Betancourt, T.; Byrne, J.D.; Sunaryo, N.; Crowder, S.W.; Kadapakkam, M.; Patel, S.; Casciato, S.; Brannon-Peppas, L. PEGylation strategies for active targeting of PLA/PLGA nanoparticles. J. Biomed. Mater. Res. Part A 2009, 91, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Hamdy, S.; Haddadi, A.; Shayeganpour, A.; Samuel, J.; Lavasanifar, A. Activation of antigen-specific T cell-responses by mannan-decorated PLGA nanoparticles. Pharm. Res. 2011, 28, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, B.M.; Schwendeman, S.P. Characterization of the initial burst release of a model peptide from poly(d,l-lactide-co-glycolide) microspheres. J. Controll. Release 2002, 82, 289–307. [Google Scholar] [CrossRef]
- Schroeder, U.; Graff, A.; Buchmeier, S.; Rigler, P.; Silvan, U.; Tropel, D.; Jockusch, B.M.; Aebi, U.; Burkhard, P.; Schoenenberger, C.-A. Peptide nanoparticles serve as a powerful platform for the immunogenic display of poorly antigenic actin determinants. J. Mol. Biol. 2009, 386, 1368–1381. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chen, Y.-H. High epitope density in a single protein molecule significantly enhances antigenicity as well as immunogenicity: A novel strategy for modern vaccine development and a preliminary investigation about B cell discrimination of monomeric proteins. Eur. J. Immunol. 2005, 35, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Dasgupta, D.; Doll, T.A.; Burkhard, P.; Lanar, D.E. Expression, purification and refolding of a self-assembling protein nanoparticle (SAPN) malaria vaccine. Methods 2013, 60, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Scheerlinck, J.-P.Y.; Greenwood, D.L.V. Virus-sized vaccine delivery systems. Drug Discov. Today 2008, 13, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Pearse, M.J.; Drane, D. ISCOMATRIX® adjuvant for antigen delivery. Adv. Drug Deliv. Rev. 2005, 57, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Gupta, M. Synthesis and surface engineering of iron oxide nanoparticles for biomedical applications. Biomaterials 2005, 26, 3995–4021. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.H.; Salabas, E.E.L.; Schüth, F. Magnetic nanoparticles: Synthesis, protection, functionalization, and application. Angew. Chem. Int. Ed. 2007, 46, 1222–1244. [Google Scholar] [CrossRef] [PubMed]
- Alexiou, C.; Arnold, W.; Klein, R.J.; Parak, F.G.; Hulin, P.; Bergemann, C.; Erhardt, W.; Wagenpfeil, S.; Lübbe, A.S. Locoregional cancer treatment with magnetic drug targeting. Cancer Res. 2000, 60, 6641–6648. [Google Scholar] [PubMed]
- Babes, L.; Denizot, B.; Amp, X.; Tanguy, G.; le Jeune, J.J.; Jallet, P. Synthesis of iron oxide nanoparticles used as MRI contrast agents: A parametric study. J. Colloid Interface Sci. 1999, 212, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Laurent, S.; Forge, D.; Port, M.; Roch, A.; Robic, C.; Vander Elst, L.; Muller, R.N. Magnetic iron oxide nanoparticles: Synthesis, stabilization, vectorization, physicochemical characterizations, and biological applications. Chem. Rev. 2008, 108, 2064–2110. [Google Scholar] [CrossRef] [PubMed]
- Luten, J.; van Nostrum, C.F.; de Smedt, S.C.; Hennink, W.E. Biodegradable polymers as non-viral carriers for plasmid DNA delivery. J. Controll. Release 2008, 126, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Graves Patricia, M.; Gelband, H. Vaccines for preventing malaria (SPf66). In Cochrane Database of Systematic Reviews; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2006. [Google Scholar]
- Newman, K.D.; Elamanchili, P.; Kwon, G.S.; Samuel, J. Uptake of poly(d,l-lactic-co-glycolic acid) microspheres by antigen-presenting cells in vivo. J. Biomed. Mater. Res. 2002, 60, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Churcher, T.S.; Blagborough, A.M.; Delves, M.; Ramakrishnan, C.; Kapulu, M.C.; Williams, A.R.; Biswas, S.; Da, D.F.; Cohuet, A.; Sinden, R.E. Measuring the blockade of malaria transmission—An analysis of the standard membrane feeding assay. Int. J. Parasitol. 2012, 42, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Purwada, A.; Roy, K.; Singh, A. Engineering vaccines and niches for immune modulation. Acta Biomater. 2013, 10, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tan, J.; Thomas, A.; Ou-Yang, D.; Muzykantov, V.R. The shape of things to come: Importance of design in nanotechnology for drug delivery. Ther. Deliv. 2012, 3, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Stellacci, F. Effect of surface properties on nanoparticle-cell interactions. Small 2010, 6, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.P.; Geckeler, K.E. Polymer nanoparticles: Preparation techniques and size-control parameters. Prog. Polymer Sci. 2011, 36, 887–913. [Google Scholar] [CrossRef]
- Byrne, J.; Telling, N.; Coker, V.; Pattrick, R.; van der Laan, G.; Arenholz, E.; Tuna, F.; Lloyd, J. Control of nanoparticle size, reactivity and magnetic properties during the bioproduction of magnetite by Geobacter sulfurreducens. Nanotechnology 2011, 22, 455709. [Google Scholar] [CrossRef] [PubMed]
- Sau, T.K.; Pal, A.; Jana, N.; Wang, Z.; Pal, T. Size controlled synthesis of gold nanoparticles using photochemically prepared seed particles. J. Nanoparticle Res. 2001, 3, 257–261. [Google Scholar] [CrossRef]
- Lin, Y.-S.; Haynes, C.L. Synthesis and characterization of biocompatible and size-tunable multifunctional porous silica nanoparticles. Chem. Mater. 2009, 21, 3979–3986. [Google Scholar] [CrossRef]
- Hou, Y.; Kondoh, H.; Ohta, T.; Gao, S. Size-controlled synthesis of nickel nanoparticles. Appl. Surf. Sci. 2005, 241, 218–222. [Google Scholar] [CrossRef]
- Xiang, S.D.; Scholzen, A.; Minigo, G.; David, C.; Apostolopoulos, V.; Mottram, P.L.; Plebanski, M. Pathogen recognition and development of particulate vaccines: Does size matter? Methods 2006, 40, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.F.; Jennings, G.T. Vaccine delivery: A matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 2010, 10, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Gu, W.; Xu, Z.P. Re-considering how particle size and other properties of antigen-adjuvant complexes impact on the immune responses. J. Colloid Interface Sci. 2013, 395, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fifis, T.; Gamvrellis, A.; Crimeen-Irwin, B.; Pietersz, G.A.; Li, J.; Mottram, P.L.; McKenzie, I.F.C.; Plebanski, M. Size-dependent immunogenicity: Therapeutic and protective properties of nano-vaccines against tumors. J. Immunol. 2004, 173, 3148–3154. [Google Scholar] [CrossRef] [PubMed]
- Oyewumi, M.O.; Kumar, A.; Cui, Z. Nano-microparticles as immune adjuvants: Correlating particle sizes and the resultant immune responses. Expert Rev. Vaccines 2010, 9, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Good, M.F.; Doolan, D.L. Malaria vaccine design: Immunological considerations. Immunity 2010, 33, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Mottram, P.L.; Leong, D.; Crimeen-Irwin, B.; Gloster, S.; Xiang, S.D.; Meanger, J.; Ghildyal, R.; Vardaxis, N.; Plebanski, M. Type 1 and 2 immunity following vaccination is influenced by nanoparticle size: Formulation of a model vaccine for respiratory syncytial virus. Mol. Pharm. 2006, 4, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.T.; Rehor, A.; Schmoekel, H.G.; Hubbell, J.A.; Swartz, M.A. In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J. Controll. Release 2006, 112, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Oussoren, C.; Zuidema, J.; Crommelin, D.J.A.; Storm, G. Lymphatic uptake and biodistribution of liposomes after subcutaneous injection: II. Influence of liposomal size, lipid composition and lipid dose. Biochim. Biophys. Acta 1997, 1328, 261–272. [Google Scholar] [CrossRef]
- Kourtis, I.C.; Hirosue, S.; de Titta, A.; Kontos, S.; Stegmann, T.; Hubbell, J.A.; Swartz, M.A. Peripherally administered nanoparticles target monocytic myeloid cells, secondary lymphoid organs and tumors in mice. PLoS ONE 2013, 8, e61646. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.N.; Vokali, E.; Lund, A.W.; Hubbell, J.A.; Swartz, M.A. Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response. Biomaterials 2014, 35, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.T.; van der Vlies, A.J.; Simeoni, E.; Angeli, V.; Randolph, G.J.; O’Neil, C.P.; Lee, L.K.; Swartz, M.A.; Hubbell, J.A. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotech. 2007, 25, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 2008, 38, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Gutierro, I.; Hernández, R.M.; Igartua, M.; Gascón, A.R.; Pedraz, J.L. Size dependent immune response after subcutaneous, oral and intranasal administration of BSA loaded nanospheres. Vaccine 2002, 21, 67–77. [Google Scholar] [CrossRef]
- Li, X.; Sloat, B.R.; Yanasarn, N.; Cui, Z. Relationship between the size of nanoparticles and their adjuvant activity: Data from a study with an improved experimental design. Eur. J. Pharm. Biopharm. 2011, 78, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Chithrani, B.D.; Chan, W.C.W. Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano Lett. 2007, 7, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Wu, S.-H.; Hung, Y.; Mou, C.-Y. Size effect on cell uptake in well-suspended, uniform mesoporous silica nanoparticles. Small 2009, 5, 1408–1413. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Li, J.; Bao, G.; Zhang, S. Variable nanoparticle-cell adhesion strength regulates cellular uptake. Phys. Rev. Lett. 2010. [Google Scholar] [CrossRef]
- Albanese, A.; Tang, P.S.; Chan, W.C.W. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu. Rev. Biomed. Eng. 2012, 14, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, J.; Watanabe, W. Physical and chemical stability of drug nanoparticles. Adv. Drug Deliv. Rev. 2011, 63, 456–469. [Google Scholar] [CrossRef] [PubMed]
- Yeung, T.; Gilbert, G.E.; Shi, J.; Silvius, J.; Kapus, A.; Grinstein, S. Membrane phosphatidylserine regulates surface charge and protein localization. Science 2008, 319, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.D.; Fuchsberger, M.; Karlson, T.D.L.; Hardy, C.L.; Selomulya, C.; Plebanski, M. Nanoparticles, Immunomodulation and Vaccine Delivery. In Handbook of Immunological Properties of Engineered Nanomaterials; Dobrovolskaia, M.A., McNeil, S.E., Eds.; World Scientific Publishing: Hackensack, NJ, USA, 2013; pp. 449–475. [Google Scholar]
- Thomas, C.; Gupta, V.; Ahsan, F. Influence of surface charge of PLGA particles of recombinant hepatitis B surface antigen in enhancing systemic and mucosal immune responses. Int. J. Pharm. 2009, 379, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Henriksen-Lacey, M.; Christensen, D.; Bramwell, V.W.; Lindenstrøm, T.; Agger, E.M.; Andersen, P.; Perrie, Y. Liposomal cationic charge and antigen adsorption are important properties for the efficient deposition of antigen at the injection site and ability of the vaccine to induce a CMI response. J. Controll. Release 2010, 145, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.C.; Xie, J.; Wurm, P.A.; Xia, Y. Understanding the role of surface charges in cellular adsorption versus internalization by selectively removing gold nanoparticles on the cell surface with a I2/KI etchant. Nano Lett. 2009, 9, 1080–1084. [Google Scholar] [CrossRef]
- Villanueva, A.; Canete, M.; Roca, A.G.; Calero, M.; Veintemillas-Verdaguer, S.; Serna, C.J.; del Puerto Morales, M.; Miranda, R. The influence of surface functionalization on the enhanced internalization of magnetic nanoparticles in cancer cells. Nanotechnology 2009. [Google Scholar] [CrossRef] [PubMed]
- Foged, C.; Brodin, B.; Frokjaer, S.; Sundblad, A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int. J. Pharm. 2005, 298, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Badiee, A.; Jaafari, M.R.; Khamesipour, A.; Samiei, A.; Soroush, D.; Kheiri, M.T.; Barkhordari, F.; McMaster, W.R.; Mahboudi, F. The role of liposome charge on immune response generated in BALB/c mice immunized with recombinant major surface glycoprotein of Leishmania (rgp63). Exp. Parasitol. 2009, 121, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Harush-Frenkel, O.; Rozentur, E.; Benita, S.; Altschuler, Y. Surface charge of nanoparticles Determines their endocytic and transcytotic pathway in Polarized MDCK cells. Biomacromolecules 2008, 9, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.T.; Swartz, M.A.; Hubbell, J.A. Targeting dendritic cells with biomaterials: Developing the next generation of vaccines. Trends Immunol. 2006, 27, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Duthie, M.S.; Windish, H.P.; Fox, C.B.; Reed, S.G. Use of defined TLR ligands as adjuvants within human vaccines. Immunol. Rev. 2011, 239, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Demento, S.L.; Siefert, A.L.; Bandyopadhyay, A.; Sharp, F.A.; Fahmy, T.M. Pathogen-associated molecular patterns on biomaterials: A paradigm for engineering new vaccines. Trends Biotechnol. 2011, 29, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.J.; James, E.; Shastri, N.; Fréchet, J.M.J. In vivo targeting of dendritic cells for activation of cellular immunity using vaccine carriers based on pH-responsive microparticles. Proc. Natl. Acad. Sci. USA 2005, 102, 18264–18268. [Google Scholar] [CrossRef] [PubMed]
- Trumpfheller, C.; Finke, J.S.; López, C.B.; Moran, T.M.; Moltedo, B.; Soares, H.; Huang, Y.; Schlesinger, S.J.; Park, C.G.; Nussenzweig, M.C.; et al. Intensified and protective CD4+ T cell immunity in mice with anti-dendritic cell HIV gag fusion antibody vaccine. J. Exp. Med. 2006, 203, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Bonifaz, L.C.; Bonnyay, D.P.; Charalambous, A.; Darguste, D.I.; Fujii, S.-I.; Soares, H.; Brimnes, M.K.; Moltedo, B.; Moran, T.M.; Steinman, R.M. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J. Exp. Med. 2004, 199, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; Tacken, P.J.; Fokkink, R.; Joosten, B.; Stuart, M.C.; Albericio, F.; Torensma, R.; Figdor, C.G. Targeted PLGA nano- but not microparticles specifically deliver antigen to human dendritic cells via DC-SIGN in vitro. J. Controll. Release 2010, 144, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Thomann, J.-S.; Heurtault, B.; Weidner, S.; Brayé, M.; Beyrath, J.; Fournel, S.; Schuber, F.; Frisch, B. Antitumor activity of liposomal ErbB2/HER2 epitope peptide-based vaccine constructs incorporating TLR agonists and mannose receptor targeting. Biomaterials 2011, 32, 4574–4583. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; Tacken, P.J.; Fokkink, R.; Figdor, C.G. The influence of PEG chain length and targeting moiety on antibody-mediated delivery of nanoparticle vaccines to human dendritic cells. Biomaterials 2011, 32, 6791–6803. [Google Scholar] [CrossRef] [PubMed]
- Cedervall, T.; Lynch, I.; Lindman, S.; Berggård, T.; Thulin, E.; Nilsson, H.; Dawson, K.A.; Linse, S. Understanding the nanoparticle-protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc. Natl. Acad. Sci. USA 2007, 104, 2050–2055. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano-bio interface. Nature materials 2009, 8, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Jansch, M.; Stumpf, P.; Graf, C.; Rühl, E.; Müller, R.H. Adsorption kinetics of plasma proteins on ultrasmall superparamagnetic iron oxide (USPIO) nanoparticles. Int. J. Pharm. 2012, 428, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Göppert, T.M.; Müller, R.H. Adsorption kinetics of plasma proteins on solid lipid nanoparticles for drug targeting. Int. J. Pharm. 2005, 302, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Saptarshi, S.R.; Duschl, A.; Lopata, A.L. Interaction of nanoparticles with proteins: Relation to bio-reactivity of the nanoparticle. J. Nanobiotechnol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Monopoli, M.P.; Walczyk, D.; Campbell, A.; Elia, G.; Lynch, I.; Baldelli Bombelli, F.; Dawson, K.A. Physical-chemical aspects of protein corona: Relevance to in vitro and in vivo biological impacts of nanoparticles. J. Am. Chem. Soc. 2011, 133, 2525–2534. [Google Scholar] [CrossRef] [PubMed]
- Klein, J. Probing the interactions of proteins and nanoparticles. Proc. Natl. Acad. Sci. USA 2007, 104, 2029–2030. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tiruppathi, C.; Minshall, R.D.; Malik, A.B. Size and dynamics of caveolae studied using nanoparticles in living endothelial cells. ACS Nano 2009, 3, 4110–4116. [Google Scholar] [CrossRef] [PubMed]
- Fadeel, B.; Garcia-Bennett, A.E. Better safe than sorry: Understanding the toxicological properties of inorganic nanoparticles manufactured for biomedical applications. Adv. Drug Deliv. Rev. 2010, 62, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Seth, A.; Wibowo, N.; Zhao, C.-X.; Mitter, N.; Yu, C.; Middelberg, A.P.J. Nanoparticle vaccines. Vaccine 2014, 32, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Chesko, J.; Kazzaz, J.; Ugozzoli, M.; O’Hagan, D.T.; Singh, M. An investigation of the factors controlling the adsorption of protein antigens to anionic PLG microparticles. J. Pharm. Sci. 2005, 94, 2510–2519. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Powles, L.; Xiang, S.D.; Selomulya, C.; Plebanski, M. The Use of Synthetic Carriers in Malaria Vaccine Design. Vaccines 2015, 3, 894-929. https://doi.org/10.3390/vaccines3040894
Powles L, Xiang SD, Selomulya C, Plebanski M. The Use of Synthetic Carriers in Malaria Vaccine Design. Vaccines. 2015; 3(4):894-929. https://doi.org/10.3390/vaccines3040894
Chicago/Turabian StylePowles, Liam, Sue D. Xiang, Cordelia Selomulya, and Magdalena Plebanski. 2015. "The Use of Synthetic Carriers in Malaria Vaccine Design" Vaccines 3, no. 4: 894-929. https://doi.org/10.3390/vaccines3040894
APA StylePowles, L., Xiang, S. D., Selomulya, C., & Plebanski, M. (2015). The Use of Synthetic Carriers in Malaria Vaccine Design. Vaccines, 3(4), 894-929. https://doi.org/10.3390/vaccines3040894