The Landscape of Tumor-Specific Antigens in Colorectal Cancer


Abstract
:1. Background
2. Immunotherapy in CRC-A Brief Overview
3. Tumor Antigens
3.1. Tumor-Associated Antigens vs Tumor-Specific Antigens
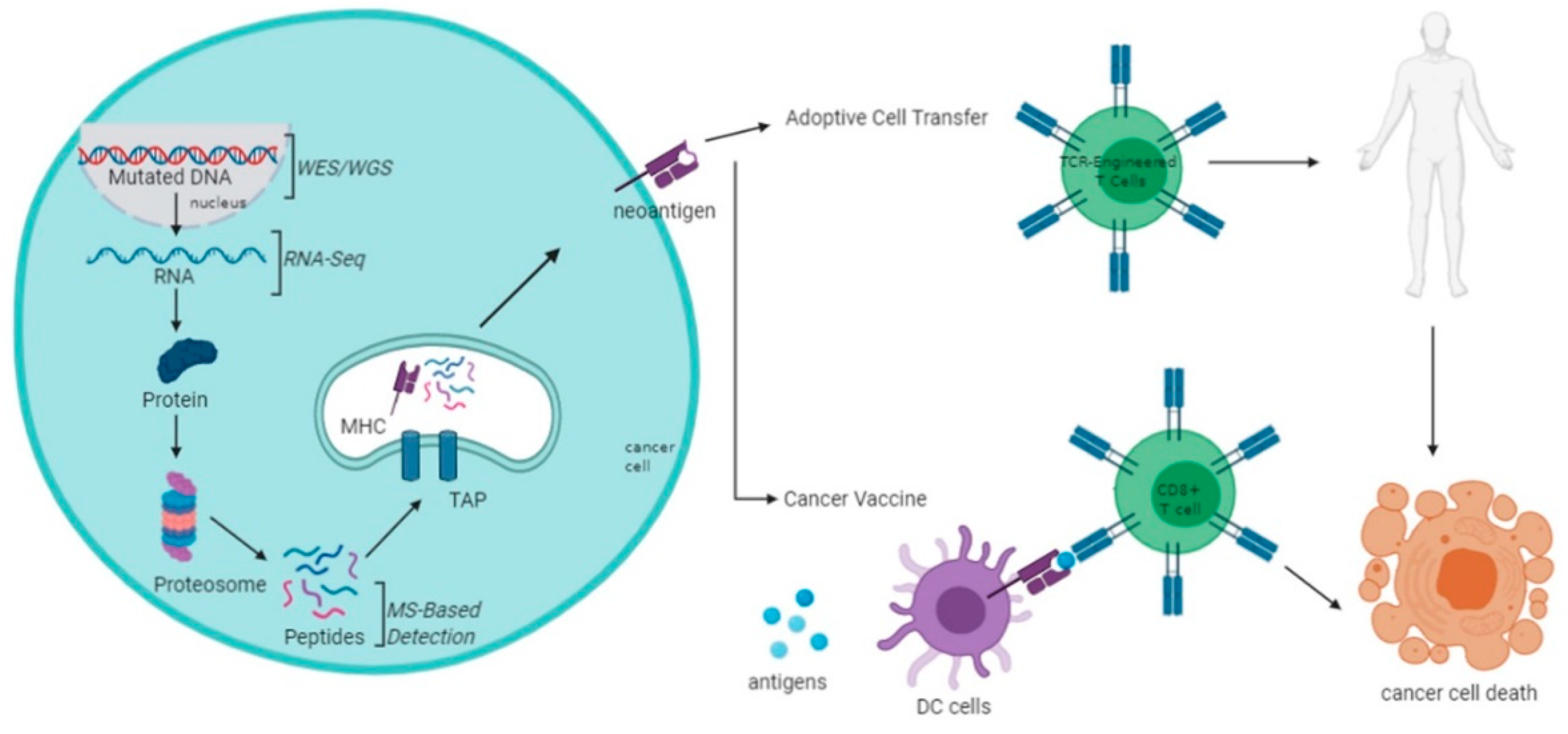
3.2. Biosynthesis of Neoantigens
4. Neoantigen Studies in CRC
4.1. SNVs-Derived Neoantigens
4.2. Frameshift/Indels-Derived Neoantigens in MSI CRC
4.3. Fusion Genes-Derived Neoantigens
4.4. Shared Neoantigens in CRC
5. Clinical Trials Utilizing Tumor-Specific Antigens in CRC
6. An Overview of Common Platforms and Protocol for Neoantigen Prediction
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, M.; Ravula, S.; Tatishchev, S.F.; Wang, H.L. Colorectal carcinoma: Pathologic aspects. J. Gastrointest Oncol. 2012, 3, 153–173. [Google Scholar]
- Rosenberg, S.A. A new era for cancer immunotherapy based on the genes that encode cancer antigens. Immunity 1999, 10, 281–287. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borst, J.; Ahrends, T.; Bąbała, N.; Melief, C.J.M.; Kastenmüller, W. CD4+ T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef]
- Martínez-Lostao, L.; Anel, A.; Pardo, J. How do cytotoxic lymphocytes kill cancer cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef] [Green Version]
- Gotwals, P.; Cameron, S.; Cipolletta, D.; Cremasco, V.; Crystal, A.; Hewes, B.; Mueller, B.; Quaratino, S.; Sabatos-Peyton, C.; Petruzzelli, L.; et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 2017, 17, 286–301. [Google Scholar] [CrossRef]
- Dranoff, G. Cytokines in cancer pathogenesis and cancer therapy. Nat. Rev. Cancer 2004, 4, 11–22. [Google Scholar] [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8 cytotoxic t lymphocytes in cancer immunotherapy: A review. J. Cell Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef]
- Volpe, E.; Sambucci, M.; Battistini, L.; Borsellino, G. Fas–fas ligand: Checkpoint of t cell functions in multiple sclerosis. Front. Immunol. 2016, 7, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, J.H.; Ley, T.J. Lymphocyte mediated cytotoxicity. Annu. Rev. Immunol. 2002, 20, 323–370. [Google Scholar] [CrossRef] [PubMed]
- Pardo, J.; Aguilo, J.I.; Anel, A.; Martin, P.; Joeckel, L.; Borner, C.; Wallich, R.; Müllbacher, A.; Froelich, C.J.; Simon, M.M. The biology of cytotoxic cell granule exocytosis pathway: Granzymes have evolved to induce cell death and inflammation. Microbes Infect. 2009, 11, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Herrick, E.J.; Nicholl, M.B. A possible role for perforin and granzyme b in resveratrol-enhanced radiosensitivity of prostate cancer. J. Androl. 2012, 33, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.D.; Simon, M.M. Are proteinases functional molecules of t lymphocytes? Immunol. Today 1987, 8, 140–142. [Google Scholar] [CrossRef]
- Dougan, M.; Dranoff, G.; Dougan, S.K. Cancer immunotherapy: Beyond checkpoint blockade. Annu. Rev. Cancer Biol. 2019, 3, 55–75. [Google Scholar] [CrossRef]
- Gong, J.; Chehrazi-Raffle, A.; Reddi, S.; Salgia, R. Development of pd-1 and pd-l1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J. Immunother. Cancer 2018, 6, 8. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the pd-1 immunoinhibitory receptor by a novel b7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Altmann, D.M. A nobel prize-worthy pursuit: Cancer immunology and harnessing immunity to tumour neoantigens. Immunology 2018, 155, 283–284. [Google Scholar] [CrossRef] [Green Version]
- Lipson, E.J.; Drake, C.G. Ipilimumab: An anti-ctla-4 antibody for metastatic melanoma. Clin. Cancer Res. 2011, 17, 6958–6962. [Google Scholar] [CrossRef] [Green Version]
- Ottaviano, M.; De Placido, S.; Ascierto, P.A. Recent success and limitations of immune checkpoint inhibitors for cancer: A lesson from melanoma. Virchows Archiv. Int. J. Pathol. 2019, 474, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Trinh, B.; Donath, M.Y.; Läubli, H. Successful treatment of immune checkpoint inhibitor–induced diabetes mellitus with infliximab. Diabetes Care 2019, 42, e153–e154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Jørgensen, L.; Gøtzsche, P.C.; Jefferson, T. Benefits and harms of the human papillomavirus (hpv) vaccines: Systematic review with meta-analyses of trial data from clinical study reports. Syst. Rev. 2020, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Sarvizadeh, M.; Ghasemi, F. Vaccines for colorectal cancer: An update. J. Cell. Biochem. 2019, 120, 8815–8828. [Google Scholar] [CrossRef] [PubMed]
- Anassi, E.; Ndefo, U.A. Sipuleucel-T (provenge) injection: The first immunotherapy agent (vaccine) for hormone-refractory prostate cancer. Pharm. Ther. 2011, 36, 197–202. [Google Scholar]
- Bilusic, M.; Madan, R.A. Therapeutic cancer vaccines: The latest advancement in targeted therapy. Am. J. Ther. 2012, 19, e172–e181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, S.; Mullins, C.S.; Linnebacher, M. Colorectal cancer vaccines: Tumor-associated antigens vs neoantigens. World J. Gastroenterol. 2018, 24, 5418–5432. [Google Scholar] [CrossRef]
- Goldman, B.; DeFrancesco, L. The cancer vaccine roller coaster. Nat. Biotechnol. 2009, 27, 129–139. [Google Scholar] [CrossRef]
- Redeker, A.; Arens, R. Improving adoptive T cell therapy: The particular role of T cell costimulation, cytokines, and post-transfer vaccination. Front. Immunol. 2016, 7, 345. [Google Scholar] [CrossRef] [Green Version]
- Besser, M.J.; Shapira-Frommer, R.; Treves, A.J.; Zippel, D.; Itzhaki, O.; Hershkovitz, L.; Levy, D.; Kubi, A.; Hovav, E.; Chermoshniuk, N.; et al. Clinical responses in a phase ii study using adoptive transfer of short-term cultured tumor infiltration lymphocytes in metastatic melanoma patients. Clin. Cancer Res. 2010, 16, 2646–2655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using t-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, Y.; Naito, M.; Yamada, T.; Aisu, N.; Daibo, K.; Mera, T.; Tanaka, T.; Naito, K.; Yasumoto, K.; Kamigaki, T.; et al. Adoptive chemoimmunotherapy using activated αβ t cells for stage iv colorectal cancer. Anticancer Res. 2016, 36, 3741–3746. [Google Scholar]
- Lurquin, C.; Van Pel, A.; Mariamé, B.; De Plaen, E.; Szikora, J.P.; Janssens, C.; Reddehase, M.J.; Lejeune, J.; Boon, T. Structure of the gene of tum- transplantation antigen P91A: The mutated exon encodes a peptide recognized with ld by cytolytic T cells. Cell 1989, 58, 293–303. [Google Scholar] [CrossRef]
- Traversari, C.; van der Bruggen, P.; Van den Eynde, B.; Hainaut, P.; Lemoine, C.; Ohta, N.; Old, L.; Boon, T. Transfection and expression of a gene coding for a human melanoma antigen recognized by autologous cytolytic t lymphocytes. Immunogenetics 1992, 35, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Van den Eynde, B.; Peeters, O.; De Backer, O.; Gaugler, B.; Lucas, S.; Boon, T. A new family of genes coding for an antigen recognized by autologous cytolytic t lymphocytes on a human melanoma. J. Exp. Med. 1995, 182, 689–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.-Q.; Brasseur, F.; Serrano, A.; De Plaen, E.; van der Bruggen, P.; Boon, T.; Van Pel, A. Cytolytic T lymphocytes recognize an antigen encoded by MAGE-A10 on a human melanoma. J. Immunol. 1999, 162, 6849–6854. [Google Scholar] [PubMed]
- Vigneron, N. Human tumor antigens and cancer immunotherapy. BioMed Res. Int. 2015, 2015, 948501. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.Y.; Looi, K.S.; Tan, E.M. Identification of tumor-associated antigens as diagnostic and predictive biomarkers in cancer. In Methods in Molecular Biology (Clifton, N.J.); Humana Press: Totowa, NJ, USA, 2009; Volume 520, pp. 1–10. [Google Scholar]
- Chen, Y.T.; Scanlan, M.J.; Sahin, U.; Türeci, O.; Gure, A.O.; Tsang, S.; Williamson, B.; Stockert, E.; Pfreundschuh, M.; Old, L.J. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc. Natl. Acad. Sci. USA 1997, 94, 1914–1918. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Yuan, Y.H.; Han, Y.; Liu, Y.X.; Yan, L.; Wang, Y.; Gu, J. Expression profile of cancer-testis genes in 121 human colorectal cancer tissue and adjacent normal tissue. Clin. Cancer Res. 2005, 11, 1809–1814. [Google Scholar] [CrossRef] [Green Version]
- Tarnowski, M.; Czerewaty, M.; Deskur, A.; Safranow, K.; Marlicz, W. Expression of cancer testis antigens in colorectal cancer: New prognostic and therapeutic implications. Dis. Markers 2016, 2016, 1987505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soh, J.E.; Abu, N.; Sagap, I.; Mazlan, L.; Yahaya, A.; Mustangin, M.; Khoo, T.S.; Saidin, S.; Ishak, M.; Ab Mutalib, N.S.; et al. Validation of immunogenic PASD1 peptides against HLA-A*24:02 colorectal cancer. Immunotherapy 2019, 11, 1205–1219. [Google Scholar] [CrossRef] [PubMed]
- Soh, J.E.C.; Abu, N.; Jamal, R. The potential immune-eliciting cancer testis antigens in colorectal cancer. Immunotherapy 2018, 10, 1093–1104. [Google Scholar]
- Wirth, T.C.; Kühnel, F. Neoantigen targeting-dawn of a new era in cancer immunotherapy? Front. Immunol. 2017, 8, 1848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.A.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Schubert, U.; Antón, L.C.; Gibbs, J.; Norbury, C.C.; Yewdell, J.W.; Bennink, J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000, 404, 770–774. [Google Scholar] [CrossRef]
- Lord, J.M.; Davey, J.; Frigerio, L.; Roberts, L.M. Endoplasmic reticulum-associated protein degradation. Semin. Cell Dev. Biol. 2000, 11, 159–164. [Google Scholar] [CrossRef]
- Etlinger, J.D.; Goldberg, A.L. A soluble atp-dependent proteolytic system responsible for the degradation of abnormal proteins in reticulocytes. Proc. Natl. Acad. Sci. USA 1977, 74, 54–58. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef]
- Rock, K.L.; Goldberg, A.L. Degradation of cell proteins and the generation of mhc class i-presented peptides. Annu. Rev. Immunol. 1999, 17, 739–779. [Google Scholar] [CrossRef]
- Vigneron, N.; Van den Eynde, B.J. Insights into the processing of mhc class i ligands gained from the study of human tumor epitopes. Cell. Mol. Life Sci. CMLS 2011, 68, 1503–1520. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, M.; Del Cid, N.; Rizvi, S.M.; Peters, L.R. Mhc class i assembly: Out and about. Trends Immunol. 2008, 29, 436–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulsson, K.; Wang, P. Chaperones and folding of mhc class i molecules in the endoplasmic reticulum. Biochim. Biophys. Acta 2003, 1641, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.-H.; Jang, S.J.; Kim, J.; Sohn, I.; Lee, J.-Y.; Cho, E.J.; Chun, S.-M.; Sung, C.O. Spontaneous mutations in the single ttn gene represent high tumor mutation burden. npj Genom. Med. 2020, 5, 33. [Google Scholar]
- Jia, Q.; Wang, J.; He, N.; He, J.; Zhu, B. Titin mutation associated with responsiveness to checkpoint blockades in solid tumors. JCI Insight 2019, 4, e127901. [Google Scholar] [CrossRef]
- Parkhurst, M.R.; Robbins, P.F.; Tran, E.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Ivey, G.; Li, Y.F.; El-Gamil, M.; Lalani, A.; et al. Unique neoantigens arise from somatic mutations in patients with gastrointestinal cancers. Cancer Discov. 2019, 9, 1022–1035. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, Q.; Wu, R.; Li, B.; Chen, Q.; Zhang, X.; Shi, C. A comprehensive survey of genomic alterations in gastric cancer reveals recurrent neoantigens as potential therapeutic targets. BioMed Res. Int. 2019, 2019, 2183510. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of unique neoantigen qualities in long term pancreatic cancer survivors. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef]
- Wu, J.; Zhao, W.; Zhou, B.; Su, Z.; Gu, X.; Zhou, Z.; Chen, S. Tsnadb: A database for tumor-specific neoantigens from immunogenomics data analysis. Genom. Proteom. Bioinform. 2018, 16, 276–282. [Google Scholar] [CrossRef]
- Mennonna, D.; Maccalli, C.; Romano, M.C.; Garavaglia, C.; Capocefalo, F.; Bordoni, R.; Severgnini, M.; Bellis, G.D.; Sidney, J.; Sette, A.; et al. T cell neoepitope discovery in colorectal cancer by high throughput profiling of somatic mutations in expressed genes. Gut 2017, 66, 454–463. [Google Scholar] [CrossRef] [Green Version]
- Newey, A.; Griffiths, B.; Michaux, J.; Pak, H.S.; Stevenson, B.J.; Woolston, A.; Semiannikova, M.; Spain, G.; Barber, L.J.; Matthews, N.; et al. Immunopeptidomics of colorectal cancer organoids reveals a sparse HLA class I neoantigen landscape and no increase in neoantigens with interferon or mek-inhibitor treatment. J. Immunother. Cancer 2019, 7, 309. [Google Scholar] [CrossRef]
- Orloff, M.; Weight, R.; Valsecchi, M.E.; Sato, T. Immune check point inhibitors combination in melanoma: Worth the toxicity? Rev. Recent Clin. Trials 2016, 11, 81–86. [Google Scholar] [CrossRef]
- Pakkala, S.; Owonikoko, T.K. Immune checkpoint inhibitors in small cell lung cancer. J. Thorac. Dis. 2018, 10, S460–S467. [Google Scholar] [CrossRef] [Green Version]
- Hamanishi, J.; Mandai, M.; Ikeda, T.; Minami, M.; Kawaguchi, A.; Murayama, T.; Kanai, M.; Mori, Y.; Matsumoto, S.; Chikuma, S.; et al. Safety and antitumor activity of anti-pd-1 antibody, nivolumab, in patients with platinum-resistant ovarian cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 4015–4022. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. Pd-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet. Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef]
- Rospo, G.; Lorenzato, A.; Amirouchene-Angelozzi, N.; Magrì, A.; Cancelliere, C.; Corti, G.; Negrino, C.; Amodio, V.; Montone, M.; Bartolini, A.; et al. Evolving neoantigen profiles in colorectal cancers with DNA repair defects. Genome Med. 2019, 11, 42. [Google Scholar] [CrossRef] [Green Version]
- Maby, P.; Tougeron, D.; Hamieh, M.; Mlecnik, B.; Kora, H.; Bindea, G.; Angell, H.K.; Fredriksen, T.; Elie, N.; Fauquembergue, E.; et al. Correlation between density of CD8+ t-cell infiltrate in microsatellite unstable colorectal cancers and frameshift mutations: A rationale for personalized immunotherapy. Cancer Res. 2015, 75, 3446–3455. [Google Scholar] [CrossRef] [Green Version]
- Tougeron, D.; Fauquembergue, E.; Rouquette, A.; Le Pessot, F.; Sesboüé, R.; Laurent, M.; Berthet, P.; Mauillon, J.; Di Fiore, F.; Sabourin, J.-C.; et al. Tumor-infiltrating lymphocytes in colorectal cancers with microsatellite instability are correlated with the number and spectrum of frameshift mutations. Mod. Pathol. 2009, 22, 1186–1195. [Google Scholar] [CrossRef] [Green Version]
- Maby, P.; Galon, J.; Latouche, J.-B. Frameshift mutations, neoantigens and tumor-specific cd8+ t cells in microsatellite unstable colorectal cancers. Oncoimmunology 2015, 5, e1115943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, K.; Taggart, M.W.; Reyes-Uribe, L.; Borras, E.; Riquelme, E.; Barnett, R.M.; Leoni, G.; Lucas, F.A.S.; Catanese, M.T.; Mori, F.; et al. Immune profiling of premalignant lesions in patients with lynch syndrome. JAMA Oncol. 2018, 4, 1085–1092. [Google Scholar] [CrossRef] [Green Version]
- Sæterdal, I.; Bjørheim, J.; Lislerud, K.; Gjertsen, M.K.; Bukholm, I.K.; Olsen, O.C.; Nesland, J.M.; Eriksen, J.A.; Møller, M.; Lindblom, A.; et al. Frameshift-mutation-derived peptides as tumor-specific antigens in inherited and spontaneous colorectal cancer. PNAS 2001, 98, 13255–13260. [Google Scholar] [CrossRef] [Green Version]
- Sæterdal, I.; Gjertsen, M.K.; Straten, P.; Eriksen, J.A.; Gaudernack, G. A tgfβrii frameshift-mutation-derived ctl epitope recognised by HLA-A2-restricted CD8+ T cells. Cancer Immunol. Immunother. 2001, 50, 469–476. [Google Scholar] [PubMed]
- Linnebacher, M.; Gebert, J.; Rudy, W.; Woerner, S.; Yuan, Y.P.; Bork, P.; Doeberitz, M.v.K. Frameshift peptide-derived t-cell epitopes: A source of novel tumor-specific antigens. Int. J. Cancer 2001, 93, 6–11. [Google Scholar] [CrossRef]
- Inderberg, E.M.; Wälchli, S.; Myhre, M.R.; Trachsel, S.; Almåsbak, H.; Kvalheim, G.; Gaudernack, G. T cell therapy targeting a public neoantigen in microsatellite instable colon cancer reduces in vivo tumor growth. OncoImmunology 2017, 6, e1302631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripberger, E.; Linnebacher, M.; Schwitalle, Y.; Gebert, J.; Doeberitz, M.V.K. Identification of an HLA-A0201-restricted ctl epitope generated by a tumor-specific frameshift mutation in a coding microsatellite of the ogt gene. J. Clin. Immunol. 2003, 23, 415–423. [Google Scholar] [CrossRef]
- Schwitalle, Y.; Linnebacher, M.; Ripberger, E.; Gebert, J.; Doeberitz, M.V.K. Immunogenic peptides generated by frameshift mutations in DNA mismatch repair-deficient cancer cells. Cancer Immun. 2004, 4, 2–10. [Google Scholar]
- Garbe, Y.; Maletzki, C.; Linnebacher, M. An msi tumor specific frameshift mutation in a coding microsatellite of MSH3 encodes for HLA-A0201-restricted CD8+ cytotoxic t cell epitopes. PLoS ONE 2011, 6, e26517. [Google Scholar] [CrossRef] [Green Version]
- Linnebacher, M.; Wienck, A.; Boeck, I.; Klar, E. Identification of an MSI-H Tumor-Specific Cytotoxic T Cell Epitope Generated by the (1) Frame of U79260 (FTO). J. Biomed. Biotechnol. 2010, 2010, 841451. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, T.; Fujita, T.; Suzuki, Y.; Okabe, S.; Yuasa, Y.; Iwai, T.; Kawakami, Y. Tumor-specific immunological recognition of frameshift-mutated peptides in colon cancer with microsatellite instability. Cancer Res. 2003, 63, 5564–5572. [Google Scholar]
- Speetjens, F.M.; Lauwen, M.M.; Franken, K.L.; Janssen-van Rhijn, C.M.; van Duikeren, S.; Bres, S.A.; van de Velde, C.J.H.; Melief, C.J.M.; Kuppen, P.J.K.; van der Burg, S.H.; et al. Prediction of the immunogenic potential of frameshift-mutated antigens in microsatellite instable cancer. Int. J. Cancer 2008, 123, 838–845. [Google Scholar] [CrossRef]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Rathe, S.K.; Popescu, F.E.; Johnson, J.E.; Watson, A.L.; Marko, T.A.; Moriarity, B.S.; Ohlfest, J.R.; Largaespada, D.A. Identification of candidate neoantigens produced by fusion transcripts in human osteosarcomas. Sci. Rep. 2019, 9, 358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Lee, K.-W.; Srivastava, R.M.; Kuo, F.; Krishna, C.; Chowell, D.; Makarov, V.; Hoen, D.; Dalin, M.G.; Wexler, L.; et al. Immunogenic neoantigens derived from gene fusions stimulate t cell responses. Nat. Med. 2019, 25, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Zhou, C.; Zhang, Z.; Guan, M.; Zhang, C.; Liu, Z.; Liu, Q. The landscape of tumor fusion neoantigens: A pan-cancer analysis. iScience 2019, 21, 249–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iiizumi, S.; Ohtake, J.; Murakami, N.; Kouro, T.; Kawahara, M.; Isoda, F.; Hamana, H.; Kishi, H.; Nakamura, N.; Sasada, T. Identification of novel hla class ii-restricted neoantigens derived from driver mutations. Cancers 2019, 11, 266. [Google Scholar] [CrossRef] [Green Version]
- Veatch, J.R.; Jesernig, B.L.; Kargl, J.; Fitzgibbon, M.; Lee, S.M.; Baik, C.; Martins, R.; Houghton, A.M.; Riddell, S.R. Endogenous CD4+ T cells recognize neoantigens in lung cancer patients, including recurrent oncogenic KRAS and ERBB2 (Her2) driver mutations. Cancer Immunol. Res. 2019, 7, 910–922. [Google Scholar] [CrossRef] [Green Version]
- Quandt, J.; Schlude, C.; Bartoschek, M.; Will, R.; Cid-Arregui, A.; Schölch, S.; Reissfelder, C.; Weitz, J.; Schneider, M.; Wiemann, S.; et al. Long-peptide vaccination with driver gene mutations in p53 and kras induces cancer mutation-specific effector as well as regulatory T cell responses. OncoImmunology 2018, 7, e1500671. [Google Scholar] [CrossRef]
- Lo, W.; Parkhurst, M.; Robbins, P.F.; Tran, E.; Lu, Y.C. Immunologic recognition of a shared p53 mutated neoantigen in a patient with metastatic colorectal cancer. Cancer Immunol. Res. 2019, 7, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanović, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral t cell responses in phase ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef]
- Robbins, P.F.; Lu, Y.-C.; El-Gamil, M.; Li, Y.F.; Gross, C.; Gartner, J.; Lin, J.C.; Teer, J.K.; Cliften, P.; Tycksen, E.; et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 2013, 19, 747–752. [Google Scholar] [CrossRef]
- Fritsch, E.F.; Rajasagi, M.; Ott, P.A.; Brusic, V.; Hacohen, N.; Wu, C.J. Hla-binding properties of tumor neoepitopes in humans. Cancer Immunol. Res. 2014, 2, 522–529. [Google Scholar] [CrossRef] [Green Version]
- Gubin, M.M.; Artyomov, M.N.; Mardis, E.R.; Schreiber, R.D. Tumor neoantigens: Building a framework for personalized cancer immunotherapy. J. Clin. Investig. 2015, 125, 3413–3421. [Google Scholar] [CrossRef]
- Lee, C.-H.; Yelensky, R.; Jooss, K.; Chan, T.A. Update on tumor neoantigens and their utility: Why it is good to be different. Trends Immunol. 2018, 39, 536–548. [Google Scholar] [CrossRef]
- Szolek, A.; Schubert, B.; Mohr, C.; Sturm, M.; Feldhahn, M.; Kohlbacher, O. Optitype: Precision hla typing from next-generation sequencing data. Bioinformatics 2014, 30, 3310–3316. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.; Lundegaard, C.; Lund, O.; Keşmir, C. The role of the proteasome in generating cytotoxic t-cell epitopes: Insights obtained from improved predictions of proteasomal cleavage. Immunogenetics 2005, 57, 33–41. [Google Scholar] [CrossRef]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large-scale validation of methods for cytotoxic t-lymphocyte epitope prediction. BMC Bioinform. 2007, 8, 424. [Google Scholar] [CrossRef] [Green Version]
- Andreatta, M.; Nielsen, M. Gapped sequence alignment using artificial neural networks: Application to the mhc class i system. Bioinformatics 2016, 32, 511–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. Netmhcpan-4.1 and netmhciipan-4.0: Improved predictions of mhc antigen presentation by concurrent motif deconvolution and integration of ms mhc eluted ligand data. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Hundal, J.; Carreno, B.M.; Petti, A.A.; Linette, G.P.; Griffith, O.L.; Mardis, E.R.; Griffith, M. Pvac-seq: A genome-guided in silico approach to identifying tumor neoantigens. Genome Med. 2016, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Gardina, P.J.; Clark, T.A.; Shimada, B.; Staples, M.K.; Yang, Q.; Veitch, J.; Schweitzer, A.; Awad, T.; Sugnet, C.; Dee, S.; et al. Alternative splicing and differential gene expression in colon cancer detected by a whole genome exon array. BMC Genom. 2006, 7, 325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.C.; Beckermann, K.E.; Bortone, D.S.; De Cubas, A.A.; Bixby, L.M.; Lee, S.J.; Panda, A.; Ganesan, S.; Bhanot, G.; Wallen, E.M.; et al. Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J. Clin. Investig. 2018, 128, 4804–4820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al Bakir, M.; et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet. Oncol. 2017, 18, 1009–1021. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.C.; Selitsky, S.R.; Chai, S.; Armistead, P.M.; Vincent, B.G.; Serody, J.S. Alternative tumour-specific antigens. Nat. Rev. Cancer 2019, 19, 465–478. [Google Scholar] [CrossRef]
- Brightman, S.E.; Naradikian, M.S.; Miller, A.M.; Schoenberger, S.P. Harnessing neoantigen specific cd4 t cells for cancer immunotherapy. J. Leukoc. Biol. 2020, 107, 625–633. [Google Scholar] [CrossRef]
- Liu, S.; Matsuzaki, J.; Wei, L.; Tsuji, T.; Battaglia, S.; Hu, Q.; Cortes, E.; Wong, L.; Yan, L.; Long, M.; et al. Efficient identification of neoantigen-specific t-cell responses in advanced human ovarian cancer. J. Immunother. Cancer 2019, 7, 156. [Google Scholar] [CrossRef]
- Garcia-Garijo, A.; Fajardo, C.A.; Gros, A. Determinants for neoantigen identification. Front. Immunol. 2019, 10, 1392. [Google Scholar] [CrossRef] [Green Version]



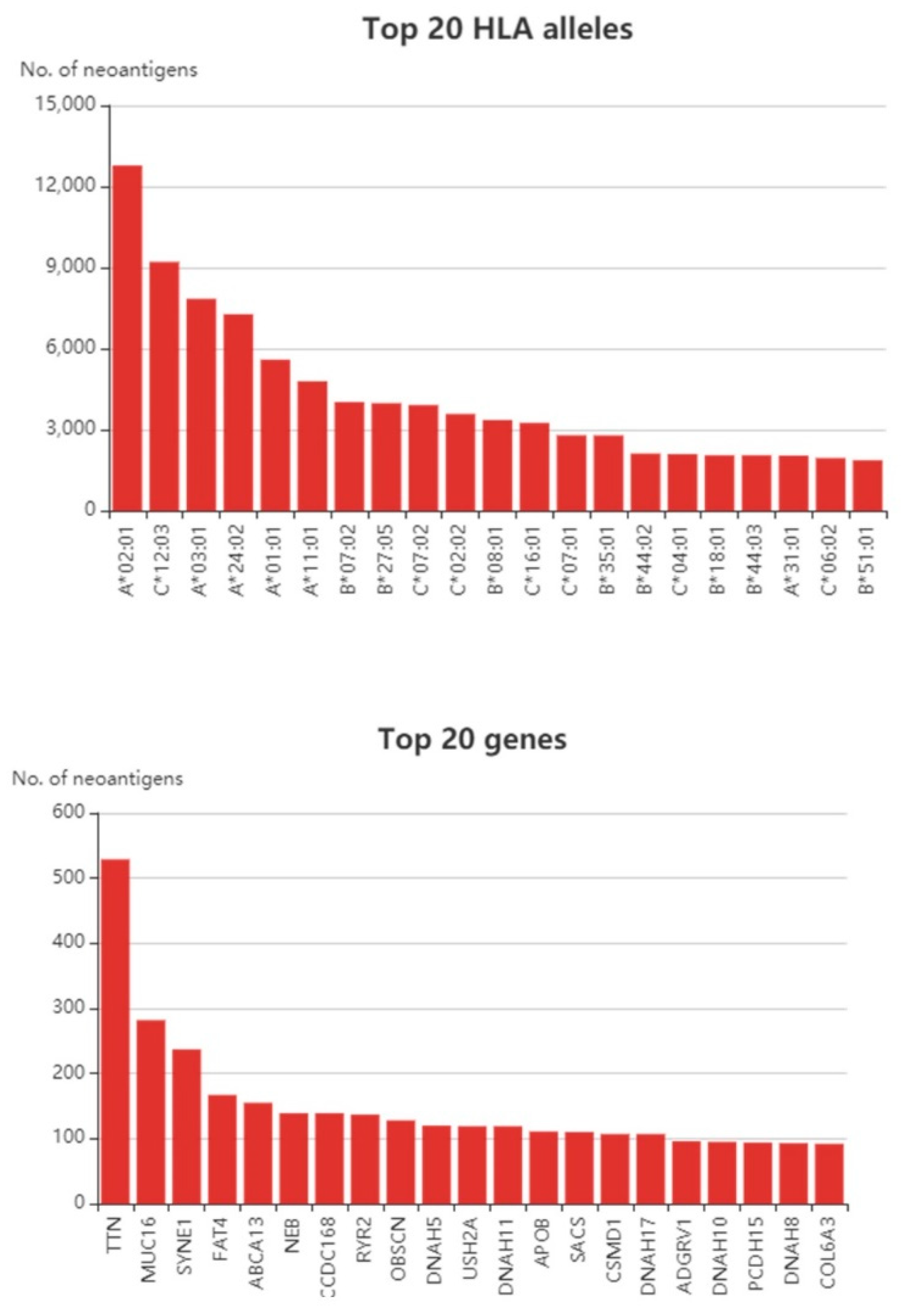{kind=link}
{kind=link}
| Source | Gene | Epitope | HLA | CD4+ or CD8+ T Cells | References |
|---|---|---|---|---|---|
| MSI-CRC | OGT | SLYKFSPFPL | HLA-A0201 | CD8+ | [79] |
| CDX2 | – | – | N/A | [83] | |
| U79260(FTO) | TLSPGWSAV | HLA-A0201 | CD8+ | [82] | |
| TGFβIIR | RLSSCVPVA | HLA-A0201 | CD8+ | [75,76,77,78] | |
| CASP5 | FLIIWQNTM | HLA-A0201 | CD8+ | [80] | |
| CASP8 | ELLVRINRL | HLA-B*08:01 | CD8+ | [85] | |
| MSH03 | – | HLA-A0201 | CD8+ | [81] | |
| MARCKS-1, MARCKS-2 CDX2-2, TAF1B-1, PCNXL2-2, TCF7L2-2 Baxα+1 | – | – | CD8+ | [84] | |
| CRC | SMAD4 | – | – | CD8+ & CD4+ | [61] |
| CRC Organoids | U2SURP | IQEERDERHKR | – | N/A | [62] |
| CRC Organoids | MED25 | SVDANTTL | – | N/A | |
| CRC Organoids | FMO5 | RYVENQRHTI | – | N/A |
| Gene | The Overall, Prevalence in CRC | Mutation Hotspots | Mutation Yielding Possible Neoantigens | Reference/Database |
|---|---|---|---|---|
| KRAS | 42% | G12, G13, Q61 | G12A, G12C, G12D, G12R, G12S, G12 V, G13C, G13D, Q61H, Q61R, Q61 K | [60,89,90] TSNAdb, TCIA |
| BRAF | 12% | V600 | V600E | TSNAdb |
| TP53 | 60% | R175, R248, R273, R213, R282 | R213 L, R213Q, R248Q, R248 W, | TSNAdb [91,92] |
| PIK3CA | 28% | E545 | E545 K | TSNAdb |
| FBXW7 | 17% | R465 | – | TSNAdb |
| Type of Therapy | Strategy | Combined Therapy | Number of Patients | Status | Trial Number |
|---|---|---|---|---|---|
| Vaccine | Personalized neoepitope yeast-based vaccine | – | 16 | Recruiting | NCT03552718 |
| Peptide Vaccine | AIM2(-1)/HT001(-1)/TAF1B(-1) FSP vaccine | Montanide® ISA-51 VG | 22 | Completed | NCT01461148 |
| Peptide vaccine | Personalized Synthetic Tumor-Associated Peptide Vaccine | Imiquimod, Pembrolizumab | 60 | Active | NCT02600949 |
| Peptide Vaccine | mutant KRAS-targeted long peptide vaccine | Nivolumab and Ipilimumab | 30 | Active | NCT04117087 |
| Adoptive Cell Therapy | TCR-engineered T cells against TGFBRII frameshift peptide | – | 1 | Terminated | NCT03431311 |
| Adoptive Cell Therapy | CD8+T cells against personalized peptide antigens | Pembrolizumab | 1 | Active | NCT02757391 |
| Adoptive Cell Therapy | neoantigen-targeting T cells | Nivolumab | 148 | Active | NCT03970382 |
| No. | Tools | Interpretations |
|---|---|---|
| 1. | OptiType [99] | HLA typing |
| 2. | NetChop [100] | Proteasomal peptide processing |
| 3. | NetCTL [101] | TAP transport, proteasomal processing, HLA binding |
| 4. | NetMHC [102] | HLA binding |
| 5. | NetMHCpan [103] | HLA binding across more HLA alleles |
| 6. | pVAC-Seq [104] | RNA expression, HLA binding, DNA mutation |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rus Bakarurraini, N.A.A.; Ab Mutalib, N.S.; Jamal, R.; Abu, N. The Landscape of Tumor-Specific Antigens in Colorectal Cancer. Vaccines 2020, 8, 371. https://doi.org/10.3390/vaccines8030371
Rus Bakarurraini NAA, Ab Mutalib NS, Jamal R, Abu N. The Landscape of Tumor-Specific Antigens in Colorectal Cancer. Vaccines. 2020; 8(3):371. https://doi.org/10.3390/vaccines8030371
Chicago/Turabian StyleRus Bakarurraini, Nurul Ainaa Adilah, Nurul Syakima Ab Mutalib, Rahman Jamal, and Nadiah Abu. 2020. "The Landscape of Tumor-Specific Antigens in Colorectal Cancer" Vaccines 8, no. 3: 371. https://doi.org/10.3390/vaccines8030371
APA StyleRus Bakarurraini, N. A. A., Ab Mutalib, N. S., Jamal, R., & Abu, N. (2020). The Landscape of Tumor-Specific Antigens in Colorectal Cancer. Vaccines, 8(3), 371. https://doi.org/10.3390/vaccines8030371