1. Introduction
The bovine respiratory disease complex (BRDC) remains a major economic problem for both beef and dairy cattle industries in North America and worldwide due to calf mortality, treatment expenses, and additional labor incurred. The United States Department of Agriculture National Animal Health Monitoring Service reported that BRD affects 12.4% of calves during the pre-weaning period, resulting in 22.5% calf mortality. Additionally, 5.9% of post-weaning animals are eventually diagnosed with BRD, causing 46.5% of the mortality during that period [
1]. The detrimental economic impact of BRD on the American beef industry is even larger than in the dairy industry. It is the most expensive disease affecting feedlot cattle, and it is estimated to cause losses of approximately one billion dollars per year in the USA [
2]. BRDC frequently involves an initial viral respiratory infection followed by a secondary bacterial infection, i.e.,
Mannheimia haemolytica (
M. haemolytica). The initial viral respiratory infection creates a favorable condition for colonization of the lungs, usually by
M. haemolytica, resulting in severe pneumonia and death of infected cattle, especially in the feedlots [
3].
Among the respiratory viral agents implicated in BRDC, BoHV-1, and BVDV play significant roles because both viruses cause immunosuppression. BoHV-1 downregulates MHC-I [
4,
5], causes abortive infection and loss of CD4
+ T lymphocytes [
6], and interferes with the migration of lymphocytes and macrophages to the site of infection by counteracting chemokine activity [
7]. BVDV causes leukopenia by infecting and killing lymphocytes and plasma cells [
8]. Consequently, initial BoHV-1 and BVDV infections facilitate secondary bacterial infections that lead to death [
8,
9,
10]. Furthermore, BoHV-1 causes lifelong latency in trigeminal ganglia (TG) with intermittent reactivation and nasal virus shedding [
9,
11], whereas BVDV causes persistently infected animals that shed large amounts of virus [
12]. As a result, both viruses are maintained in the cattle population [
13].
BoHV-1 encodes at least two immunosuppressive envelope proteins, U
L49.5 and glycoprotein (g) G. The U
L49.5, a non-glycosylated alphaherpesvirus gN homolog, transiently down-regulates MHC class I antigen presentation, which allows the virus to escape T cell recognition and clearance of the infected cells [
4,
5,
14]. Similarly, BoHV-1 gG and its homologs in alphaherpesviruses bind to different chemokines secreted by the infected cells and interfere with activated migration of lymphocytes and neutrophils to the site of infection [
7]. Consequently, gG disrupts chemokine gradients allowing survival of the infected cell. BVDV is also well-skilled in evading the host’s innate and adaptive immunity. Most viruses have only one possibility when they infect a host: either “hit and run” or “infect and persist”. The BVDV has mastered both strategies: (i) it counteracts innate immunity, primarily by inhibiting interferon production; (ii) it causes a transient leukopenia by infecting and killing the T-lymphocytes and macrophages; (iii) it doesn’t harm its persistently infected (PI), immunotolerant host for its survival and maintenance; (iv) the PI animals shed large amounts of virus, which infects naïve animals, usually subclinically, over a short time; and (v) it can mutate rapidly [
15,
16]. These properties of both viruses are retained in the current modified-live virus (MLV) vaccine strains [
9,
11,
17].
Current vaccinations against these viral diseases utilize polyvalent vaccines containing BoHV-1, BVDV, and BRSV in two formats: MLV or killed virus (KV) [
18] vaccines. Additionally, in the EU countries, live and killed BoHV-1 envelope glycoprotein E (gE) gene-deleted vaccines are mandated instead of the traditional MLV and KV vaccines [
9,
11]. Like the BoHV-1 wild-type (wt) virus, BoHV-1 MLV vaccines establish lifelong latency in the TG and cause nasal virus shedding following latent reactivation. Similarly, BVDV MLV vaccines can also persistently infect calves and have an added risk of mutating or reverting to virulence. Also, BoHV-1 and BVDV MLV vaccine strains retain the immunosuppressive traits of their respective parental wt strains [
9,
11,
17]. Assessment of the effect of widespread BVDV vaccination over several decades is disappointing since this effort has failed to lower BVDV prevalence [
15,
19]. This failure is due to the unique biology of BVDV infection, which was not fully understood for a long time, and is still widely underestimated [
20]. Together, these problems associated with the current vaccines have further complicated the BRDC epidemiology in the field and perhaps contributed to outbreaks of abortion and/or respiratory infections in the vaccinated animals [
9,
11,
21,
22,
23,
24].
While MLV vaccines’ safety is of concern, the efficacy of inactivated vaccines is not adequate because they do not induce a cellular immune response. In one instance, an effort to influence the cellular immune response of the BVDV inactivated vaccine resulted in bovine neonatal pancytopenia [
25,
26]. BoHV-1 glycoprotein E deleted [Δ] marker vaccine is safer than the MLV because it is not transmitted from the vaccinated to the non-vaccinated animals, rarely shed following latency reactivation, and the vaccinated animals are distinguishable from the infected animals. However, based on the protective efficacy measured by neutralization antibody titers following vaccination, the gEΔ marker vaccine was less efficacious than the traditional MLV, glycoprotein C (gC)-, and Thymidine kinase (TK)-deleted vaccines [
27]. To improve the gEΔ marker vaccine’s vaccine efficacy, we engineered a BoHV-1 triple gene-mutated virus (BoHV-1 TMV). In the BoHV-1 TMV; (i) the coding sequences of the U
L49.5 ectodomain residues 30–32 and the entire cytoplasmic tail residues 80–96 were deleted, and (ii) the coding sequences for the gE cytoplasmic tail residues 452–575 (372 bp), the gE-Us9 intergenic region and the entire Us9 ORF (541 bp) were deleted [
28]. We compared the vaccine efficacy of the BoHV-1 TMV with that of a gEΔ virus against a virulent BoHV-1 wt challenge and determined that its protective efficacy was significantly better than the gEΔ vaccine [
28] while retaining the safety and serological marker properties of the gEΔ virus [
29]. Our results showed that both after vaccination and challenge, BoHV-1 TMV generated a considerably better cellular immune response in calves. After the BoHV-1 wt challenge and compared with that of the sham- and gEΔ-vaccinated calves, BoHV-1 TMV-vaccinated calves; (i) had more rapid and significant increases in neutralizing antibody titers and (ii) had a markedly reduced and shorter duration of nasal virus shedding [
28].
In the present study, our goal was to use the BoHV-1 TMV as a delivery vector for a BVDV subunit vaccine. Initially, we generated a construct designated BoHV-1 TMV-BVDV.E21, encoding a BVDV-1 envelope glycoprotein E2. However, BoHV-1 TMV-BVDV-1.E2 vaccine virus was less effective than a commercial MLV (Bovi-Shield Gold
® IBR-BVD; Zoetis) against a virulent BVDV-1 challenge, and thus, it needed improvement. Therefore, we additionally deleted the chemokine binding, gG envelope protein. In the resulting quadruple gene-deleted BoHV-1 (BoHV-1 QMV), the gG-dependent blockade of chemokine signaling for immune evasion was eliminated. The modified BoHV-1 QMV vector was used to generate novel construct whereby the chimeric genes encoding BVDV-2 E2 and Flag-tagged Erns-bovine granulocyte-macrophage colony-stimulating factor (GM-CSF) fusion (Erns-GM-CSF) proteins were inserted in the gE cytoplasmic tail (CT)-Us9 and gG deletion loci, respectively. GM-CSF is known to enhance both humoral and cellular immune responses in viral vaccines [
30]. The results presented here demonstrate that BoHV-1 QMV expressing the BVDV-2 E2 and Erns-GM-CSF (QMV-E2/Erns-GM-CSF), hereafter designated as QMV-BVD2*, is a safe and effective vaccine for the protection of calves against BVDV-2. The QMV-BVD2* prototype subunit vaccine induced the BoHV-1 and BVDV-2 neutralizing antibody responses along with BVDV-1 and -2 cross-reactive cellular immune responses. Moreover, after a virulent BVDV-2 challenge, the QMV-BVD2* prototype subunit vaccine conferred a more rapid recall BVDV-2-specific neutralizing antibody response and a considerably better recall BVDV types 1 and 2-cross protective cellular immune responses than that of a commercial MLV trivalent (BoHV-1, BVDV-1 and -2 strains) MLV (Zoetis Bovi-shield Gold 3).
2. Materials and Methods
2.1. Cells
The Madin Darby bovine kidney (MDBK) cell line was maintained in Dulbecco’s modified Eagles medium (DMEM # 10-017-CV, Corning®, Corning, NY, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; EquaFETAL, Atlas Biologicals, Fort Collins, CO, USA) and 1× antibiotic/antimycotic solution (cat. # 30-004-CI; Corning®).
2.2. Viruses
BoHV-1 wt Cooper (Colorado-1) strain was obtained from the American Type Culture Collection (#VR-864, ATCC
®, Manassas, VA, USA), and low passage viral stocks were maintained at −80 °C. BoHV-1 TMV was generated previously [
28]. The cytopathic (cp) BVDV-1a Singer strain was received from LSU Louisiana Animal Disease Diagnostic Laboratory (LAADL). BVDV-1b cp strain TGAC was received from Dr. C. Chase from South Dakota State University [
18]. BVDV-2a (cp) strain 125 was kindly provided by Dr. Clayton Keling, the University of Nebraska, at Lincoln, Nebraska. BVDV-1b non-cytopathic (ncp) strain CA04011866a (designated hereafter as CA), and ncp BVDV-2a strains 890 and 1373 were obtained from USDA/APHIS, Ames, IA, USA.
2.3. Antibodies
BVDV types 1 and 2, E2-specific monoclonal antibody (mAb; # 348) and BVDV-2 E2-specific mAb (# BA-2) were from vmrd® (Pullman, WA, USA). Anti-Flag-specific mAb (# F1804) was from Sigma-Aldrich® (St. Louis, MO, USA). Donkey anti-mouse highly cross-absorbed secondary antibody conjugated, Alexa Fluor 488 (# A-21202) was from Invitrogen (Carlsbad, CA, USA).
2.4. Virus Titrations
Virus titration, in the cases of BoHV-1 and cytopathic BVDV-2 strain 125 (125) was performed by plaque assay. Each viral stock solution was serially diluted ten-fold in DMEM supplemented with 5% FBS and 1× antibiotic/antimycotic solution. 200 µL of each virus-dilution was applied in duplicate onto the wells of 24-well cell culture plates over confluent MDBK cells. The plates were incubated for 2 h at 37 °C in a CO
2 incubator before cells were overlaid with 1.6% carboxyl methylcellulose (CMC-high viscosity, Sigma-Aldrich
®, # C5013) in DMEM. After 48 h (BoHV-1) and 72 h (BVDV-2), cells were fixed with 10% formalin solution for 1 h at room temperature (RT) and stained with 0.35% crystal violet. Plaques were counted under a surgical microscope. Virus titer was expressed as plaque-forming units (PFUs)/mL by using the following calculation: Reciprocal of the highest virus dilution × average number of plaques (5–20 plaques) counted in the two wells × 5. The viral plaque assay of BVDV-2 (ncp) strain 890 was performed similarly as above (for 125), but the cells were fixed at RT for 20 min (3% paraformaldehyde solution in PBS), and the viral plaques were visualized by immunofluorescence [
31] assay using the BVDV-specific (both types 1 and 2) mAb # 348 (vmrd
®).
2.5. Construction of BoHV-1 QMV Vector Virus
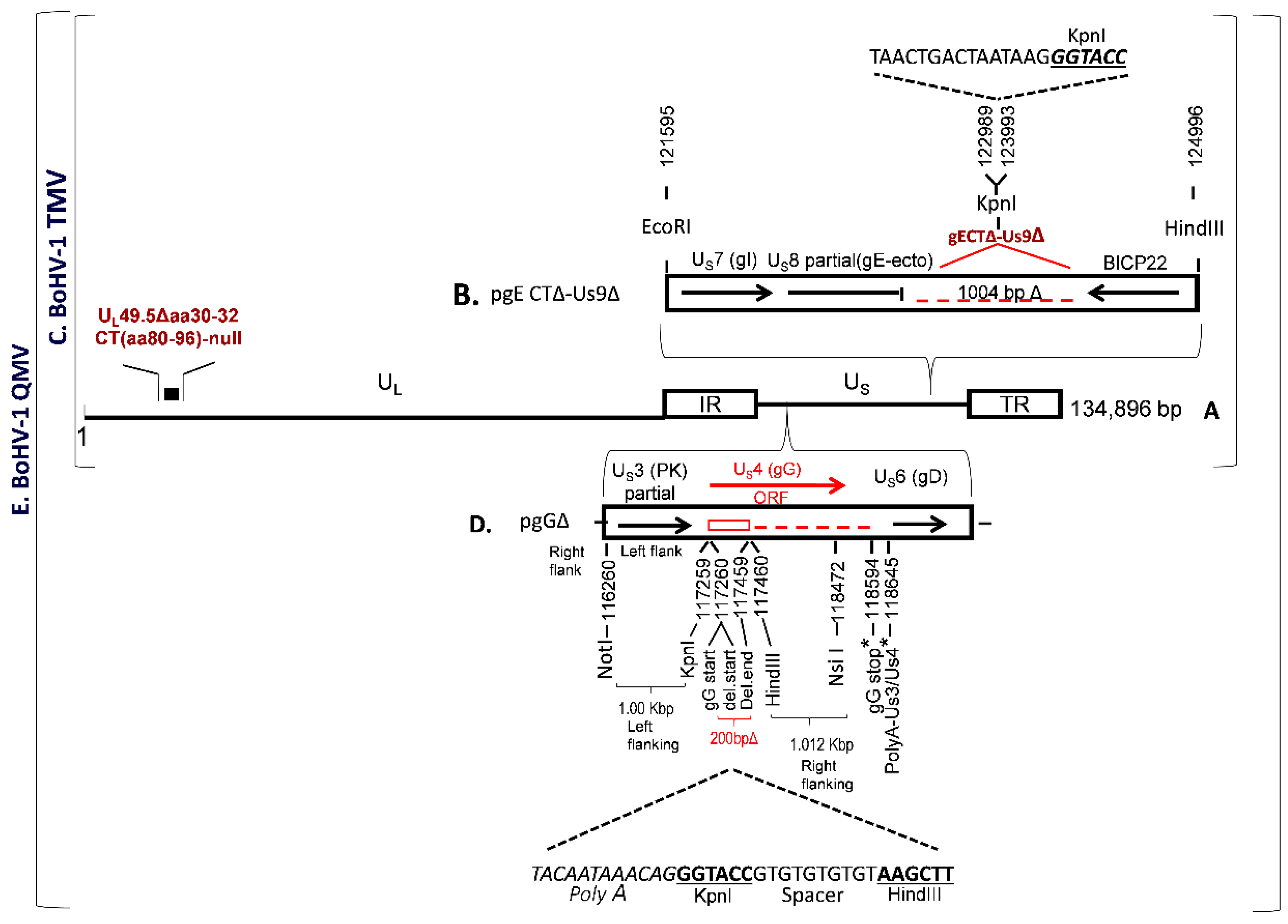
BoHV-1 TMV was constructed earlier, in which (i) UL49.5 residues 30–32 and CT residues 80–96 were deleted and (ii) the entire gE CT-Us9 coding regions were deleted (
Figure 1A–C). To further improve the immunogenicity of BoHV–1 TMV, we deleted/null–mutated the gG gene in the BoHV–1 TMV genome and generated a BoHV–1 QMV. The BoHV–1 gG (Us4) is flanked by Us3 and Us6 (gD) genes, on the left and right, respectively (
Figure 1D). The nucleotide numbers are shown in
Figure 1D, which corresponds to the GenBank accession number J×898220.
Earlier, we could not isolate a viable gG ORF–deleted virus when the entire gG ORF coding region was deleted. We suspected that the putative gD gene promoter sequence (an essential viral gene) might be partially overlapping with the gG ORF sequences at the carboxy end. Alternatively, it could be that the deletion might have affected the shared Us3/Us4 Poly A site (
Figure 1D), situated down–stream of the gG stop codon (nt 118640–118645). Therefore, our strategy was (i) to delete only the coding region of amino–terminal 67 amino acids, including the start codon of gG (
Figure 1D); and (ii) to incorporate a synthetic Poly A, replacement, sequence in the deletion site to compensate for the authentic Us3 Poly–A, and the KpnI and HindIII restriction sites (
Figure 1D) for the insertion of a BVDV–2 Erns–GM–CSF chimeric gene (see below). We designed a 2055 bp long sequence, containing 5’–3’ as follows: The 1000 bp US3 (partial) and US3–gG intergenic sequence (nt 116260–117259) with a NotI restriction site at the 5’ end, followed by 34 bp sequence containing 5’–3’, a replacement Us3 PolyA sequence as it appears in the BoHV–1 genomic sequence with its 3 bp flanking sequence on either side (nt 1186637 to 118648), a KpnI restriction site, a 10 bp spacer sequence and a HindIII restriction site. The HindIII restriction site is flanked on the right by the 1012 bp carboxy–terminal gG ORF sequence (nt 117460–118472) followed by a NsiI restriction site at the 3’ end (
Figure 1D). According to this configuration of the designed 2055 bp long sequence, a 200 bp gG ORF sequence coding for the gG amino–terminal 67 amino acids, including the start codon (nt 117260–117459) were deleted, and the downstream gG amino acid residues 68–444 (nt 117460–118620) were not translated. Further, the gG deletion locus was flanked by 1000 bp on the left and 1160 bp BoHV–1 genomic sequences on the right sides, respectively, for homologous recombination into the BoHV–1 genome (
Figure 1D). The 2055 bp NotI–Nsi fragment designed above was synthesized and cloned into the corresponding sites of the plasmid pBME–amp (Biomatik Corporation, Kitchener, ON, Canada). The resulting plasmid clone’s integrity, pgGΔ, was verified by sequencing (Biomatik Corporation). A BoHV–1 QMV was generated by cotransfection/ homologous recombination of pgGΔ with the full–length BoHV–1 TMV DNA using Lipofectamine (Invitrogen) as described earlier [
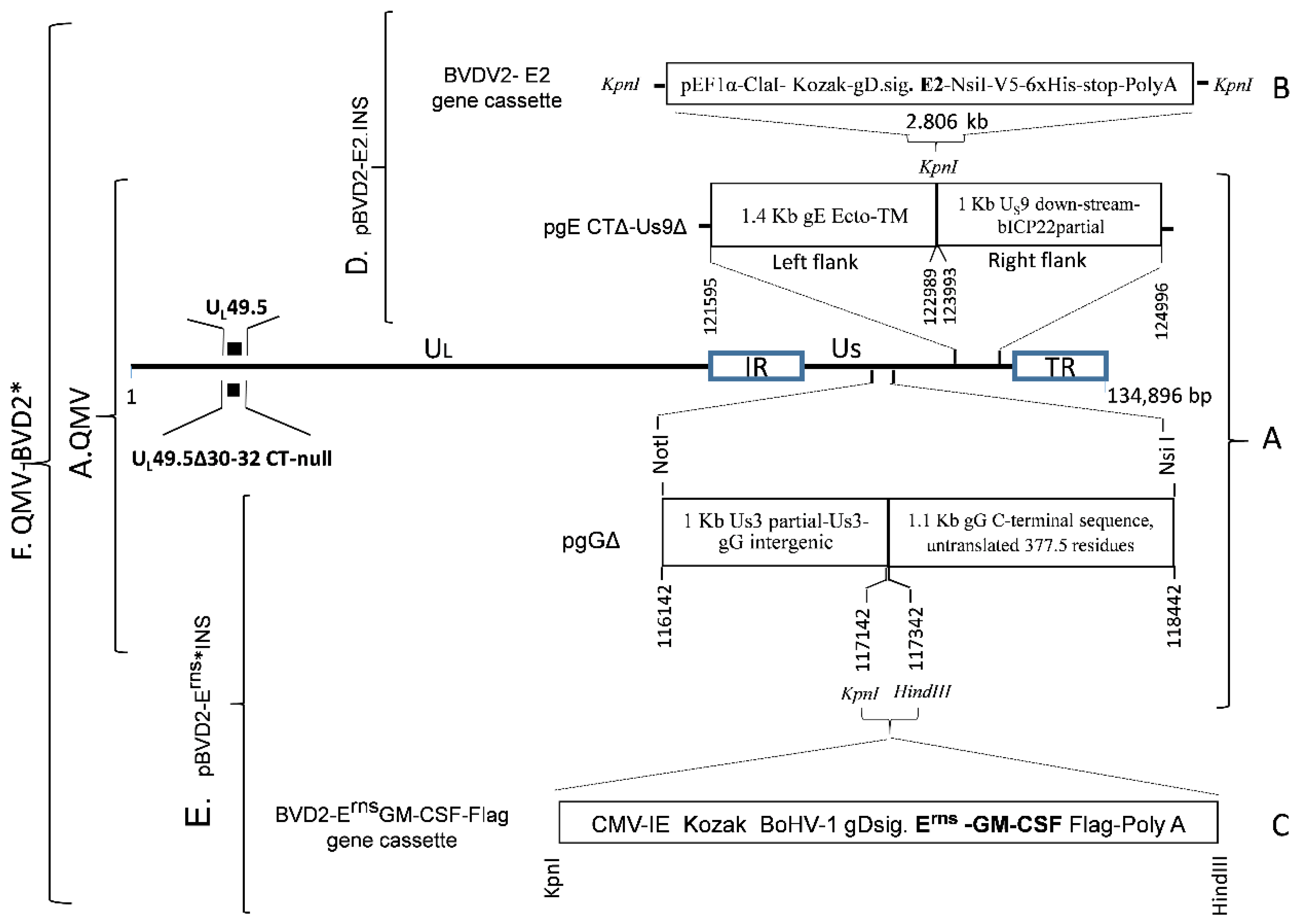
32]. Several putative BoHV–1 QMV recombinants were analyzed by sequencing the genomic region spanning the Us3–gD genes. One of the recombinants was selected for the insertion of the BVDV–2 E2 and Erns–GM–CSF chimeric genes (
Figure 2).
2.6. Incorporation of Chimeric BVDV 2–E2 Gene Cassette in the BoHV–1 QMV Genome to Generate BoHV–1 QMV–BVD2.E2
2.6.1. Construction of BVDV–2 E2 Insertion Plasmid
The plasmid pgE CTΔ–Us9Δ was generated previously [
28]. Briefly, it contains a 2400 bp BoHV–1 genomic sequence inserted into the EcoRI–HindIII sites of plasmid pGEM–7Z (
Figure 1B). The EcoRI–HindIII fragment consisted of gE ORF sequence, nt 121595–122989 bp coding for 451 gE amino–terminal, gE Ecto– and gE transmembrane amino acids flanked by EcoRI and KpnI at the 5’ and 3’ ends, respectively, were fused to 1004 nt of partial BoHV-1 infected cell protein (bICP) 22 gene sequence flanked by KpnI and HindIII at the 5’ and 3’ ends respectively (
Figure 1B). In this configuration, a 1004 bp BoHV–1 genomic sequence (nt 122989–123993) comprised of gE CT amino acids 452–575 (approx. 372 bp), 106 bp gE–Us9 intergenic region, the entire Us9 ORF coding sequence (435 bp), and 88 bp of the Us9–bICP22 intergenic region were deleted (GenBank accession # JX898220). Further, a 21 bp sequence containing four stop codons and a Poly A signal, as well as a KpnI restriction site, was incorporated in the gE CT–Us9 deletion locus (
Figure 1B).
To construct a BVDV–2 E2 insertion plasmid, first, a 2806 bp BVDV–2 E2 chimeric gene cassette (pBVD2–E2 gene cassette) was synthesized (GenScript, Piscataway, NJ, USA), which consisted of the following: A 1286 bp sequence for human elongation factor– 1α (EF–1α) promotor flanked by KpnI (5’) and ClaI (3’) restriction sites, followed by a 1183 bp chimeric sequence containing, the Kozak sequence (
CGCCGCCACC), BoHV–1 gD signal sequence (nt 118819 to 118875, #JX898220; aa 1–19, GenBank accession # AFB76672.1), and BVDV–2 E2 ORF coding sequence, codon–optimized for
Bos Taurus (GenBank accession #AAC72814.1), followed by a 337 bp NsiI–KpnI fragment containing the V5 epitope, 6xHis coding sequence, a stop codon (TGA) and bovine growth hormone (BGH) Poly A sequence (
Figure 2B;
Figure S1). The chimeric BVDV–2 E2 gene cassette was cloned into the KpnI site of pUC 57, and the integrity of the inserted sequence was verified (GenScript).
The 2806 bp chimeric BVDV–2 E2 gene cassette was then recloned into the KpnI site of the pgE CTΔ–Us9Δ plasmid clone described above (
Figure 2A). The resulting plasmid clone, pBoHV–1 gEΔCT US9Δ–BVDV2 E2–INS (pBVD2–E2.INS) (
Figure 2D), contains the 2806 bp BVDV–2 E2 chimeric gene flanked by 1400 bp (on the left) and 1000 bp (on the right) BoHV–1 genomic sequences for recombination and incorporation of the BVDV–2 chimeric E2 gene into the gE CT–Us9 deletion site of BoHV–1 genome (
Figure 2A,B,D). The integrity of the flanking BoHV–1 and the inserted chimeric E2 sequences was verified (Genelab, Pathobiological Sciences, Louisiana State University, Baton Rouge, LA, USA).
2.6.2. Construction of BoHV–1 QMV–E2.2 (QMV–BVD2.E2) Virus by Homologous Recombination
To generate a BoHV–1 QMV–E2 virus, linearized pBVD2–E2.INS insertion vector DNA was transfected [
32] with the full–length BoHV–1 QMV genomic DNA. Several putative recombinant viruses were identified by PCR. One putative BoHV–1 QMV–E2 recombinant was plaque purified, and the integrity of the flanking BoHV–1 genomic and the chimeric E2 gene sequences were verified by sequencing (Genelab, LSU).
2.7. Incorporation of Chimerc BVDV–2 Erns–GM–CSF.Flag Gene Cassette in the QMV–BVD2.E2 Genome to Generate QMV–BVD2– E2.Erns–GM–CSF.Flag (Designated Here After as QMV–BVD2*)
2.7.1. Construction of BVDV2–Erns–GM–CSF.Flag Insertion Plasmid
To construct a BVDV–2 E
rns–GMCSF.Flag insertion plasmid (pBVD2–Erns*–INS), first a 2037 bp BVDV–2 Erns–GMCSF–Flag chimeric gene cassette (
Figure 2C) was synthesized (Biomatik) as follows: A 605 bp human cytomegalovirus (HCMV) promotor sequence (nt 1 to 605, GenBank # CVU55763) with a KpnI restriction site at the 5’ end, followed by a 1183 bp nucleotide sequence containing a kozak sequence (
CGCCGCCACC), BoHV–1 gD signal sequence (nt 118819 to 118875, # JX898220; aa 1–19, # AFB76672.1), codon–optimized (
Bos Taurus) nucleotide sequences for BVDV–2 1373 Erns (aa 271 to 497, # AAD38683) and bovine GM–CSF, lacking the signal sequence (Methionine and residues 18 to 143, #NP776452), the nucleotide sequence coding for a flag tag (GACTACAAAGACGATGA–CGACAAG), a stop codon (TAA), the simian virus 40 (SV40) termination/polyadenylation (Poly A) site (nt 1411 to 1640, # CVU55763), and the restriction site for HindIII (
Figure 2C;
Figure S2). The chimeric BVDV2 ErnsGM–CSF.Flag gene cassette was cloned into the KpnI–HindIII restriction sites of plasmid pgGΔ (
Figure 2A), and the integrity of the inserted sequence was verified (Biomatik). In the resulting plasmid clone pBVD2 Erns–GM–CSF.Flag insertion plasmid (pBVD2–Erns*–INS), the 2037 bp BVDV–2 Erns–GM–CSF chimeric gene sequence (
Figure 2E) was flanked by 1000 bp on the left and 1160 bp BoHV–1 genomic sequences on the right sides, respectively. The integrity of the flanking BoHV–1 and the inserted chimeric Erns–GM–CSF sequences was verified (Genelab, LSU).
2.7.2. Construction of BoHV–1 QMV–E2/Erns–GM–CSF Virus (QMV–BVD2*)
To generate a recombinant QMV–BVD2* vaccine virus, linearized pBVD–2 Erns*INS was cotransfected with full–length QMV–BVDV–2.E2 recombinant (constructed above in
Section 2.6.2) genomic DNA. Two putative QMV–BVD2* recombinant viruses were plaque purified (3x) and verified by PCR and sequence analyses (Genelab, LSU). To determine the stability of QMV–BVD2*, the recombinant virus was passaged several times in MDBK cells and tested again for the expression of BVDV–2 E2 and Erns–GMCSF.Flag (Flag–tagged Erns–GMCSF). Low passage QMV–BVD2* viral stocks were maintained at −80 °C.
2.8. Mock–And Virus–Infected Cell Lysates, SDS PAGE, and Immunoblotting
For Western blot analysis of chimeric E2 and Erns–GM–CSF expression by QMV–BVD2*, MDBK cells were mock–infected or infected with QMV–BVD2*, BoHV–1 wt, and non–cytopathic (ncp) BVDV–2 890. Cells were harvested by centrifugation/pelleting (2095×
g at 4° C) in a Beckman Coulter centrifuge (Avanti J–54) after 24–36 h post–infection (hpi) when the cytopathic effect was 95–100% (QMV–BVD2* and BoHV–1 wt) or 5 days post–infection (BVDV–2 strain 890). Each cell pellet was resuspended in a lysis buffer (50 mM Tris, pH 8.0, 0.5 M NaCl, 5 mM EDTA, 1% Triton X–100 and 0.1% SDS) at 40% weight/volume to adjust/ normalize the differences between the amounts of the samples of cells and proteins. After 1 hr incubation of the cell lysates on ice, the solubilized proteins in the supernatant were collected by centrifugation (21,130×
g for 15 min, at 4 °C). The supernatants were aliquoted and stored at −80° C. For SDS–PAGE/ immuno–blotting analysis, 25 μL of each cell lysate was mixed 1:1 with 2× sample buffer (250 mM Tris, pH 6.8, 4% SDS, 10% glycerol, 0.2% Bromophenol Blue and 2% β–mercaptoethanol), boiled for 5 min and loaded on the 10% SDS–PAGE gel. To detect the chimeric BVDV E2 (
Figure 2B) and Flag–tagged Erns–GMCSF (
Figure 2C) expressed by the QMV–BVD2* recombinant virus (
Figure 2F), immunoblotting with the BVDV–2 E2–specific Mab (vmrd
®, # BA–2) and anti–Flag–specific Mab (Sigma–Aldrich # F1804) respectively, was performed as described earlier [
33].
2.9. Comparison of QMV–BVD2* Growth Characteristics with That of BoHV–1 wt in MDBK Cells
To compare the growth characteristics of QMV–BVD2* with that of BoHV–1 wt, we determined average plaque morphologies and one–step growth curves of QMV–BVD2* and BoHV–1 wt. Two wells of a six–well plate containing a confluent monolayer of MDBK cells were infected with 80–100 PFU of QMV–BVD2* or BoHV–1 wt viruses and overlaid with 1.6% CMC at 2 h post–infection (2 hpi). At 48 hpi, the cells were fixed (10% formaldehyde) and stained with crystal violet. The average plaque size of wt and mutant viruses was determined by measuring approx. 50 randomly selected plaques for each virus under a microscope with a graduated ocular objective, as described earlier [
14]. The one–step virus growth property of the QMV–BVD2* was compared with wt, as described earlier [
34]. Virus titers were determined by standard plaque assay as described above in 2.4.
2.10. Animals and Experimental Design
Animal infection, handling, sample collection, and euthanasia protocols were previously approved by the LSU Institutional Animal Care and Use Committee (Protocol # 14-078). Fifteen, four to five–month–old cross–bred steer, bull, or heifer calves were obtained from a BVDV free supplier. Before inclusion in the study, the calves were tested for BoHV–1 and BVDV serum neutralizing (SN) antibody titers (4–<4) and nasal BVDV shedding to ensure BoHV–1/BVDV free status. Five calves with >4 BVDV–2 maternal SN antibody titers were selected for the control group. Five calves of the remaining 10 were allocated randomly to each of the two treatment groups. Group 1 (QMV–BVD* group) and group 2 (Bovi–Shield Gold® IBR–BVD; Zoetis; designated hereafter as the “Bovi” group) were housed in pens located in the School of Veterinary Medicine (closed) large animal isolation barn. Two pens, holding either two or three calves from each of the two vaccine groups, were well isolated (more than 100 feet apart). Foot baths were located at the main entry and in front of the entrance to each pen. Five calves selected for the control group (group 3) had slightly higher maternal serum neutralizing titers (16–32). They were housed individually in separate pens at an open–air barn with a concrete floor and restricted access. The barn housing the control calves was approx. 100 yards away from the other barn, and a foot bath was located at the main entrance.
2.11. Vaccination and Challenge
Vaccination, challenge, and sample collection scheme are shown in
Figure 3. After one week of acclimatization, QMV–BVD2* group was vaccinated both intranasally (IN) with 2 × 10
7 PFU/nostril and subcutaneously (SQ) with 1 × 10
7 PFU. The calves in the “Bovi” group were vaccinated SQ according to manufactures recommendations, and calves in the control group were sham vaccinated intranasally with 1.0 mL of cell culture media. The calves in QMV–BVD2* and “Bovi” groups received the Micotil
® 300 (Tilmicosin; 20 mg/kg body weight) by SQ injection to prevent secondary bacterial infection. At 34 days post–vaccination (dpv), animals of all groups were challenged intranasally with a total of 2 × 10
6 PFU (1 × 10
6 PFU/mL/nostril) of ncp BVDV–2 890. The animals received Microtil
® 300 subcutaneously as above. The experiment was terminated at 54 dpv/20 days post–challenge (dpc). Upon euthanasia, a complete necropsy was performed to investigate any gross pathological lesions and collect tissue samples.
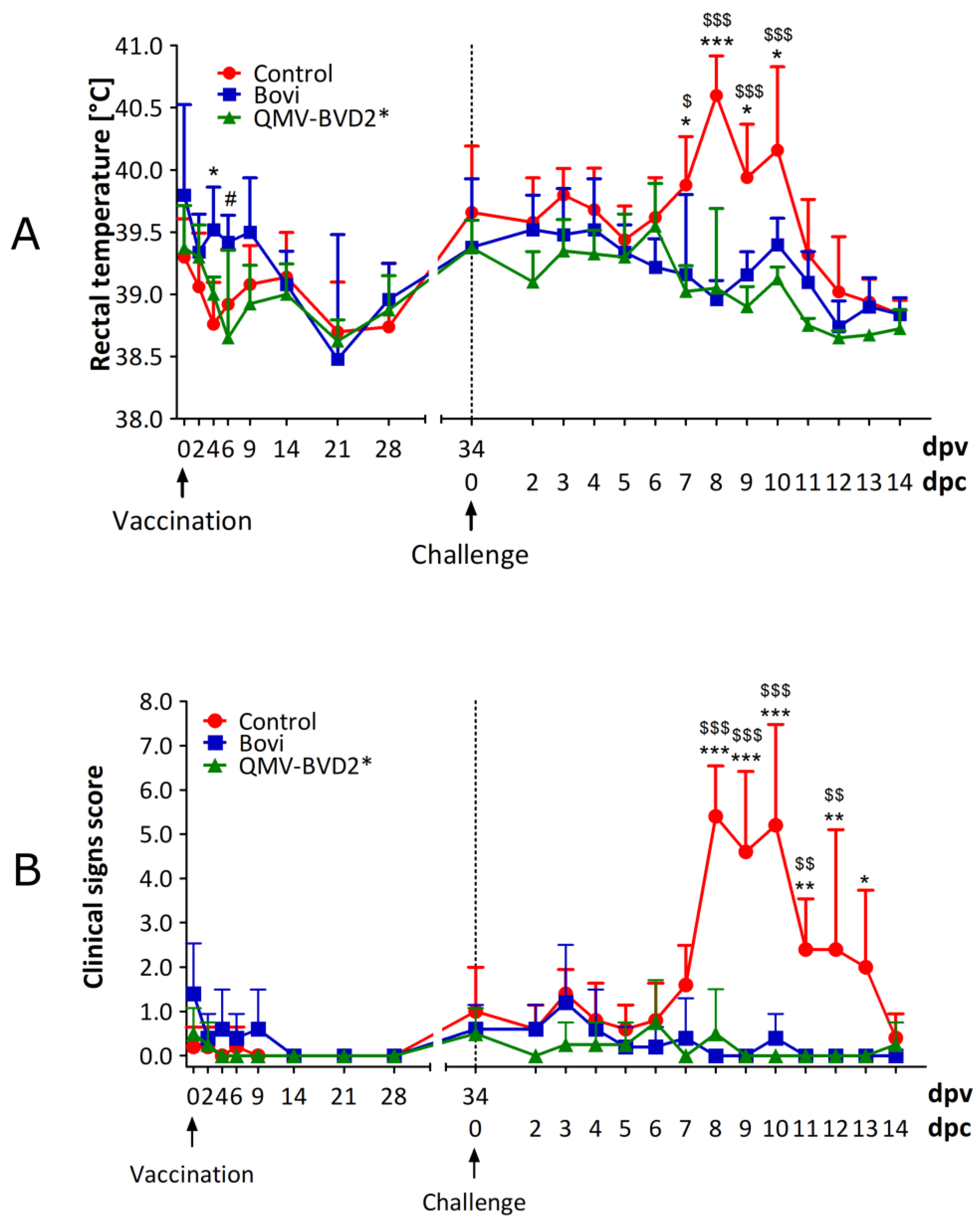
2.12. Clinical Assessment of Calves
Calves were clinically assessed for the rectal temperature, feed, and water intake, on the day of vaccination (0 dpv) and 2, 4, 6, 9, 14, 21, and 28 dpv and on 34 dpv/0 dpc (
Figure 3). Following the challenge with ncp BVDV–2 890, clinical signs were recorded daily until 14 dpc (
Figure 3). The clinical assessment included rectal temperature, nasal and ocular discharge, dyspnea, coughing, lethargy, anorexia, mucosal and oral lesions, and diarrhea. Clinical scores for each animal were calculated based on the criteria listed in
Table S1.
2.13. Sample Collection and Processing
The schedule of EDTA–blood, serum, and nasal swab collection is shown in
Figure 3. Nasopharyngeal swabs were collected in 1 ml of cell culture media, supplemented with 2× antibiotic/antimycotic solution, on 0, 2, 4, 6, 7, 9, 14, and 21 dpv, and 0, 2, 4, 6, 8, and 11 dpc (
Figure 3). The samples were processed and stored at −80 °C. Virus titrations by plaque assay were performed as described above in
Section 2.4.
2.14. Isolation and Freezing of PBMC
PBMCs were isolated using Ficoll–Paque (Ficoll–PaqueTM PLUS, GE healthcare, Chicago, IL, USA) density–gradient centrifugation as previously described [
35]. For freezing, isolated PBMCs were resuspended in 10% FBS–RPMI–1640 medium containing 10% dimethyl sulfoxide (DMSO; Sigma–Aldrich) at a concentration of 5 × 10
6 cells/ml. Aliquots of PBMCs were subjected to slow freezing at −80 °C (overnight) before transferring to a liquid nitrogen tank for long–term storage.
2.15. Leukocyte Counting in Whole Blood–EDTA Samples
For counting the leukocytes in whole blood–EDTA samples, an automatic hematological analyzer (Advia 120; Siemens Healthcare Diagnostics, Tarrytown, NY, USA) was used. On the day of challenge (0 dpc) and on 4, 6, 8, 11, and 14 dpc, total leukocyte counts were determined and recorded. In addition, the percent decline in leukocyte numbers in each calf was calculated as follows and described earlier [
36]:
A decline in the leukocyte count of more than 25% was considered leukopenia [
37].
2.16. Virus–Neutralization Assay
Sera were heat–inactivated at 56 °C for 30 min. 250 µL of BoHV–1 wt Cooper or BVDV–2 125 virus suspension containing approx. 100 PFUs/100 µL were preincubated with 250 µL of serial four–fold serum dilutions (for BVDV–2) or serial two–fold serum dilutions (for BoHV–1) at 37 °C for 2 h. Similarly, 250 µL of plain cell culture media was incubated with 250 µL of the respective virus suspensions in 6–8 tubes (virus control) and incubated at 37 °C for 2 h. 200 Two hundred microliters of the serum–virus mixture from each serum dilution were added to two wells (duplicate) of 24–well cell culture plates containing confluent MDBK cells. For the virus control, 200 µL of virus–media mixtures were added to 6–8 wells of 24–well plates. After 2 h incubation at 37 °C, 0.8 mL of 1.6% CMC in DMEM was added to each well. The plates were incubated in a CO2 incubator at 37 °C for two days for the BoHV–1 and 4 days for the BVDV–2 plaque assays. After fixing the cells with 10% formalin (1–2 h) and washing with tap water, the cells were stained with 0.35% crystal violet solution (20 min). The viral plaques in the serum–virus mixture wells and their respective virus control wells were counted under a microscope. The reciprocal of the highest dilution of each serum that inhibited/neutralized 50% of the average number of the respective control virus plaques, but not less than 40–45 plaques, was reported as the virus–neutralization titer.
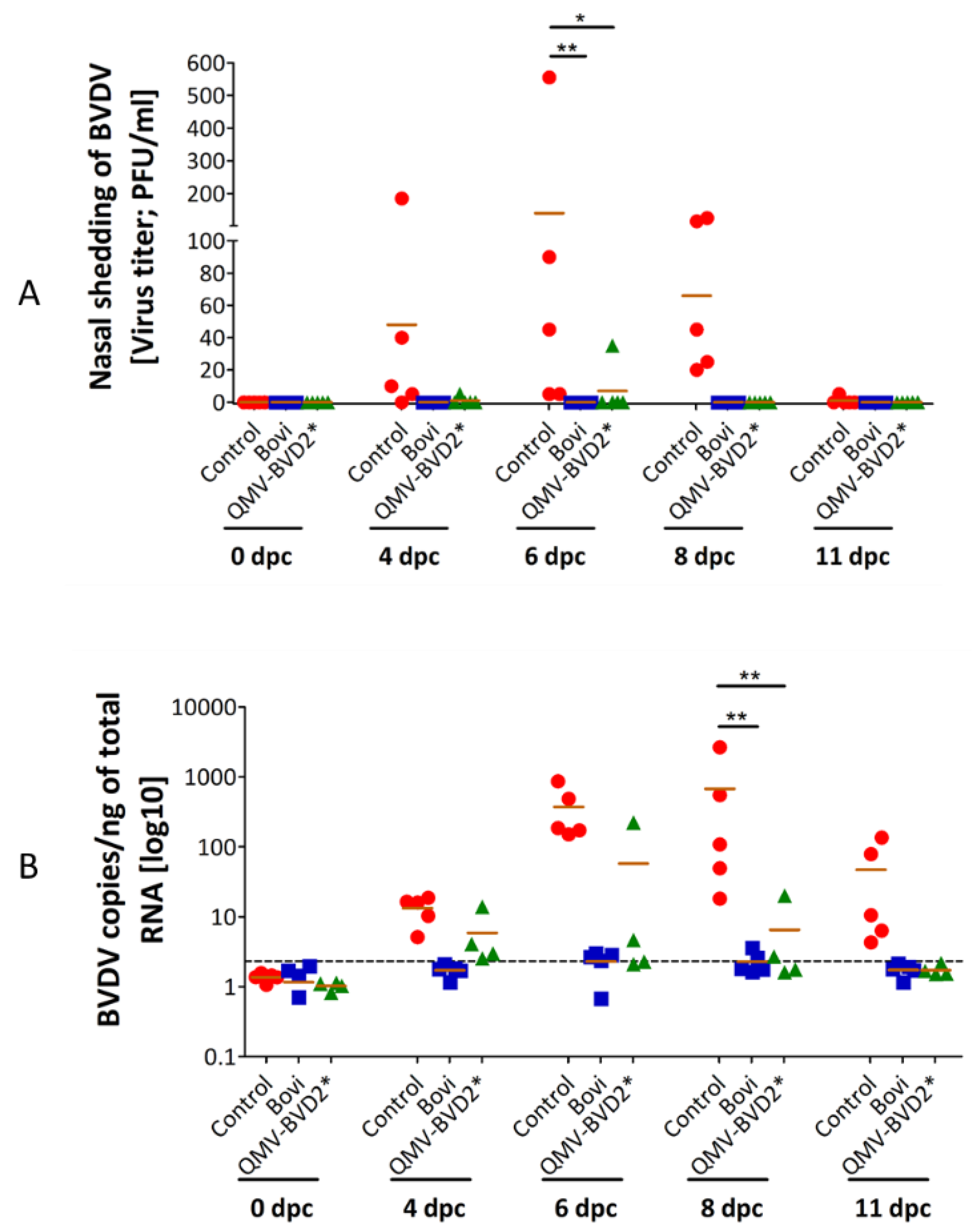
2.17. Reverse Transcription Quantitative Real–Time PCR (RT–qPCR) for BVDV Viremia
To detect BVDV, we performed RT–qPCR on post–challenge PBMCs samples. Briefly, total RNAs were extracted from the PBMCs of calves at 0, 4, 6, 8, and 11 dpc, using RNA easy extraction kit (Qiagen®, Hilden, North Rhine-Westphalia, Germany) according to the manufacturer’s recommendations. cDNA was generated from 250 ng of total RNA followed by RT–qPCR using the VetMax–Gold BVDV detection kit (Thermo Fisher Scientific, Waltham, MA, USA, # 4413938). RNA isolation was performed two times, and the RT–qPCR analysis was repeated three times in duplicate for each sample. BVDV genome load was calculated following the manufacturer’s instruction. According to the manufacturer’s instruction, 1 µL of the positive control (25 × BVDV RNA) contains 10,000 copies of BVDV. To generate a standard curve, 8 µL of the positive control were serial diluted 10–fold. Standard samples corresponding to 4, 40, 400, and 4000 copies were included in each PCR analysis. BVDV copy numbers in each sample were calculated according to the standard curve’s CT–values, divided by 250 to BVDV genome in one ng total RNA. All samples that had a copy number lower than the highest copy number detected in samples from 0 dpc (Threshold of 2.32 copies/ng total RNA) were evaluated as negative.
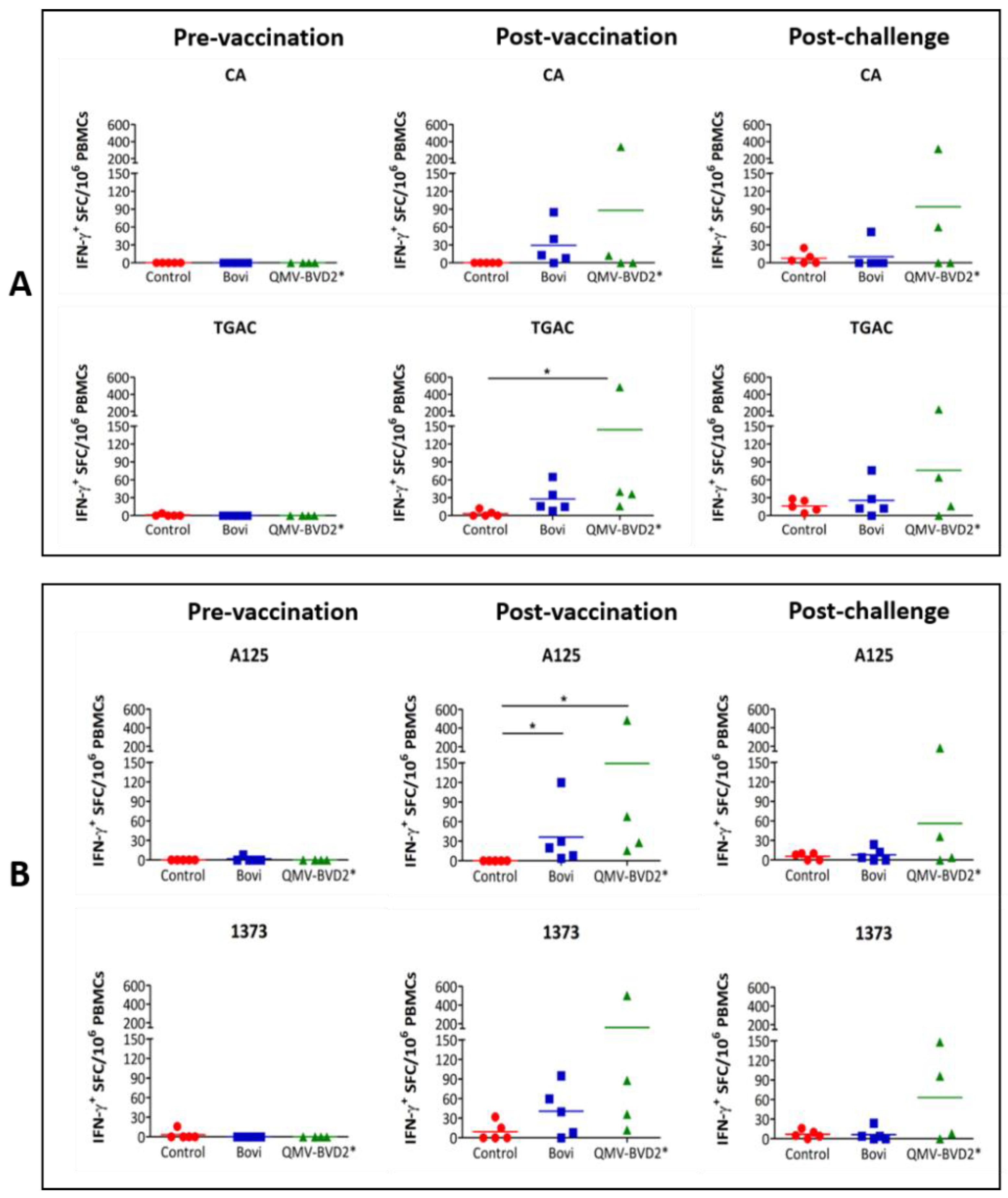
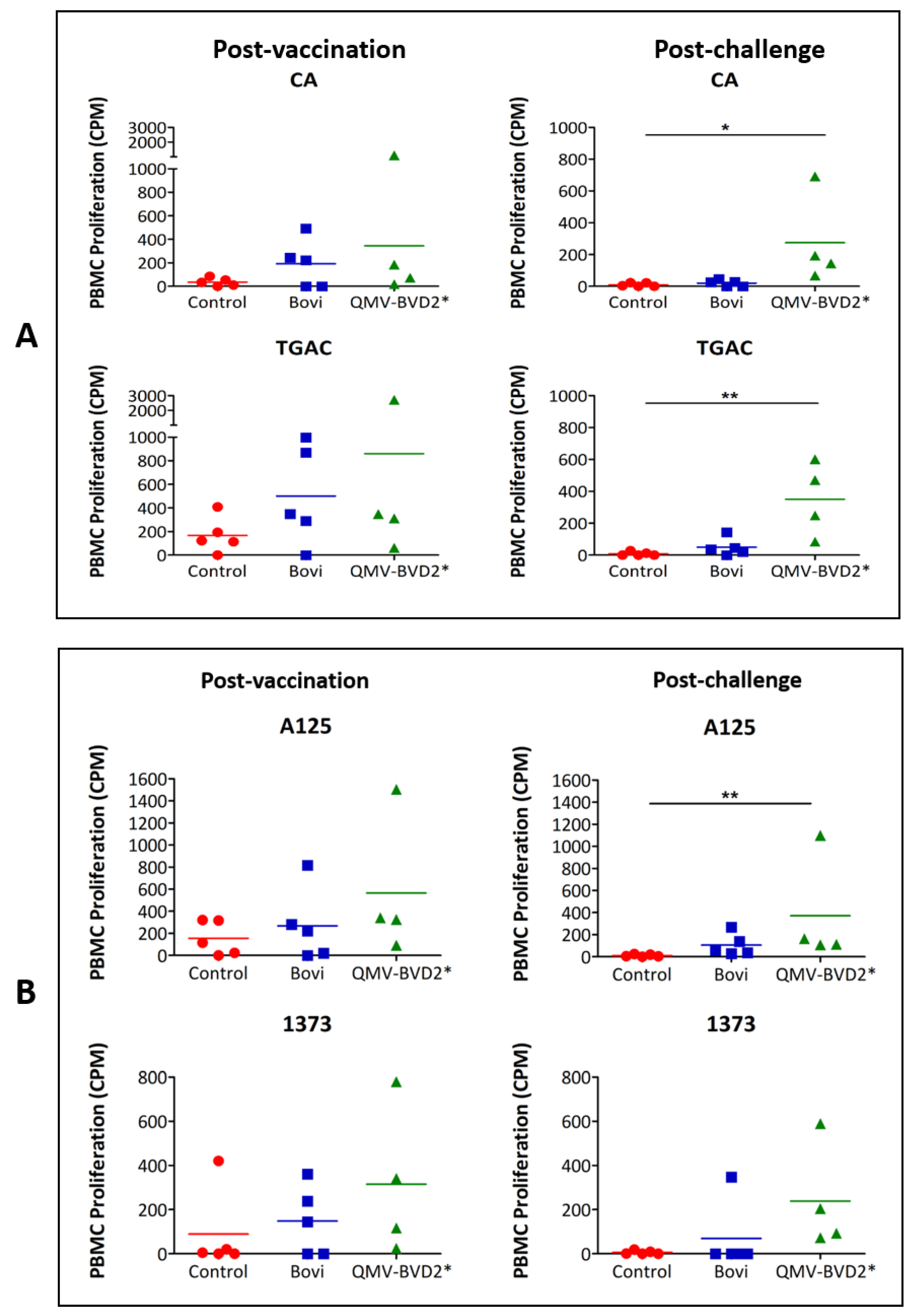
2.18. BVDV–Specific Cellular IFN–γ and Proliferation Responses
At day 0 and 14 post–vaccination and day 6 post–challenge, IFN–γ responses in PBMCs were evaluated by enzyme–linked immunospot (ELISPOT) assay. The assay was performed using Bovine IFN–γ ELISpot
BASIC (ALP) kit (Mabtech, Stockholm, Sweden, # 3119–2A) as per the manufacturer’s instruction and as described previously [
31,
38]. Briefly, 0.25 × 10
6 whole–blood–derived PBMCs were seeded in triplicate wells of MultiScreen–IP plates (MilliporeSigma™, # MAIPS4510) with whole heat–killed BVDV virus [CA0401186a (CA), TGAC, A125 or 1373] in a final volume of 100 µL complete RPMI 1640 medium. The positive control was 2.5 µg/ml concanavalin A (ConA), whereas medium alone was used as a negative control. The spots were quantified with an ELISPOT reader, Cellular Technology Limited (CTL, Shaker Heights, OH, USA) ImmunoSpot
® S6 Analyzer. The results were presented as the mean number of BVDV–specific IFN–γ
+ spot–forming cells (SFC) per 10
6 PBMCs after deducted background medium counts.
BVDV–specific PBMC proliferation responses on 14 dpv and 4 dpc were determined using
3H–Thymidine incorporation assay as previously described [
31,
38]. Briefly, 0.5 × 10
6 whole–blood–derived PBMCs were cultured for 72 h at 37 °C in triplicate wells of round–bottom 96–well plates in a total volume of 100 μL of complete RPMI 1640 medium containing 10 µg/mL of whole heat–killed BVDV virus. The positive control was 1.25 μg/mLConA, whereas medium alone was used as a negative control. Cells were labeled with 0.25 μCi of
3H–thymidine for 12 h and then harvested using a semi–automatic cell harvester (Perkin Elmer, Waltham, MA, USA), and the incorporated
3H–thymidine was counted with a Micro–Beta liquid scintillation counter (Perkin Elmer). The incorporation of
3H–thymidine by the proliferating PBMCs was presented as mean counts per minute (CPM) of triplicate wells after deducting the background medium counts.
2.19. Euthanasia, Necropsy and Pathology
Calves were euthanatized with xylazine and pentabarbitol 20 dpc (
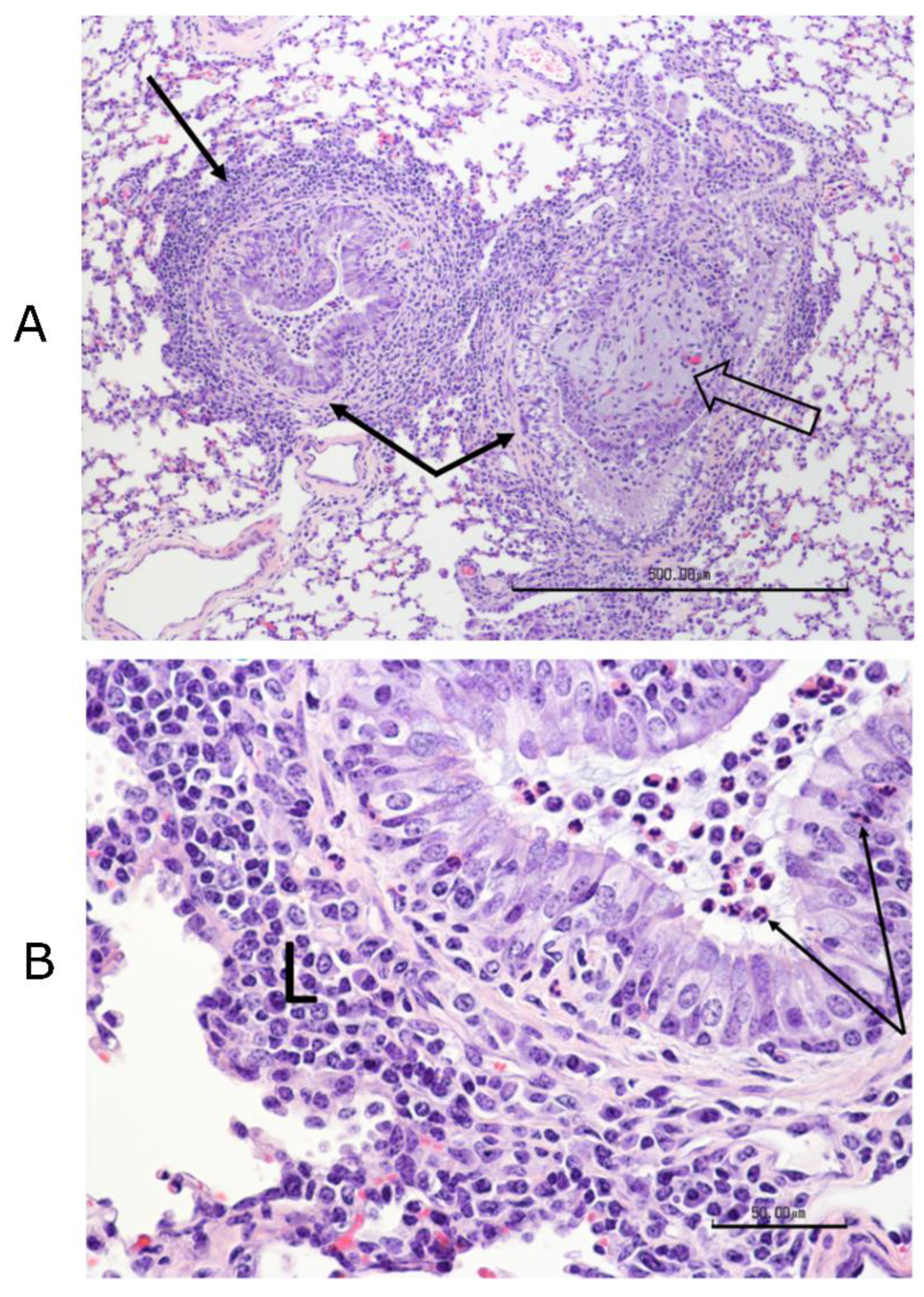
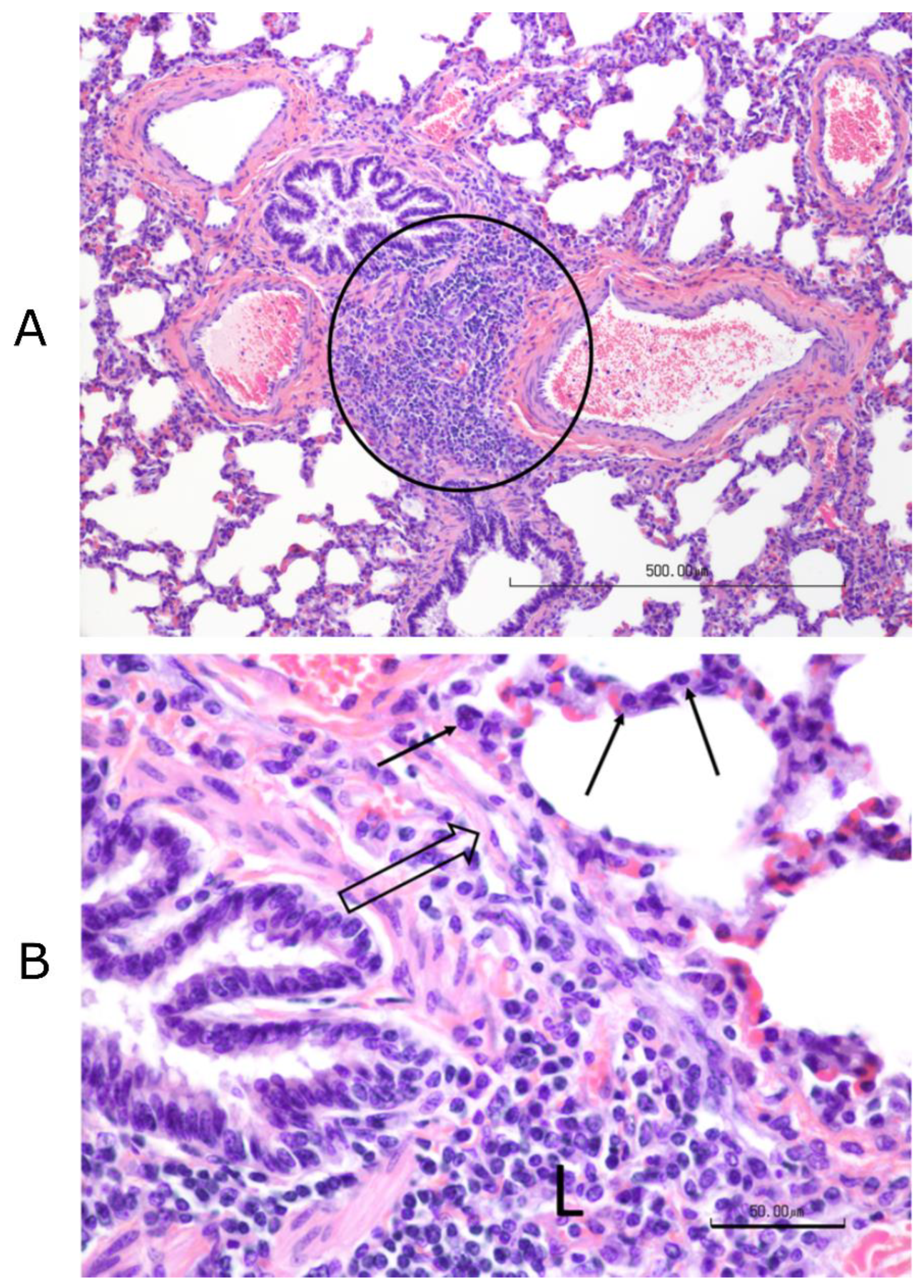
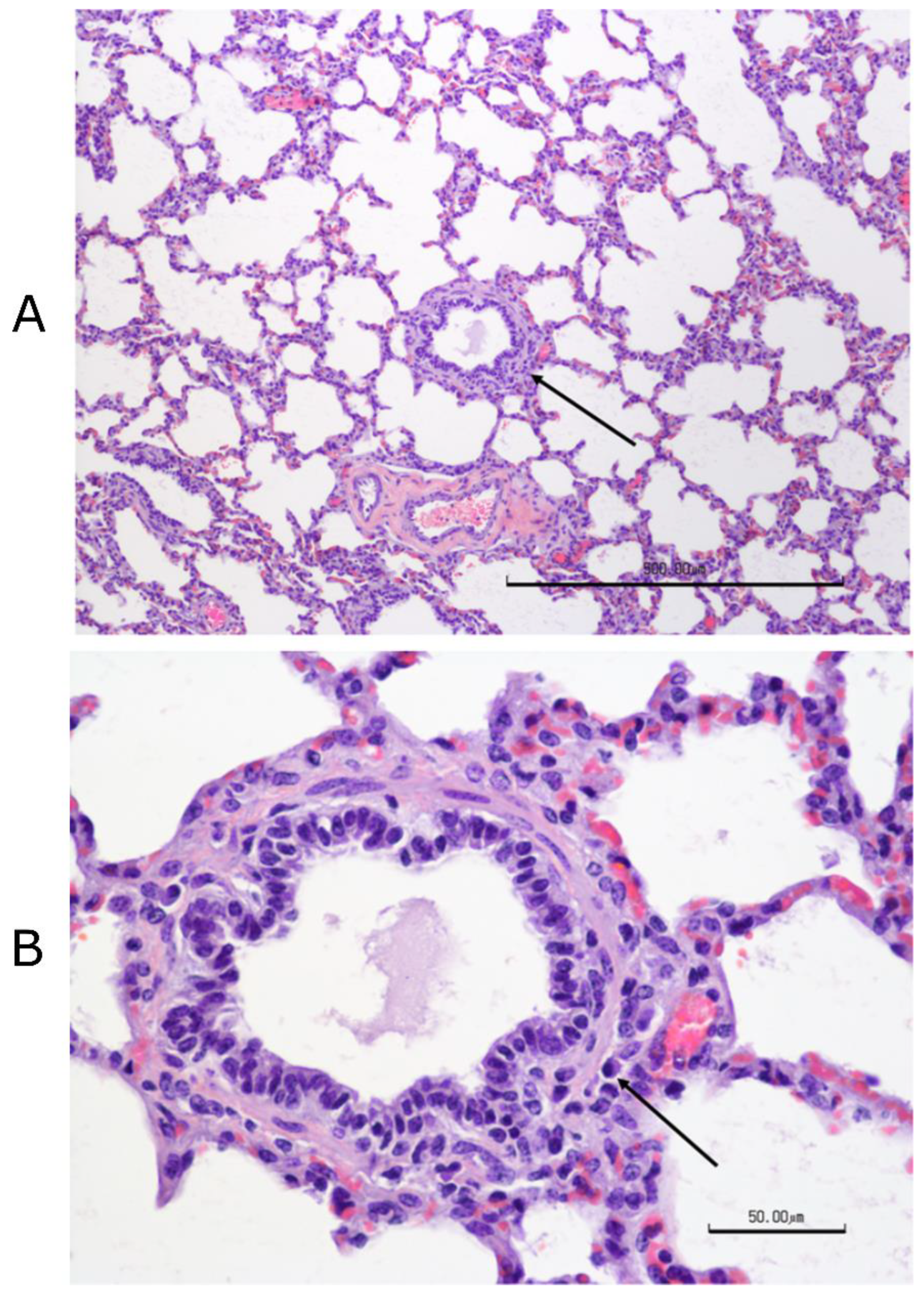
Figure 3). Complete necropsies were performed. Samples were collected from the lungs, systematically from cranial and caudal lobes with additional sections from gross lesions. Lungs were photographed, and lesions were recorded. Samples were also collected from the kidney, liver, spleen, mesenteric lymph node, tonsil, bronchial lymph node, cervical lymph node, and bone marrow. Samples were fixed in 10% buffered–neutral formalin. Hematoxylin–eosin (HE) stained slides were obtained from paraffin–embedded tissues by routine methods. All tissues were evaluated and scored by a single veterinary pathologist, who was blinded to treatment.
Tissues, except lungs, were scored on a scale of 0–4 (0 = normal, 1 = minimal, 2 = mild, 3 = moderate, and 4 = severe) on multiple parameters. Parameters included acute inflammation, chronic inflammation, and necrosis for all tissues with additional tissue–specific parameters, such as glomerular changes for kidneys, lymphoid depletion, and hyperplasia for all lymphoid organs, sinus histiocytosis for lymph nodes, and myeloid/erythroid hyperplasia in the bone marrow. All lung sections were individually scored on a 0–3 scale (0 = normal, 1 = mild, 2 = moderate, and 3 = severe). Bonchi/bronchioles, parenchyma, and septa/pleura were evaluated in each section.
2.20. Statistical Analysis
2.20.1. Nasal Virus Shedding and Viremia
All data were expressed as means ± standard deviation. Statistical analyses were performed using GraphPad PRISM® software version 5.04 (GraphPad Software, San Diego, CA, USA). The two–way analysis of variance (ANOVA) followed by Bonferroni posts–tests to compare replicate means by row were performed. A value of p ˂ 0.05 was considered statistically significant.
2.20.2. Calculation of Outliers
Outliers in data points that differ significantly from other observations were estimated by Grubb’s test (generalized extreme studentized deviate method) with alpha level of 0.05 using GraphPad PRISM® software.
2.20.3. Cellular Immune Response
The Nonparametric Kruskal–Wallis test with Dunn’s multiple comparisons test was used to analyze the significant differences between groups. Post–vaccination and post–challenge, the significance of the differences in BVDV–specific immune responses (cellular IFN–γ and proliferation responses) were compared among all groups. Statistical analysis was performed using GraphPad Prism 7 (Version 7.04, GraphPad Software). A significance level of <0.05 was used for all analyses.
2.20.4. Histopathology of Lung Sections
A nonparametric rank test of factorial ANOVA with repeated measures was performed to detect the differences in the three vaccinated groups’ efficacy levels, adjusted by Aligned Rank Transformation [
22]. The rationale of this approach is to allow us to perform nonparametric factorial analyses when handling repeated measures [
39]. This approach is more robust to test sophisticated data structure than other traditional nonparametric tests [
40]. The adjustment method aligned rank transform (ART) relies on alignment and ranking step before using F–tests. Therefore, ART is similar to the parametric ANOVA, except that the response variable may be continuous or ordinal and is not required to be normally distributed. Post–hoc pairwise comparisons were conducted, and the alpha levels were adjusted by Tukey method. All statistical analyses were performed by R.
4. Discussion
Current vaccination practices against the viruses causing BRDC include trivalent attenuated, BoHV–1, BVDV–1 and –2 live vaccines. While these vaccines protect against the severity of BoHV–1 and BVDV infections, these vaccines were linked to outbreaks of abortion (BoHV–1) in dairy cattle industries, respiratory diseases (BoHV–1 and BVDV) in the beef and dairy cattle industries, and persistent infections (BVDV) in dairy cattle industries. In several cases, the causal agent(s) was traced back to the vaccine strain of BoHV–1 used in the polyvalent vaccine because the traditional BoHV–1–MLV vaccine virus establishes latency in the TG, reactivates with stress, and be shed in the nasal secretions. Therefore, only the gE–deleted BoHV–1 marker vaccine is allowed in several EU countries for vaccination against BoHV–1. The BoHV–1 gE–deleted marker vaccine is distinguishable from the BoHV–1 MLV strains serologically. Under field conditions, the gE marker vaccine virus in most cases was not shed from the nose of vaccinated animals following reactivation from the latency. However, a low–level gE marker virus shedding occurred in some instances of latency–reactivation (
http://ec.europa.eu/food/fs/sc/scah/out49_en.pdf) [
43].
The live attenuated BVDV strains used in the multivalent BRD vaccines are suspected in BVDV–associated problems in the cattle industry because of its RNA genome’s inherent ability to mutate under the field conditions. Additionally, like the wt BVDV, the vaccine virus also causes immunosuppression and vertical transmission in pregnant cows and persistent infection of calves [
19,
44,
45,
46,
47]. Recently, the single Npro and double Npro–Erns live BVDV mutants were also developed to avoid the traditional BVDV MLV vaccine–associated problems. However, both the mutant viruses can cross the placental barrier and established persistent infection [
19,
48]. Therefore, traditional MLV and genetically engineered BVDV vaccines are not allowed in many EU countries or discouraged. Instead, eradication of BVDV by (i) testing and identifying newborn calves for persistent BVDV infection, (ii) removing PI calves, and (iii) taking hygienic, and biosecurity control measures have been implemented. However, this latter approach renders the naïve cattle population vulnerable to severe and widespread BVDV infection if the virus is introduced into the cattle population.
This study constructed a BoHV–1 quadruple mutant virus, BoHV–1QMV, which lacks the BoHV–1 UL49.5 Ecto–domain residues (30–32) plus the CT residues (80–96), the entire gE CT and Us9, and gG. Further, we inserted the chimeric BVDV–2 E2 and E
rns–GM–CSF genes in the gE CT–Us9 and gG deletion loci, respectively. We determined that the resulting QMV–BVD2* vaccine elicited higher cross–reactive IFN–γ and proliferation responses in the vaccinated calves against BVDV–1 and –2 before and after the virulent BVDV–2 challenge when compared with the "Bovi"–vaccinated group. The QMV–BVD2* vaccine also induced BVDV–2–specific seroconversion at 34 dpv, but the “Bovi” generated a 50–fold higher BVDV–2 serum–neutralizing titer by that time. Nevertheless, QMV–BVD2* vaccinated calves had a better recall neutralizing antibody response after the challenge (6 dpc), and the BVDV–2–specific SN titer increased four–fold in the QMV–BVD2*–vaccinated calves (17 to 64). In contrast, the SN titers in the “Bovi” group remained the same (878 on 0 dpc and 885 on 6dpc). Notably, by 14dpc, the SN titer in the QMV–BVD2* sky–rocketed 5500–fold relative to that on the day of the challenge, whereas the corresponding increase in SN titer in the “Bovi” group was 19–fold. Taken together, upon BVDV–2 challenge, QMV–BVD2* vaccinated claves had stronger and higher levels of BVDV cross–reactive (types 1 and 2) T cell as well as BVDV–2–specific recall SN neutralizing responses compared with that of “Bovi” (commercial trivalent MLV). Most notably, the calves in both the control–unvaccinated and the “Bovi” vaccinated groups had visible gross lung–lesions. Consistent with this finding, the animals in the “Bovi” and control unvaccinated groups had also lung histopathological lesions; however, the QMV–BVD2* vaccinated calves had either mild or no lesion. Therefore, we believe that the QMV–BVD2* primed and induced a better BVDV–specific memory T cell response than that of “Bovi” (MLV). Consistent with this assumption, previous attempts to use BoHV–1 vectored BVDV subunit E2 vaccines were not adequately protective even though they induced BVDV–specific neutralizing antibody response [
49,
50]. Notably, these BoHV–1 vectors still had the two immunosuppressive genes, U
L49.5 and gG, intact in their genome. Also, in those instances, only the BVDV E2 was used as a subunit antigen. In contrast, we additionally used BVDV2 Erns fused with the bovine GM–CSF chimeric protein as a second subunit antigen.
Taken together, we believe that deleting both the immunosuppressive BoHV–1 genes in the vaccine vector combined with the inclusion of GM–CSF together with Erns most likely contributed towards improved cellular and memory neutralizing antibody responses against BVDV. Remarkably, even though the subunit antigens expressed by the QMV–BVD2* were type 2 BVDV–specific, the cellular immune response induced by the prototype vaccine was reactive against both BVDV–1 and –2. Earlier, we also determined that the BoHV–1 TMV was equally attenuated as a gE–deleted virus but induced a better protective immune response against the virulent BoHV–1 challenge compared with the gE–deleted virus with respect to both the cellular immune and neutralizing antibody responses. In the case of QMV–BVD2*, in which the BoHV–1 gG gene was additionally deleted, the efficiency of virus replication in the nasal mucosa was reduced slightly compared with that of TMV [
28]. Nevertheless, BoHV–1 QMV induced slightly higher BoHV–1–specific neutralizing antibody response compared with that of BoHV–1 TMV [
28]. While in this study, we did not determine its vaccine efficacy against BoHV–1, but based on its comparable neutralizing antibody response to that of "Bovi"–vaccinated animals, we believe that the QMV–BVD2* will be equally or better protective against BoHV–1 than the BoHV–1 TMV. Taken together, we believe the QMV–BVD2* vaccine is similarly or slightly better protective against BoHV–1, BVDV–1, and BVDV–2 compared with that of “Bovi” vaccine. By using the QMV–BVD2* vaccine, we will get comparable or equal protection against the three viruses and avoid the MLV BoHV–1 and BVDV vaccines associated problems in the field.
Further, from the manufacturing perspective, the vaccine would be cost–effective. This is because instead of growing the three different viruses (BoHV–1, BVDV–types 1 and 2) to formulate the vaccine, only the QMV–BVD2*, which grows with a much higher titer in the MDBK cells compared with that of BVDV, will provide the BVDV protective antigens. Furthermore, based on the gE CT–based marker assay [
29], the QMV–BVD2* vaccinated animals can be distinguished from the wt BoHV–1–infected animals in the field. Also, a commercial BVDV NS3 based blocking ELISA test kit (BIO K 230; Bio-X Diagnostics, Rochefort, Nouvelle-Aquitaine, Belgium) exists. This could be used as a serological marker test to distinguish the QMV–BVD–2* vaccinated animals from the BVDV–infected animals. Therefore, QMV–BVD2* will fulfill the DIVA property against both BoHV–1 and BVDV to distinguish the vaccinated animals from the infected animals under field conditions.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}














