Immune Responses Induced by mRNA Vaccination in Mice, Monkeys and Humans


{kind=link}
{kind=link}
Abstract
:1. Introduction
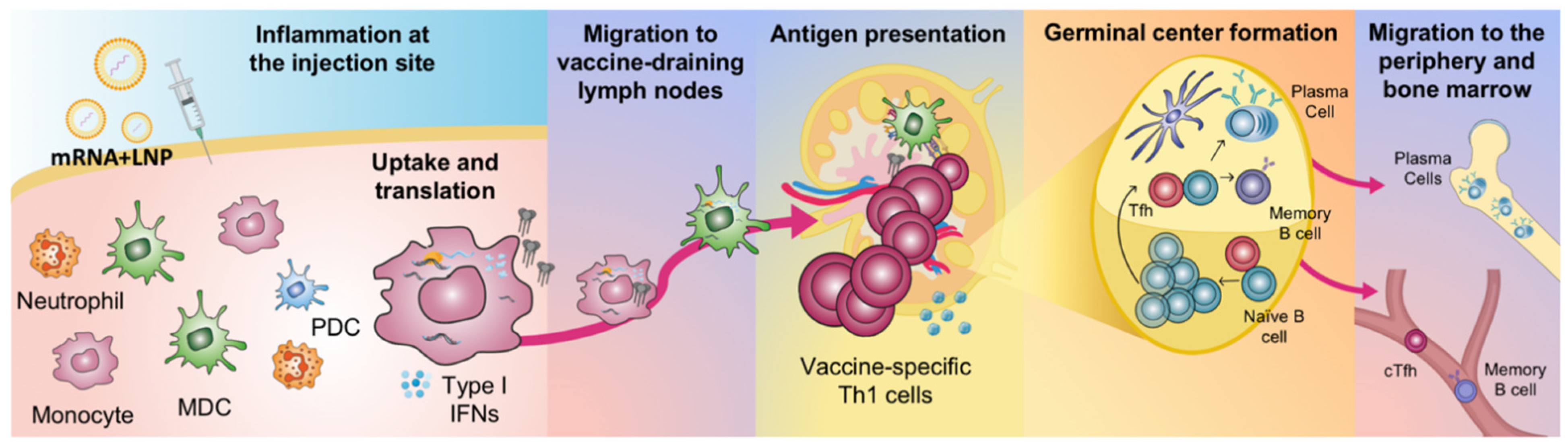
2. Innate Immune Activation of mRNA Vaccines
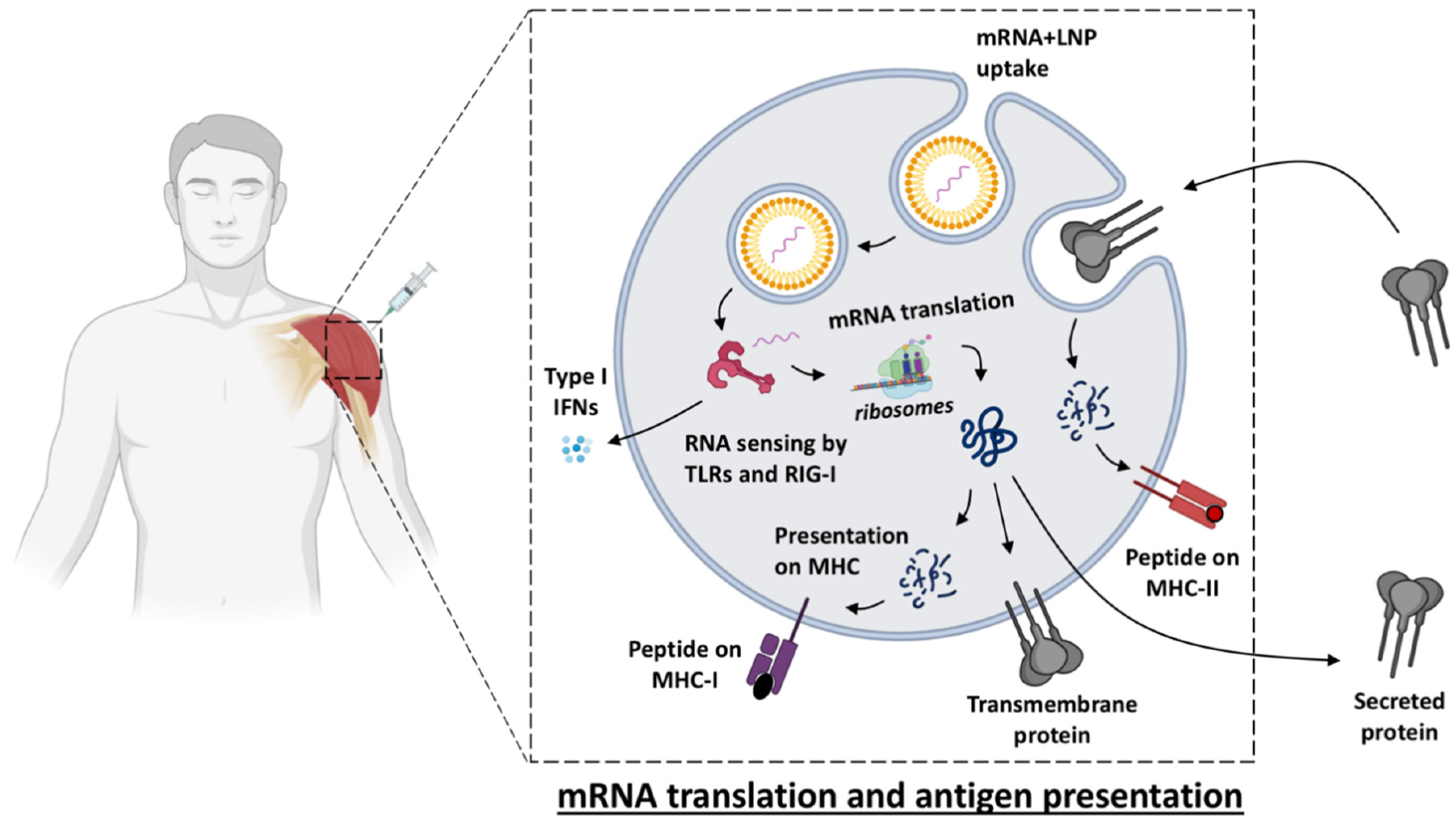
Vaccine Uptake, Translation and Biodistribution after mRNA Vaccination
3. Antibody Responses by mRNA Vaccines to Infectious Diseases
3.1. Rabies
3.2. Influenza
3.3. Zika
3.4. CMV, HIV-1, RSV and Ebola
3.5. SARS-CoV-2
4. T Cell Responses by mRNA Vaccines
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wolff, J.; Malone, R.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, J.B.; Donnelly, J.J.; Parker, S.E.; Rhodes, G.H.; Felgner, P.L.; Dwarki, V.J.; Gromkowski, S.H.; Deck, R.R.; DeWitt, C.M.; Friedman, A.; et al. Heterologous protection against influenza by injection of DNA encoding a viral protein. Science 1993, 259, 1745–1749. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Berglund, P.; Rhodes, G.; Parker, S.E.; Jondal, M.; Liljestrom, P. Self-replicating Semliki Forest virus RNA as recombinant vaccine. Vaccine 1994, 12, 1510–1514. [Google Scholar] [CrossRef]
- Martinon, F.; Krishnan, S.; Lenzen, G.; Magne, R.; Gomard, E.; Guillet, J.G.; Levy, J.P.; Meulien, P. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur. J. Immunol. 1993, 23, 1719–1722. [Google Scholar] [CrossRef]
- Kariko, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef]
- Schlake, T.; Thess, A.; Fotin-Mleczek, M.; Kallen, K.J. Developing mRNA-vaccine technologies. RNA Biol. 2012, 9, 1319–1330. [Google Scholar] [CrossRef] [Green Version]
- Graham, B.S.; Mascola, J.R.; Fauci, A.S. Novel Vaccine Technologies: Essential Components of an Adequate Response to Emerging Viral Diseases. JAMA 2018, 319, 1431–1432. [Google Scholar] [CrossRef]
- Liang, F.; Lindgren, G.; Lin, A.; Thompson, E.A.; Ols, S.; Rohss, J.; John, S.; Hassett, K.; Yuzhakov, O.; Bahl, K.; et al. Efficient Targeting and Activation of Antigen-Presenting Cells In Vivo after Modified mRNA Vaccine Administration in Rhesus Macaques. Mol. Ther. 2017, 25, 2635–2647. [Google Scholar] [CrossRef] [Green Version]
- Hassett, K.J.; Benenato, K.E.; Jacquinet, E.; Lee, A.; Woods, A.; Yuzhakov, O.; Himansu, S.; Deterling, J.; Geilich, B.M.; Ketova, T.; et al. Optimization of Lipid Nanoparticles for Intramuscular Administration of mRNA Vaccines. Mol. Ther. Nucleic. Acids. 2019, 15, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, J.A.; Cullis, P.R.; van der Meel, R. Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic. Acid. Ther. 2018, 28, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Sands, H.; Gorey-Feret, L.J.; Cocuzza, A.J.; Hobbs, F.W.; Chidester, D.; Trainor, G.L. Biodistribution and metabolism of internally 3H-labeled oligonucleotides. I. Comparison of a phosphodiester and a phosphorothioate. Mol. Pharmacol. 1994, 45, 932–943. [Google Scholar] [PubMed]
- Reichmuth, A.M.; Oberli, M.A.; Jaklenec, A.; Langer, R.; Blankschtein, D. mRNA vaccine delivery using lipid nanoparticles. Ther. Deliv. 2016, 7, 319–334. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Tam, Y.Y.C.; Lin, P.J.C.; Sung, M.M.H.; Tam, Y.K.; Cullis, P.R. Influence of particle size on the in vivo potency of lipid nanoparticle formulations of siRNA. J. Control. Release 2016, 235, 236–244. [Google Scholar] [CrossRef]
- Eygeris, Y.; Patel, S.; Jozic, A.; Sahay, G. Deconvoluting Lipid Nanoparticle Structure for Messenger RNA Delivery. Nano Lett. 2020, 20, 4543–4549. [Google Scholar] [CrossRef]
- Lee, S.; Cheng, Q.; Yu, X.; Liu, S.; Johnson, L.; Siegwart, D.J. A Systematic Study of Unsaturation in Lipid Nanoparticles Leads to Improved mRNA Transfection In Vivo. Angew Chem. Int. Ed. Engl. 2020. [Google Scholar] [CrossRef]
- Persano, S.; Guevara, M.L.; Li, Z.; Mai, J.; Ferrari, M.; Pompa, P.P.; Shen, H. Lipopolyplex potentiates anti-tumor immunity of mRNA-based vaccination. Biomaterials 2017, 125, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amanna, I.J.; Carlson, N.E.; Slifka, M.K. Duration of humoral immunity to common viral and vaccine antigens. N. Engl. J. Med. 2007, 357, 1903–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monath, T.P.; Gershman, M.; Erin Staples, J.; Barrett, A.D.T. Yellow fever vaccine. In Vaccines, 6th ed.; Plotkin, S.A., Orenstein, W.A., Offit, P.A., Eds.; W.B. Saunders: London, UK, 2013; pp. 870–968. [Google Scholar] [CrossRef]
- Medical Advisory Committee of the Immune Deficiency Foundation; Shearer, W.T.; Fleisher, T.A.; Buckley, R.H.; Ballas, Z.; Ballow, M.; Blaese, R.M.; Bonilla, F.A.; Conley, M.E.; Cunningham-Rundles, C.; et al. Recommendations for live viral and bacterial vaccines in immunodeficient patients and their close contacts. J. Allergy. Clin. Immunol 2014, 133, 961–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francica, J.R.; Sheng, Z.; Zhang, Z.; Nishimura, Y.; Shingai, M.; Ramesh, A.; Keele, B.F.; Schmidt, S.D.; Flynn, B.J.; Darko, S.; et al. Analysis of immunoglobulin transcripts and hypermutation following SHIV(AD8) infection and protein-plus-adjuvant immunization. Nat. Commun. 2015, 6, 6565. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, G.; Ols, S.; Liang, F.; Thompson, E.A.; Lin, A.; Hellgren, F.; Bahl, K.; John, S.; Yuzhakov, O.; Hassett, K.J.; et al. Induction of Robust B Cell Responses after Influenza mRNA Vaccination Is Accompanied by Circulating Hemagglutinin-Specific ICOS+ PD-1+ CXCR3+ T Follicular Helper Cells. Front. Immunol. 2017, 8, 1539. [Google Scholar] [CrossRef] [Green Version]
- McNamara, M.A.; Nair, S.K.; Holl, E.K. RNA-Based Vaccines in Cancer Immunotherapy. J. Immunol. Res. 2015, 2015, 794528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- van den Elzen, P.; Garg, S.; Leon, L.; Brigl, M.; Leadbetter, E.A.; Gumperz, J.E.; Dascher, C.C.; Cheng, T.Y.; Sacks, F.M.; Illarionov, P.A.; et al. Apolipoprotein-mediated pathways of lipid antigen presentation. Nature 2005, 437, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Lazzaro, S.; Giovani, C.; Mangiavacchi, S.; Magini, D.; Maione, D.; Baudner, B.; Geall, A.J.; De Gregorio, E.; D’Oro, U.; Buonsanti, C. CD8 T-cell priming upon mRNA vaccination is restricted to bone-marrow-derived antigen-presenting cells and may involve antigen transfer from myocytes. Immunology 2015, 146, 312–326. [Google Scholar] [CrossRef]
- Maugeri, M.; Nawaz, M.; Papadimitriou, A.; Angerfors, A.; Camponeschi, A.; Na, M.; Holtta, M.; Skantze, P.; Johansson, S.; Sundqvist, M.; et al. Linkage between endosomal escape of LNP-mRNA and loading into EVs for transport to other cells. Nat. Commun. 2019, 10, 4333. [Google Scholar] [CrossRef] [Green Version]
- Brito, L.A.; Chan, M.; Shaw, C.A.; Hekele, A.; Carsillo, T.; Schaefer, M.; Archer, J.; Seubert, A.; Otten, G.R.; Beard, C.W.; et al. A cationic nanoemulsion for the delivery of next-generation RNA vaccines. Mol. Ther. 2014, 22, 2118–2129. [Google Scholar] [CrossRef] [Green Version]
- Pardi, N.; Tuyishime, S.; Muramatsu, H.; Kariko, K.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Weissman, D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control. Release 2015, 217, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Bahl, K.; Senn, J.J.; Yuzhakov, O.; Bulychev, A.; Brito, L.A.; Hassett, K.J.; Laska, M.E.; Smith, M.; Almarsson, O.; Thompson, J.; et al. Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses. Mol. Ther. 2017, 25, 1316–1327. [Google Scholar] [CrossRef] [Green Version]
- Kowalczyk, A.; Doener, F.; Zanzinger, K.; Noth, J.; Baumhof, P.; Fotin-Mleczek, M.; Heidenreich, R. Self-adjuvanted mRNA vaccines induce local innate immune responses that lead to a potent and boostable adaptive immunity. Vaccine 2016, 34, 3882–3893. [Google Scholar] [CrossRef]
- Oberli, M.A.; Reichmuth, A.M.; Dorkin, J.R.; Mitchell, M.J.; Fenton, O.S.; Jaklenec, A.; Anderson, D.G.; Langer, R.; Blankschtein, D. Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett. 2017, 17, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.B.; Lambert, L.; Kinnear, E.; Busse, D.; Erbar, S.; Reuter, K.C.; Wicke, L.; Perkovic, M.; Beissert, T.; Haas, H.; et al. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol. Ther. 2018, 26, 446–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geall, A.J.; Verma, A.; Otten, G.R.; Shaw, C.A.; Hekele, A.; Banerjee, K.; Cu, Y.; Beard, C.W.; Brito, L.A.; Krucker, T.; et al. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl. Acad. Sci. USA 2012, 109, 14604–14609. [Google Scholar] [CrossRef] [Green Version]
- Lindsay, K.E.; Bhosle, S.M.; Zurla, C.; Beyersdorf, J.; Rogers, K.A.; Vanover, D.; Xiao, P.; Arainga, M.; Shirreff, L.M.; Pitard, B.; et al. Visualization of early events in mRNA vaccine delivery in non-human primates via PET-CT and near-infrared imaging. Nat. Biomed. Eng. 2019, 3, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Ols, S.; Lore, K. Imaging the early fate of mRNA vaccines. Nat. Biomed. Eng. 2019, 3, 331–332. [Google Scholar] [CrossRef]
- Asselin-Paturel, C.; Brizard, G.; Chemin, K.; Boonstra, A.; O’Garra, A.; Vicari, A.; Trinchieri, G. Type I interferon dependence of plasmacytoid dendritic cell activation and migration. J. Exp. Med. 2005, 201, 1157–1167. [Google Scholar] [CrossRef] [Green Version]
- Simmons, D.P.; Wearsch, P.A.; Canaday, D.H.; Meyerson, H.J.; Liu, Y.C.; Wang, Y.; Boom, W.H.; Harding, C.V. Type I IFN drives a distinctive dendritic cell maturation phenotype that allows continued class II MHC synthesis and antigen processing. J. Immunol. 2012, 188, 3116–3126. [Google Scholar] [CrossRef] [Green Version]
- Lutz, J.; Lazzaro, S.; Habbeddine, M.; Schmidt, K.E.; Baumhof, P.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Heidenreich, R.; et al. Unmodified mRNA in LNPs constitutes a competitive technology for prophylactic vaccines. NPJ Vaccine 2017, 2, 29. [Google Scholar] [CrossRef] [Green Version]
- Liang, F.; Lindgren, G.; Sandgren, K.J.; Thompson, E.A.; Francica, J.R.; Seubert, A.; De Gregorio, E.; Barnett, S.; O’Hagan, D.T.; Sullivan, N.J.; et al. Vaccine priming is restricted to draining lymph nodes and controlled by adjuvant-mediated antigen uptake. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Bourquin, C.; Schmidt, L.; Hornung, V.; Wurzenberger, C.; Anz, D.; Sandholzer, N.; Schreiber, S.; Voelkl, A.; Hartmann, G.; Endres, S. Immunostimulatory RNA oligonucleotides trigger an antigen-specific cytotoxic T-cell and IgG2a response. Blood 2007, 109, 2953–2960. [Google Scholar] [CrossRef]
- De Beuckelaer, A.; Pollard, C.; Van Lint, S.; Roose, K.; Van Hoecke, L.; Naessens, T.; Udhayakumar, V.K.; Smet, M.; Sanders, N.; Lienenklaus, S.; et al. Type I Interferons Interfere with the Capacity of mRNA Lipoplex Vaccines to Elicit Cytolytic T Cell Responses. Mol. Ther. 2016, 24, 2012–2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, C.; Rejman, J.; De Haes, W.; Verrier, B.; Van Gulck, E.; Naessens, T.; De Smedt, S.; Bogaert, P.; Grooten, J.; Vanham, G.; et al. Type I IFN counteracts the induction of antigen-specific immune responses by lipid-based delivery of mRNA vaccines. Mol. Ther. 2013, 21, 251–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Beuckelaer, A.; Grooten, J.; De Koker, S. Type I Interferons Modulate CD8 (+) T Cell Immunity to mRNA Vaccines. Trends Mol. Med. 2017, 23, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Espeseth, A.S.; Cejas, P.J.; Citron, M.P.; Wang, D.; DiStefano, D.J.; Callahan, C.; Donnell, G.O.; Galli, J.D.; Swoyer, R.; Touch, S.; et al. Modified mRNA/lipid nanoparticle-based vaccines expressing respiratory syncytial virus F protein variants are immunogenic and protective in rodent models of RSV infection. NPJ Vaccine 2020, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Bachmann, M.F.; Jennings, G.T. Vaccine delivery: A matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 2010, 10, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Mesin, L.; Ersching, J.; Victora, G.D. Germinal Center B Cell Dynamics. Immunity 2016, 45, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Heesters, B.A.; van der Poel, C.E.; Das, A.; Carroll, M.C. Antigen Presentation to B Cells. Trends Immunol. 2016, 37, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Naradikian, M.S.; Parkhouse, K.; Cain, D.W.; Jones, L.; Moody, M.A.; Verkerke, H.P.; Myles, A.; Willis, E.; et al. Nucleoside-modified mRNA vaccines induce potent T follicular helper and germinal center B cell responses. J. Exp. Med. 2018, 215, 1571–1588. [Google Scholar] [CrossRef]
- Tam, H.H.; Melo, M.B.; Kang, M.; Pelet, J.M.; Ruda, V.M.; Foley, M.H.; Hu, J.K.; Kumari, S.; Crampton, J.; Baldeon, A.D.; et al. Sustained antigen availability during germinal center initiation enhances antibody responses to vaccination. Proc. Natl. Acad. Sci. USA 2016, 113, E6639–E6648. [Google Scholar] [CrossRef] [Green Version]
- Schnee, M.; Vogel, A.B.; Voss, D.; Petsch, B.; Baumhof, P.; Kramps, T.; Stitz, L. An mRNA Vaccine Encoding Rabies Virus Glycoprotein Induces Protection against Lethal Infection in Mice and Correlates of Protection in Adult and Newborn Pigs. PLoS Negl. Trop. Dis. 2016, 10, e0004746. [Google Scholar] [CrossRef]
- Stitz, L.; Vogel, A.; Schnee, M.; Voss, D.; Rauch, S.; Mutzke, T.; Ketterer, T.; Kramps, T.; Petsch, B. A thermostable messenger RNA based vaccine against rabies. PLoS Negl. Trop. Dis. 2017, 11, e0006108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberer, M.; Gnad-Vogt, U.; Hong, H.S.; Mehr, K.T.; Backert, L.; Finak, G.; Gottardo, R.; Bica, M.A.; Garofano, A.; Koch, S.D.; et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: An open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet 2017, 390, 1511–1520. [Google Scholar] [CrossRef]
- Hekele, A.; Bertholet, S.; Archer, J.; Gibson, D.G.; Palladino, G.; Brito, L.A.; Otten, G.R.; Brazzoli, M.; Buccato, S.; Bonci, A.; et al. Rapidly produced SAM((R)) vaccine against H7N9 influenza is immunogenic in mice. Emerg. Microbes. Infect. 2013, 2, e52. [Google Scholar] [CrossRef] [PubMed]
- Petsch, B.; Schnee, M.; Vogel, A.B.; Lange, E.; Hoffmann, B.; Voss, D.; Schlake, T.; Thess, A.; Kallen, K.J.; Stitz, L.; et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat. Biotechnol. 2012, 30, 1210–1216. [Google Scholar] [CrossRef]
- Feldman, R.A.; Fuhr, R.; Smolenov, I.; Mick Ribeiro, A.; Panther, L.; Watson, M.; Senn, J.J.; Smith, M.; Almarsson, Ö.; Pujar, H.S.; et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 2019, 37, 3326–3334. [Google Scholar] [CrossRef]
- Richner, J.M.; Himansu, S.; Dowd, K.A.; Butler, S.L.; Salazar, V.; Fox, J.M.; Julander, J.G.; Tang, W.W.; Shresta, S.; Pierson, T.C.; et al. Modified mRNA Vaccines Protect against Zika Virus Infection. Cell 2017, 169, 176. [Google Scholar] [CrossRef] [Green Version]
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef]
- Richner, J.M.; Jagger, B.W.; Shan, C.; Fontes, C.R.; Dowd, K.A.; Cao, B.; Himansu, S.; Caine, E.A.; Nunes, B.T.D.; Medeiros, D.B.A.; et al. Vaccine Mediated Protection Against Zika Virus-Induced Congenital Disease. Cell 2017, 170, 273–283.e12. [Google Scholar] [CrossRef] [Green Version]
- Erasmus, J.H.; Khandhar, A.P.; Guderian, J.; Granger, B.; Archer, J.; Archer, M.; Gage, E.; Fuerte-Stone, J.; Larson, E.; Lin, S.; et al. A Nanostructured Lipid Carrier for Delivery of a Replicating Viral RNA Provides Single, Low-Dose Protection against Zika. Mol. Ther. 2018, 26, 2507–2522. [Google Scholar] [CrossRef] [Green Version]
- John, S.; Yuzhakov, O.; Woods, A.; Deterling, J.; Hassett, K.; Shaw, C.A.; Ciaramella, G. Multi-antigenic human cytomegalovirus mRNA vaccines that elicit potent humoral and cell-mediated immunity. Vaccine 2018, 36, 1689–1699. [Google Scholar] [CrossRef]
- Van Craenenbroeck, A.H.; Smits, E.L.; Anguille, S.; Van de Velde, A.; Stein, B.; Braeckman, T.; Van Camp, K.; Nijs, G.; Ieven, M.; Goossens, H.; et al. Induction of cytomegalovirus-specific T cell responses in healthy volunteers and allogeneic stem cell recipients using vaccination with messenger RNA-transfected dendritic cells. Transplantation 2015, 99, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Van Gulck, E.; Vlieghe, E.; Vekemans, M.; Van Tendeloo, V.F.; Van De Velde, A.; Smits, E.; Anguille, S.; Cools, N.; Goossens, H.; Mertens, L.; et al. mRNA-based dendritic cell vaccination induces potent antiviral T-cell responses in HIV-1-infected patients. Aids 2012, 26, F1–F12. [Google Scholar] [CrossRef] [PubMed]
- Routy, J.P.; Boulassel, M.R.; Yassine-Diab, B.; Nicolette, C.; Healey, D.; Jain, R.; Landry, C.; Yegorov, O.; Tcherepanova, I.; Monesmith, T.; et al. Immunologic activity and safety of autologous HIV RNA-electroporated dendritic cells in HIV-1 infected patients receiving antiretroviral therapy. Clin. Immunol. 2010, 134, 140–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, S.D.; De Keersmaecker, B.; de Goede, A.L.; Verschuren, E.J.; Koetsveld, J.; Reedijk, M.L.; Wylock, C.; De Bel, A.V.; Vandeloo, J.; Pistoor, F.; et al. A phase I/IIa immunotherapy trial of HIV-1-infected patients with Tat, Rev and Nef expressing dendritic cells followed by treatment interruption. Clin. Immunol. 2012, 142, 252–268. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, R.T.; Kwon, D.S.; Macklin, E.A.; Shopis, J.R.; McLean, A.P.; McBrine, N.; Flynn, T.; Peter, L.; Sbrolla, A.; Kaufmann, D.E.; et al. Immunization of HIV-1-Infected Persons With Autologous Dendritic Cells Transfected With mRNA Encoding HIV-1 Gag and Nef: Results of a Randomized, Placebo-Controlled Clinical Trial. J. Acquir. Immune. Defic. Syndr. 2016, 71, 246–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, M.; Huang, E.; Yuzhakov, O.; Ramanathan, P.; Ciaramella, G.; Bukreyev, A. Modified mRNA-Based Vaccines Elicit Robust Immune Responses and Protect Guinea Pigs From Ebola Virus Disease. J. Infect. Dis. 2018, 217, 451–455. [Google Scholar] [CrossRef]
- Mulligan, M.J.; Lyke, K.E.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Raabe, V.; Bailey, R.; Swanson, K.A.; et al. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 2020, 586, 589–593. [Google Scholar] [CrossRef]
- Walsh, E.E.; Frenck, R.W., Jr.; Falsey, A.R.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Mulligan, M.J.; Bailey, R.; et al. Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates. N. Engl. J. Med. 2020, 383, 2439–2450. [Google Scholar] [CrossRef]
- Corbett, K.S.; Edwards, D.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schafer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 mRNA Vaccine Development Enabled by Prototype Pathogen Preparedness. Nature 2020, 586, 567–571. [Google Scholar] [CrossRef]
- Corbett, K.S.; Flynn, B.; Foulds, K.E.; Francica, J.R.; Boyoglu-Barnum, S.; Werner, A.P.; Flach, B.; O’Connell, S.; Bock, K.W.; Minai, M.; et al. Evaluation of the mRNA-1273 Vaccine against SARS-CoV-2 in Nonhuman Primates. N. Engl. J. Med. 2020, 383, 1544–1555. [Google Scholar] [CrossRef]
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An mRNA Vaccine against SARS-CoV-2-Preliminary Report. N. Engl. J. Med. 2020, 383, 1920–1931. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.J.; Rouphael, N.G.; Widge, A.T.; Jackson, L.A.; Roberts, P.C.; Makhene, M.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; Pruijssers, A.J.; et al. Safety and Immunogenicity of SARS-CoV-2 mRNA-1273 Vaccine in Older Adults. N. Engl. J. Med. 2020, 383, 2427–2438. [Google Scholar] [CrossRef] [PubMed]
- Rauch, S.; Roth, N.; Schwendt, K.; Fotin-Mleczek, M.; Mueller, S.O.; Petsch, B. mRNA based SARS-CoV-2 vaccine candidate CVnCoV induces high levels of virus neutralizing antibodies and mediates protection in rodents. bioRxiv 2020. [Google Scholar] [CrossRef]
- Rauch, S.; Gooch, K.; Hall, Y.; Salguero, F.J.; Dennis, M.J.; Gleeson, F.V.; Harris, D.; Ho, C.; Humphries, H.E.; Longet, S.; et al. mRNA vaccine CVnCoV protects non-human primates from SARS-CoV-2 challenge infection. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kremsner, P.; Mann, P.; Bosch, J.; Fendel, R.; Gabor, J.J.; Kreidenweiss, A.; Kroidl, A.; Leroux-Roels, I.; Leroux-Roels, G.; Schindler, C.; et al. Phase 1 Assessment of the Safety and Immunogenicity of an mRNA- Lipid Nanoparticle Vaccine Candidate Against SARS-CoV-2 in Human Volunteers. medRxiv 2020. [Google Scholar] [CrossRef]
- Weissman, D.; Alameh, M.G.; de Silva, T.; Collini, P.; Hornsby, H.; Brown, R.; LaBranche, C.C.; Edwards, R.J.; Sutherland, L.; Santra, S.; et al. D614G Spike Mutation Increases SARS CoV-2 Susceptibility to Neutralization. Cell Host Microbe 2020, 29, 23–31.e4. [Google Scholar] [CrossRef]
- Xie, X.; Zou, J.; Fontes-Garfias, C.R.; Xia, H.; Swanson, K.A.; Cutler, M.; Cooper, D.; Menachery, V.D.; Weaver, S.; Dormitzer, P.R.; et al. Neutralization of N501Y mutant SARS-CoV-2 by BNT162b2 vaccine-elicited sera. bioRxiv 2021. [Google Scholar] [CrossRef]
- Sekine, T.; Perez-Potti, A.; Rivera-Ballesteros, O.; Stralin, K.; Gorin, J.B.; Olsson, A.; Llewellyn-Lacey, S.; Kamal, H.; Bogdanovic, G.; Muschiol, S.; et al. Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell 2020, 183, 158–168 e114. [Google Scholar] [CrossRef]
- Lipsitch, M.; Grad, Y.H.; Sette, A.; Crotty, S. Cross-reactive memory T cells and herd immunity to SARS-CoV-2. Nat. Rev. Immunol. 2020, 20, 709–713. [Google Scholar] [CrossRef]
- Sahin, U.; Muik, A.; Derhovanessian, E.; Vogler, I.; Kranz, L.M.; Vormehr, M.; Baum, A.; Pascal, K.; Quandt, J.; Maurus, D.; et al. COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature 2020, 586, 594–599. [Google Scholar] [CrossRef]
- Laczko, D.; Hogan, M.J.; Toulmin, S.A.; Hicks, P.; Lederer, K.; Gaudette, B.T.; Castano, D.; Amanat, F.; Muramatsu, H.; Oguin, T.H., 3rd; et al. A Single Immunization with Nucleoside-Modified mRNA Vaccines Elicits Strong Cellular and Humoral Immune Responses against SARS-CoV-2 in Mice. Immunity 2020, 53, 724–732.e7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.N.; Li, X.F.; Deng, Y.Q.; Zhao, H.; Huang, Y.J.; Yang, G.; Huang, W.J.; Gao, P.; Zhou, C.; Zhang, R.R.; et al. A Thermostable mRNA Vaccine against COVID-19. Cell 2020, 182, 1271–1283.e16. [Google Scholar] [CrossRef] [PubMed]
- Kose, N.; Fox, J.M.; Sapparapu, G.; Bombardi, R.; Tennekoon, R.N.; de Silva, A.D.; Elbashir, S.M.; Theisen, M.A.; Humphris-Narayanan, E.; Ciaramella, G.; et al. A lipid-encapsulated mRNA encoding a potently neutralizing human monoclonal antibody protects against chikungunya infection. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cagigi, A.; Loré, K. Immune Responses Induced by mRNA Vaccination in Mice, Monkeys and Humans. Vaccines 2021, 9, 61. https://doi.org/10.3390/vaccines9010061
Cagigi A, Loré K. Immune Responses Induced by mRNA Vaccination in Mice, Monkeys and Humans. Vaccines. 2021; 9(1):61. https://doi.org/10.3390/vaccines9010061
Chicago/Turabian StyleCagigi, Alberto, and Karin Loré. 2021. "Immune Responses Induced by mRNA Vaccination in Mice, Monkeys and Humans" Vaccines 9, no. 1: 61. https://doi.org/10.3390/vaccines9010061
APA StyleCagigi, A., & Loré, K. (2021). Immune Responses Induced by mRNA Vaccination in Mice, Monkeys and Humans. Vaccines, 9(1), 61. https://doi.org/10.3390/vaccines9010061