
Localized and Systemic Immune Responses against SARS-CoV-2 Following Mucosal Immunization

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
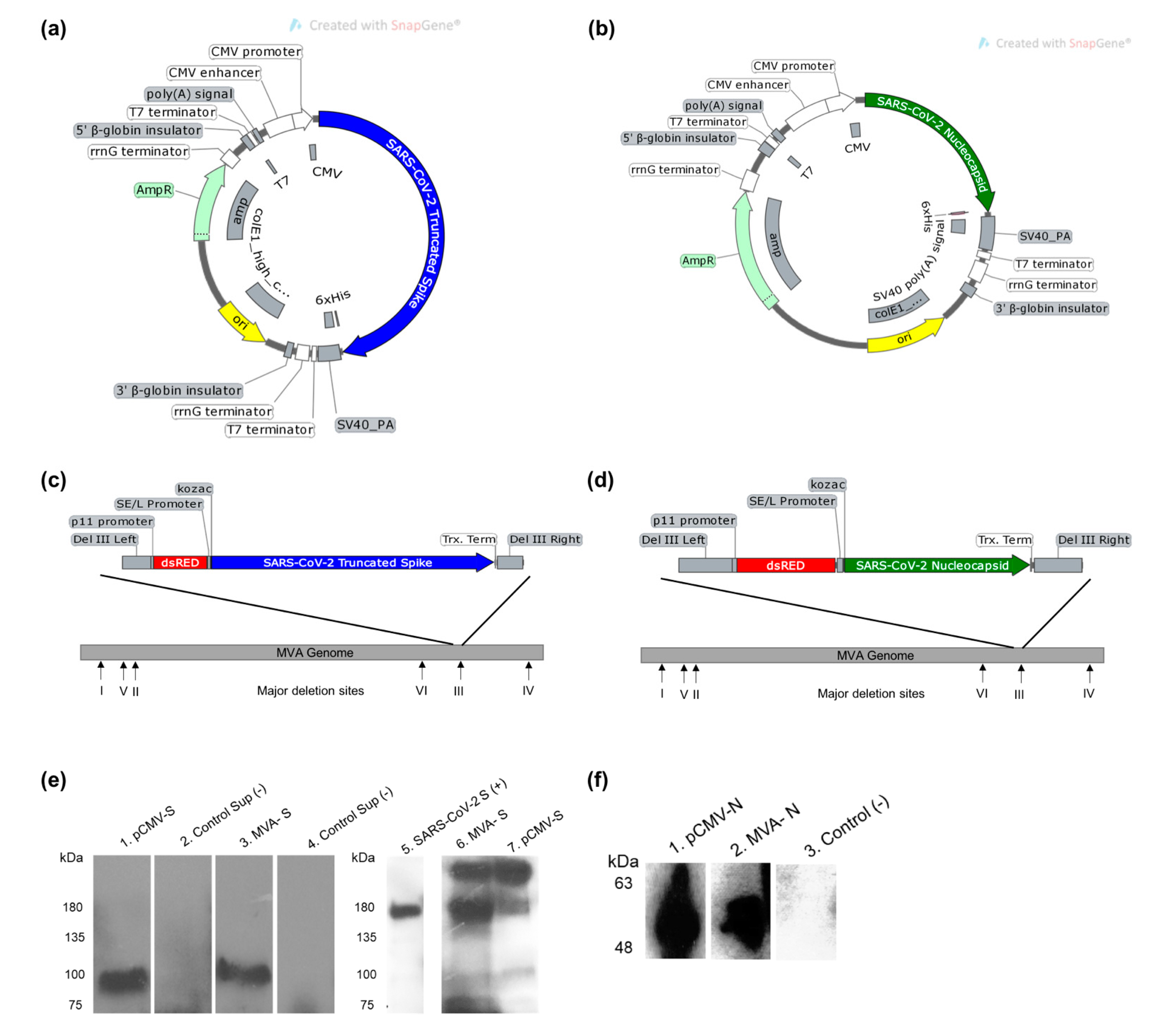
2.2. Preparation of SARS-CoV-2 Vaccine Constructs
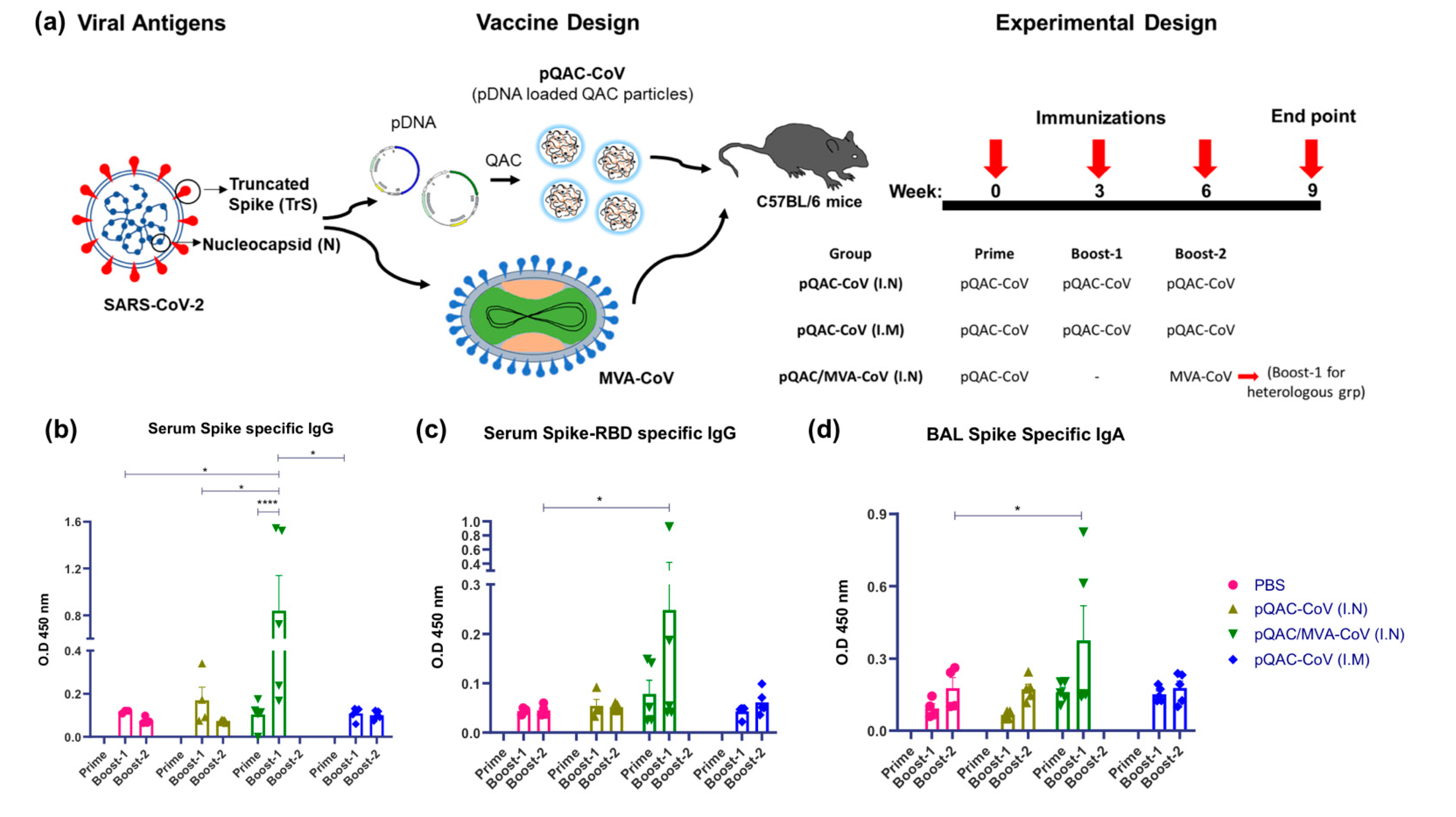
2.3. Vaccine Efficacy Study
2.4. SARS-CoV-2-Specific ELISA
2.5. SARS-CoV-2 Pseudovirus Neutralization Assay
2.6. SARS-CoV-2 Wild-Type Virus Neutralization Assay
2.7. Flow Cytometric Assessment of SARS-CoV-2-Specific Intracellular Cytokine Assay
2.8. Statistical Analysis
3. Results
3.1. Design and Construction of SARS-CoV-2 Vaccine Constructs
3.2. Induction of SARS-CoV-2-Specific Humoral Responses in Vaccinated Mice
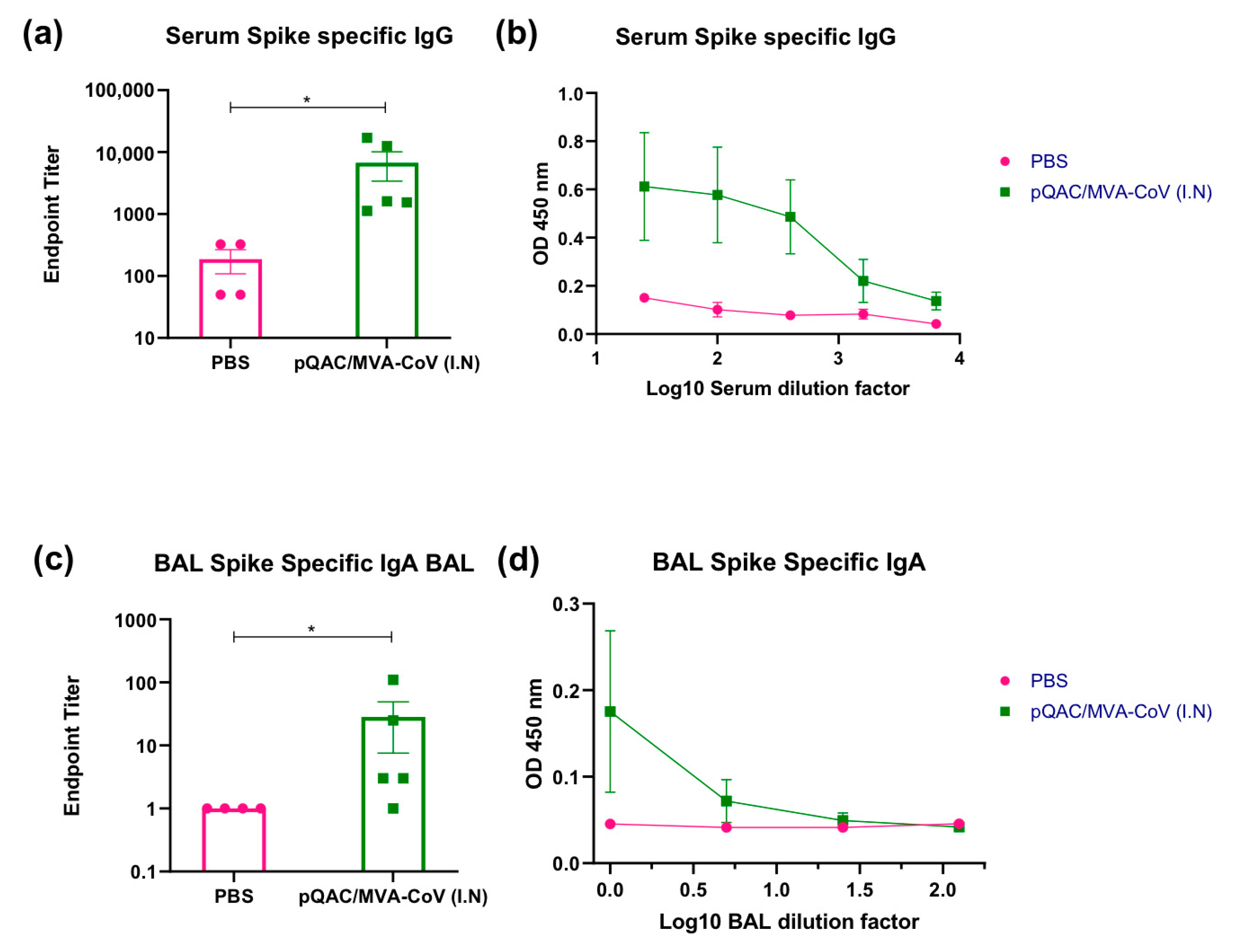
3.3. Heterologous Vaccine Strategy Elicits Neutralizing and Binding Antibody Responses
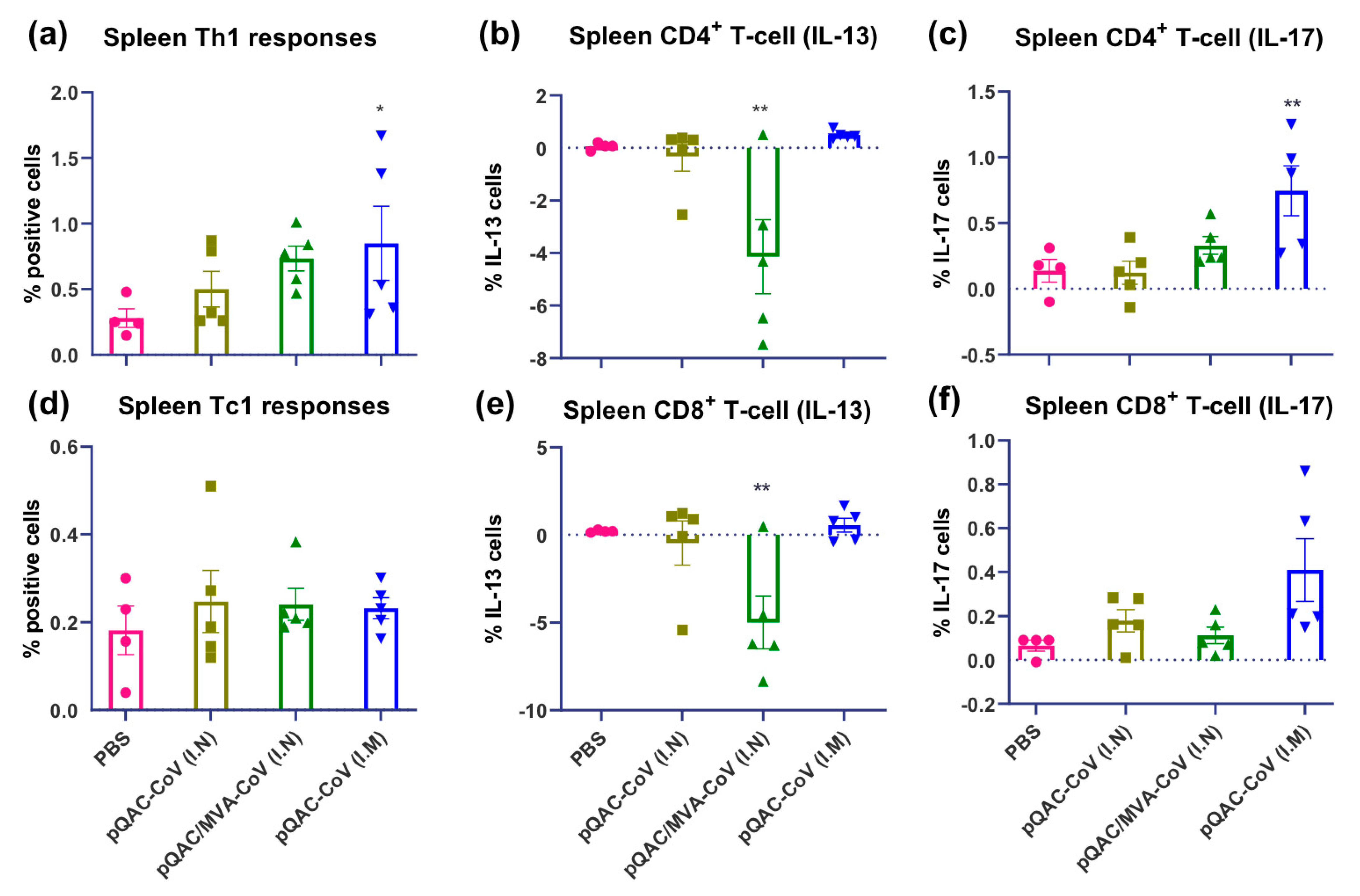
3.4. SARS-CoV-2-Specific Cellular Responses in Vaccinated Mice
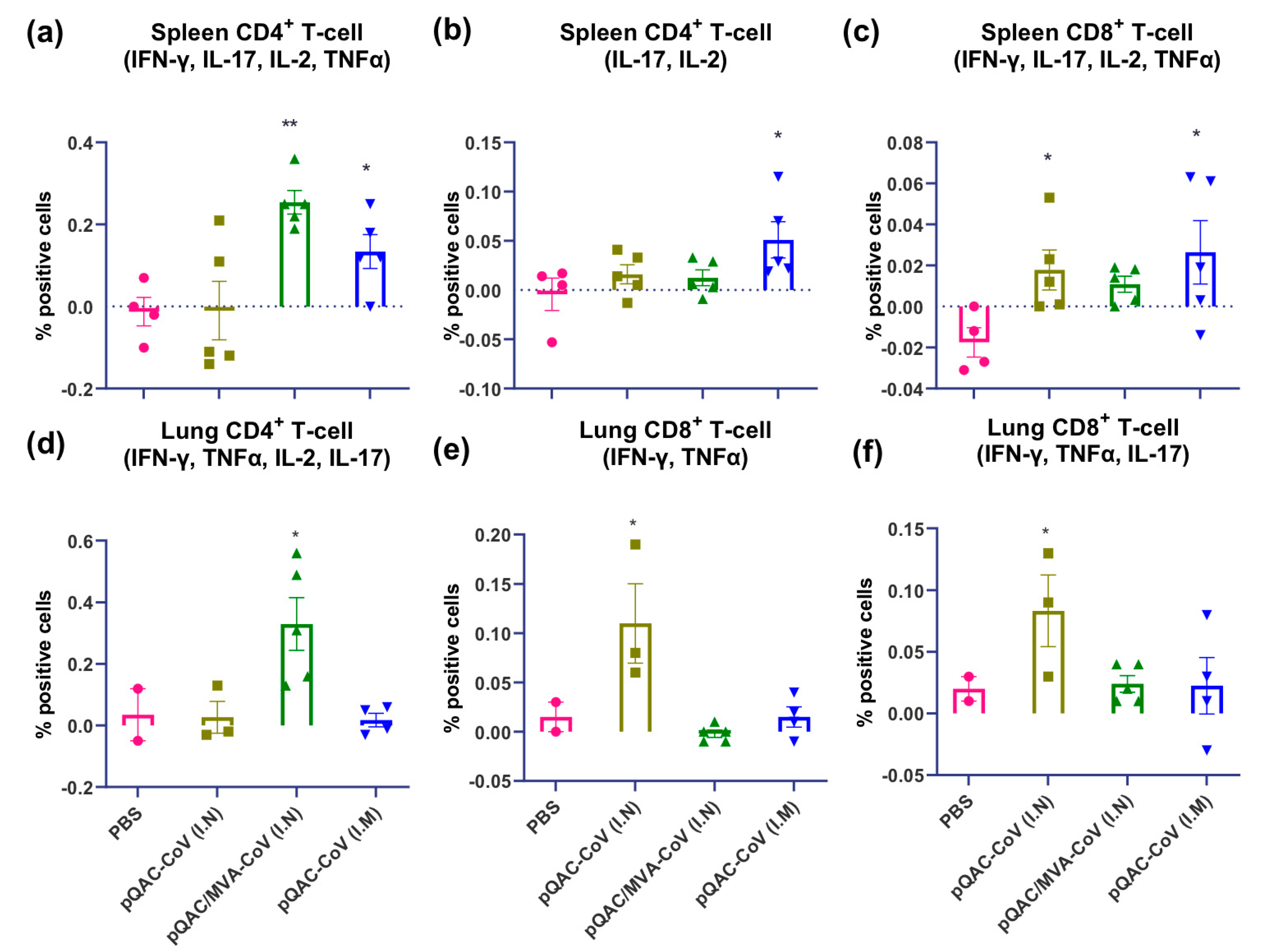
3.5. SARS-CoV-2 Vaccines Induce Polyfunctional T-Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Zhang, N.; Jiang, S.; Du, L. Current advancements and potential strategies in the development of MERS-CoV vaccines. Expert Rev. Vaccines 2014, 13, 761–774. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.; Liao, F.L.; Wang, H.; Tang, H.B.; Yang, Z.Q.; Hou, W. Evaluation of Antibody-Dependent Enhancement of SARS-CoV Infection in Rhesus Macaques Immunized with an Inactivated SARS-CoV Vaccine. Virol. Sin. 2018, 33, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Shang, J.; Jiang, S.; Du, L. Subunit Vaccines Against Emerging Pathogenic Human Coronaviruses. Front. Microbiol. 2020, 11, 298. [Google Scholar] [CrossRef] [PubMed]
- Graham, B.S.; Mascola, J.R.; Fauci, A.S. Novel Vaccine Technologies: Essential Components of an Adequate Response to Emerging Viral Diseases. JAMA 2018, 319, 1431–1432. [Google Scholar] [CrossRef]
- Altenburg, A.F.; Kreijtz, J.H.C.M.; de Vries, R.D.; Song, F.; Fux, R.; Rimmelzwaan, G.F.; Sutter, G.; Volz, A. Modified Vaccinia Virus Ankara (MVA) as Production Platform for Vaccines against Influenza and Other Viral Respiratory Diseases. Viruses 2014, 6, 2735–2761. [Google Scholar] [CrossRef] [PubMed]
- Paules, C.I.; Marston, H.D.; Fauci, A.S. Coronavirus Infections-More Than Just the Common Cold. JAMA 2020, 323, 707–708. [Google Scholar] [CrossRef] [Green Version]
- Talaat, A.M.; Lyons, R.; Johnston, S.A. A combination vaccine confers full protection against co-infections with influenza, herpes simplex and respiratory syncytial viruses. Vaccine 2001, 20, 538–544. [Google Scholar] [CrossRef]
- Cai, P.; Zhang, X.; Wang, M.; Wu, Y.L.; Chen, X. Combinatorial Nano-Bio Interfaces. ACS Nano 2018, 12, 5078–5084. [Google Scholar] [CrossRef] [PubMed]
- Hobernik, D.; Bros, M. DNA Vaccines—How Far From Clinical Use? Int. J. Mol. Sci. 2018, 19, 3605. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekar, S.S.; Kingstad-Bakke, B.A.; Wu, C.W.; Suresh, M.; Talaat, A.M. A Novel Mucosal Adjuvant System for the Immunization Against Avian Coronavirus Causing Infectious Bronchitis. J. Virol. 2020. [Google Scholar] [CrossRef]
- Sogias, I.A.; Williams, A.C.; Khutoryanskiy, V.V. Why is chitosan mucoadhesive? Biomacromolecules 2008, 9, 1837–1842. [Google Scholar] [CrossRef]
- Rajput, Z.I.; Hu, S.H.; Xiao, C.W.; Arijo, A.G. Adjuvant effects of saponins on animal immune responses. J. Zhejiang Univ. Sci. B 2007, 8, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wit, E.; Van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.S.; Flynn, B.; Foulds, K.E.; Francica, J.R.; Boyoglu-Barnum, S.; Werner, A.P.; Flach, B.; O’Connell, S.; Bock, K.W.; Minai, M.; et al. Evaluation of the mRNA-1273 Vaccine against SARS-CoV-2 in Nonhuman Primates. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- van Doremalen, N.; Lambe, T.; Spencer, A.; Belij-Rammerstorfer, S.; Purushotham, J.N.; Port, J.R.; Avanzato, V.; Bushmaker, T.; Flaxman, A.; Ulaszewska, M.; et al. ChAdOx1 nCoV-19 vaccination prevents SARS-CoV-2 pneumonia in rhesus macaques. BioRxiv 2020. [Google Scholar] [CrossRef]
- Smith, T.R.F.; Patel, A.; Ramos, S.; Elwood, D.; Zhu, X.Z.; Yan, J.; Gary, E.N.; Walker, S.N.; Schultheis, K.; Purwar, M.; et al. Immunogenicity of a DNA vaccine candidate for COVID-19. Nat. Commun. 2020, 11, 2601. [Google Scholar] [CrossRef]
- McKay, P.F.; Hu, K.; Blakney, A.K.; Samnuan, K.; Brown, J.C.; Penn, R.; Zhou, J.; Bouton, C.R.; Rogers, P.; Polra, K.; et al. Self-amplifying RNA SARS-CoV-2 lipid nanoparticle vaccine candidate induces high neutralizing antibody titers in mice. Nat. Commun. 2020, 11, 3523. [Google Scholar] [CrossRef]
- Tang, F.; Quan, Y.; Xin, Z.T.; Wrammert, J.; Ma, M.J.; Lv, H.; Wang, T.B.; Yang, H.; Richardus, J.H.; Liu, W.; et al. Lack of Peripheral Memory B Cell Responses in Recovered Patients with Severe Acute Respiratory Syndrome: A Six-Year Follow-Up Study. J. Immunol. 2011, 186, 7264–7268. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Yang, L.T.; Wang, L.Y.; Li, J.; Huang, J.; Lu, Z.Q.; Koup, R.A.; Bailer, R.T.; Wu, C.Y. Long-lived memory T lymphocyte responses against SARS coronavirus nucleocapsid protein in SARS-recovered patients. Virology 2006, 351, 466–475. [Google Scholar] [CrossRef] [Green Version]
- Seow, J.; Graham, C.; Merrick, B.; Acors, S.; Steel, K.J.A.; Hemmings, O.; O’Bryne, A.; Kouphou, N.; Pickering, S.; Galao, R.; et al. Longitudinal evaluation and decline of antibody responses in SARS-CoV-2 infection. MedRxiv 2020. [Google Scholar] [CrossRef]
- Hernandez, R.; Brown, D.T. Growth and Maintenance of Chick Embryo Fibroblasts (CEF). Curr. Protoc. Microbiol. 2010. [Google Scholar] [CrossRef]
- Stading, B.R.; Osorio, J.E.; Velasco-Villa, A.; Smotherman, M.; Kingstad-Bakke, B.; Rocke, T.E. Infectivity of attenuated poxvirus vaccine vectors and immunogenicity of a raccoonpox vectored rabies vaccine in the Brazilian Free-tailed bat (Tadarida brasiliensis). Vaccine 2016, 34, 5352–5358. [Google Scholar] [CrossRef] [Green Version]
- Valosky, J.; Hishiki, H.; Zaoutis, T.E.; Coffin, S.E. Induction of mucosal B-cell memory by intranasal immunization of mice with respiratory syncytial virus. Clin. Diagn. Lab. Immunol. 2005, 12, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, K.H.D.; Eguia, R.; Dingens, A.S.; Loes, A.N.; Malone, K.D.; Wolf, C.R.; Chu, H.Y.; Tortorici, M.A.; Veesler, D.; Murphy, M.; et al. Protocol and Reagents for Pseudotyping Lentiviral Particles with SARS-CoV-2 Spike Protein for Neutralization Assays. Viruses 2020, 12, 513. [Google Scholar] [CrossRef]
- Maeto, C.; Rodriguez, A.M.; Holgado, M.P.; Falivene, J.; Gherardi, M.M. Novel mucosal DNA-MVA HIV vaccination in which DNA-IL-12 plus cholera toxin B subunit (CTB) cooperates to enhance cellular systemic and mucosal genital tract immunity. PLoS ONE 2014, 9, e107524. [Google Scholar] [CrossRef] [PubMed]
- Manrique, M.; Kozlowski, P.A.; Wang, S.W.; Wilson, R.L.; Micewicz, E.; Montefiori, D.C.; Mansfield, K.G.; Carville, A.; Aldovini, A. Nasal DNA-MVA SIV vaccination provides more significant protection from progression to AIDS than a similar intramuscular vaccination. Mucosal Immunol. 2009, 2, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.X.; Rotzschke, O.; Tan, E.K. The role of IgA in COVID-19. Brain Behav. Immun. 2020, 87, 182–183. [Google Scholar] [CrossRef]
- Sterlin, D.; Mathian, A.; Miyara, M.; Mohr, A.; Anna, F.; Claer, L.; Quentric, P.; Fadlallah, J.; Ghillani, P.; Gunn, C.; et al. IgA dominates the early neutralizing antibody response to SARS-CoV-2. MedRxiv 2020. [Google Scholar] [CrossRef]
- Corthesy, B. Multi-faceted functions of secretory IgA at mucosal surfaces. Front. Immunol. 2013, 4, 185. [Google Scholar] [CrossRef] [Green Version]
- Kamlangdee, A.; Kingstad-Bakke, B.; Osorio, J.E. Mosaic H5 Hemagglutinin Provides Broad Humoral and Cellular Immune Responses against Influenza Viruses. J. Virol. 2016, 90, 6771–6783. [Google Scholar] [CrossRef] [Green Version]
- Kamlangdee, A.; Kingstad-Bakke, B.; Anderson, T.K.; Goldberg, T.L.; Osorio, J.E. Broad Protection against Avian Influenza Virus by Using a Modified Vaccinia Ankara Virus Expressing a Mosaic Hemagglutinin Gene. J. Virol. 2014, 88, 13300–13309. [Google Scholar] [CrossRef] [Green Version]
- Wolfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Muller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020, 581, 465–469. [Google Scholar] [CrossRef] [Green Version]
- Mubarak, A.; Alturaiki, W.; Hemida, M.G. Middle East Respiratory Syndrome Coronavirus (MERS-CoV): Infection, Immunological Response, and Vaccine Development. J. Immunol. Res. 2019, 2019, 6491738. [Google Scholar] [CrossRef]
- Hsueh, P.R.; Huang, L.M.; Chen, P.J.; Kao, C.L.; Yang, P.C. Chronological evolution of IgM, IgA, IgG and neutralisation antibodies after infection with SARS-associated coronavirus. Clin. Microbiol. Infect. 2004, 10, 1062–1066. [Google Scholar] [CrossRef] [Green Version]
- Borges, O.; Borchard, G.; Verhoef, J.C.; de Sousa, A.; Junginger, H.E. Preparation of coated nanoparticles for a new mucosal vaccine delivery system. Int. J. Pharm. 2005, 299, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Kingstad-Bakke, B.A.; Chandrasekar, S.S.; Phanse, Y.; Ross, K.A.; Hatta, M.; Suresh, M.; Kawaoka, Y.; Osorio, J.E.; Narasimhan, B.; Talaat, A.M. Effective mosaic-based nanovaccines against avian influenza in poultry. Vaccine 2019, 37, 5051–5058. [Google Scholar] [CrossRef] [PubMed]
- Saade, F.; Petrovsky, N. Technologies for enhanced efficacy of DNA vaccines. Expert Rev. Vaccines 2012, 11, 189–209. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Y.; Kong, W.P.; Huang, Y.; Roberts, A.; Murphy, B.R.; Subbarao, K.; Nabel, G.J. A DNA vaccine induces SARS coronavirus neutralization and protective immunity in mice. Nature 2004, 428, 561–564. [Google Scholar] [CrossRef] [Green Version]
- Zhi, Y.; Wilson, J.M.; Shen, H. SARS vaccine: Progress and challenge. Cell. Mol. Immunol. 2005, 2, 101–105. [Google Scholar] [PubMed]
- Prévost, J.; Gasser, R.; Beaudoin-Bussières, G.; Richard, J.; Duerr, R.; Laumaea, A.; Anand, S.P.; Goyette, G.; Ding, S.; Medjahed, H.; et al. Cross-sectional evaluation of humoral responses against SARS-CoV-2 Spike. BioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Siracusano, G.; Pastori, C.; Lopalco, L. Humoral Immune Responses in COVID-19 Patients: A Window on the State of the Art. Front. Immunol. 2020, 11, 1049. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.T.; Sbrana, E.; Iwata-Yoshikawa, N.; Newman, P.C.; Garron, T.; Atmar, R.L.; Peters, C.J.; Couch, R.B. Immunization with SARS coronavirus vaccines leads to pulmonary immunopathology on challenge with the SARS virus. PLoS ONE 2012, 7, e35421. [Google Scholar] [CrossRef]
- Chandrashekar, A.; Liu, J.; Martinot, A.J.; McMahan, K.; Mercado, N.B.; Peter, L.; Tostanoski, L.H.; Yu, J.; Maliga, Z.; Nekorchuk, M.; et al. SARS-CoV-2 infection protects against rechallenge in rhesus macaques. Science 2020, 369, 812–817. [Google Scholar] [CrossRef]
- Yu, J.; Tostanoski, L.H.; Peter, L.; Mercado, N.B.; McMahan, K.; Mahrokhian, S.H.; Nkolola, J.P.; Liu, J.; Li, Z.; Chandrashekar, A.; et al. DNA vaccine protection against SARS-CoV-2 in rhesus macaques. Science 2020, 369, 806–811. [Google Scholar] [CrossRef]
- Bagri, P.; Anipindi, V.C.; Nguyen, P.V.; Vitali, D.; Stampfli, M.R.; Kaushic, C. Novel Role for Interleukin-17 in Enhancing Type 1 Helper T Cell Immunity in the Female Genital Tract following Mucosal Herpes Simplex Virus 2 Vaccination. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anipindi, V.C.; Bagri, P.; Roth, K.; Dizzell, S.E.; Nguyen, P.V.; Shaler, C.R.; Chu, D.K.; Jimenez-Saiz, R.; Liang, H.; Swift, S.; et al. Estradiol Enhances CD4+ T-Cell Anti-Viral Immunity by Priming Vaginal DCs to Induce Th17 Responses via an IL-1-Dependent Pathway. PLoS Pathog. 2016, 12, e1005589. [Google Scholar] [CrossRef]
- Acharya, D.; Wang, P.; Paul, A.M.; Dai, J.; Gate, D.; Lowery, J.E.; Stokic, D.S.; Leis, A.A.; Flavell, R.A.; Town, T.; et al. Interleukin-17A Promotes CD8+ T Cell Cytotoxicity To Facilitate West Nile Virus Clearance. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Saravia, J.; You, D.; Shaw, A.J.; Cormier, S.A. Impaired gamma delta T cell-derived IL-17A and inflammasome activation during early respiratory syncytial virus infection in infants. Immunol. Cell Biol. 2015, 93, 126–135. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Chan, C.C.; Yang, M.; Deng, J.; Poon, V.K.; Leung, V.H.; Ko, K.H.; Zhou, J.; Yuen, K.Y.; Zheng, B.J.; et al. A critical role of IL-17 in modulating the B-cell response during H5N1 influenza virus infection. Cell. Mol. Immunol. 2011, 8, 462–468. [Google Scholar] [CrossRef] [Green Version]
- Riteau, N.; Sher, A. Chitosan: An Adjuvant with an Unanticipated STING. Immunity 2016, 44, 522–524. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Ma, K.; Chen, M.; Ko, K.H.; Zheng, B.J.; Lu, L. IL-17A Promotes Pulmonary B-1a Cell Differentiation via Induction of Blimp-1 Expression during Influenza Virus Infection. PLoS Pathog. 2016, 12, e1005367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.T.; Yao, X.T.; Peng, Q.; Chen, D.K. The protective and pathogenic roles of IL-17 in viral infections: Friend or foe? Open Biol. 2019, 9, 190109. [Google Scholar] [CrossRef] [Green Version]
- Novelli, F.; Casanova, J.L. The role of IL-12, IL-23 and IFN-gamma in immunity to viruses. Cytokine Growth Factor Rev. 2004, 15, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lorenzi, J.C.C.; Muecksch, F.; Finkin, S.; Viant, C.; Gaebler, C.; Cipolla, M.; Hoffmann, H.H.; Oliveira, T.Y.; Oren, D.A.; et al. Enhanced SARS-CoV-2 neutralization by dimeric IgA. Sci. Transl. Med. 2020. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandrasekar, S.S.; Phanse, Y.; Hildebrand, R.E.; Hanafy, M.; Wu, C.-W.; Hansen, C.H.; Osorio, J.E.; Suresh, M.; Talaat, A.M. Localized and Systemic Immune Responses against SARS-CoV-2 Following Mucosal Immunization. Vaccines 2021, 9, 132. https://doi.org/10.3390/vaccines9020132
Chandrasekar SS, Phanse Y, Hildebrand RE, Hanafy M, Wu C-W, Hansen CH, Osorio JE, Suresh M, Talaat AM. Localized and Systemic Immune Responses against SARS-CoV-2 Following Mucosal Immunization. Vaccines. 2021; 9(2):132. https://doi.org/10.3390/vaccines9020132
Chicago/Turabian StyleChandrasekar, Shaswath S., Yashdeep Phanse, Rachel E. Hildebrand, Mostafa Hanafy, Chia-Wei Wu, Chungyi H. Hansen, Jorge E. Osorio, M. Suresh, and Adel M. Talaat. 2021. "Localized and Systemic Immune Responses against SARS-CoV-2 Following Mucosal Immunization" Vaccines 9, no. 2: 132. https://doi.org/10.3390/vaccines9020132
APA StyleChandrasekar, S. S., Phanse, Y., Hildebrand, R. E., Hanafy, M., Wu, C. -W., Hansen, C. H., Osorio, J. E., Suresh, M., & Talaat, A. M. (2021). Localized and Systemic Immune Responses against SARS-CoV-2 Following Mucosal Immunization. Vaccines, 9(2), 132. https://doi.org/10.3390/vaccines9020132