
Prediction of Adsorption and Diffusion Behaviors of CO2 and CH4 in All-Silica Zeolites Using Molecular Simulation


Abstract
:1. Introduction
2. Theory
3. Materials and Methods
3.1. Synthesis of All-Silica Zeolites
3.1.1. BEA-Type Zeolite
3.1.2. CDO-Type Zeolite
3.1.3. CHA-Type Zeolite
3.1.4. DDR-Type Zeolite
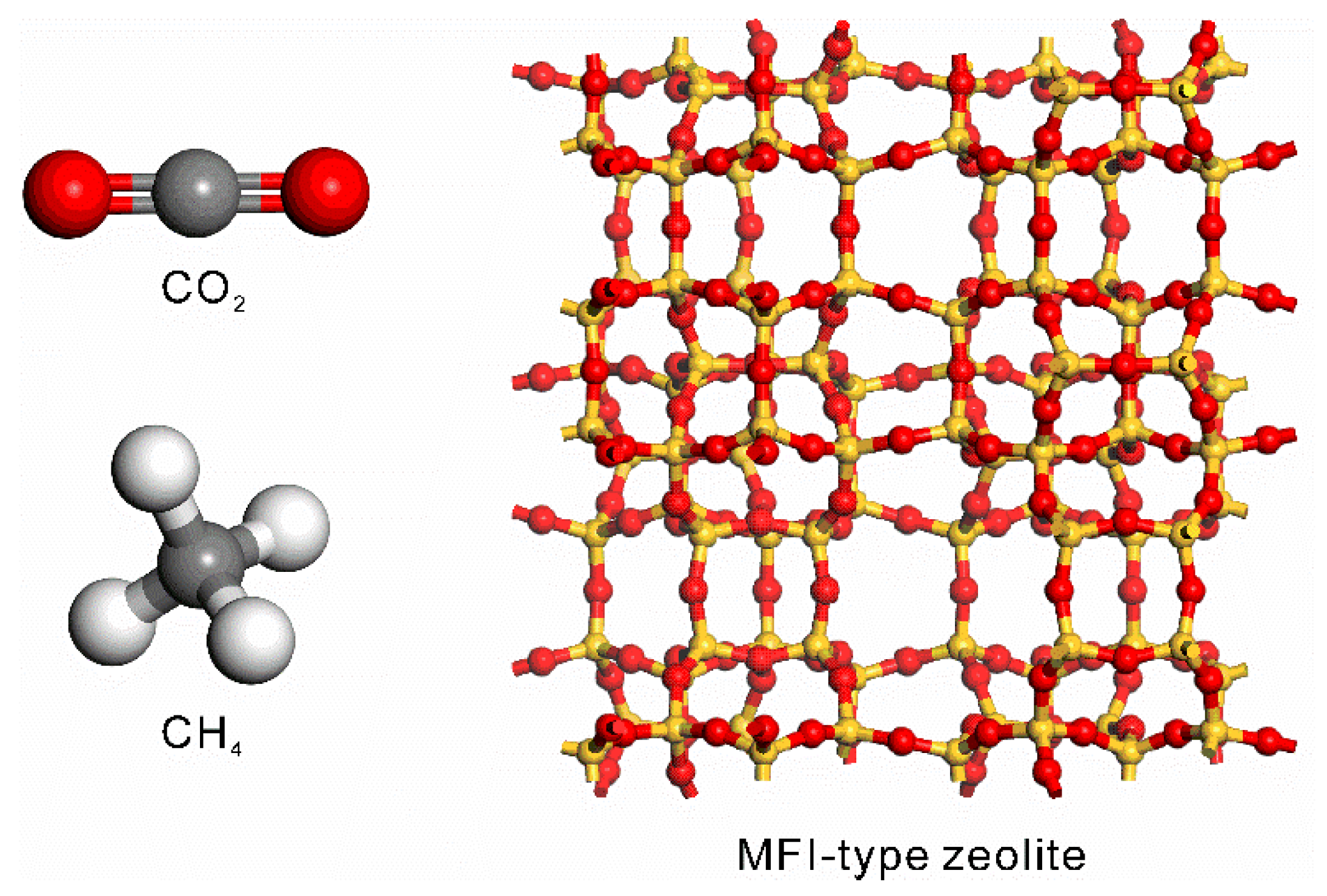
3.1.5. MFI-Type Zeolite
3.1.6. STT-Type Zeolite
3.2. Characterization
3.3. Adsorption Experiment
3.4. Simulation
4. Results and Discussion
4.1. Characterization
4.2. Adsorption Isotherm
4.3. Self-Diffusivity Adsorption Isotherm
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Nomenclatures
| E | potential energy | (kJ mol−1) |
| kb | force constant for bond stretching | (kJ mo−1 m−2) |
| kθ | force constant for angle bending | (kJ mo−1 rad−2) |
| q | partial atomic charge | (e) |
| r | distance | (m) |
| Greeks | ||
| ε | depth of potential | (kJ mol−1) |
| ε0 | vacuum permittivity | (=8.85 × 10−12 F m−1) |
| θ | bonding angle | (rad) |
| σ | distance at zero-potential energy | (m) |
| Subscripts | ||
| 0 | original position | |
| i | component i | |
| ij | pair of atoms i and j | |
References
- Sano, T.; Kiyozumi, Y.; Kawamura, M.; Mizukami, F.; Takaya, H.; Mouri, T.; Inaoka, W.; Toida, Y.; Watanabe, M.; Toyoda, K. Preparation and characterization of ZSM-5 zeolite films. Zeolites 1991, 11, 842–845. [Google Scholar] [CrossRef]
- Geus, E.R.; den Exter, M.J.; van Bekkum, H. Synthesis and characterization of zeolite (MFI) membranes on porous ceramic supports. J. Chem. Soc. Faraday Trans. 1992, 88, 3101–3109. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Matsuura, W.; Abe, C.; Ikeda, A. Influence of organic solvent species on dehydration behaviors of NaA-type zeolite membrane. Membranes 2021, 11, 347. [Google Scholar] [CrossRef]
- Inami, H.; Abe, C.; Hasegawa, Y. Development of ammonia selective permeable zeolite membrane for sensor in sewer system. Membranes 2021, 11, 348. [Google Scholar] [CrossRef]
- Imasaka, S.; Itakura, M.; Yano, K.; Fujita, S.; Okada, M.; Hasegawa, Y.; Abe, C.; Araki, S.; Yamamoto, H. Rapid preparation of high-silica CHA-type zeolite membranes and their separation properties. Sep. Purif. Technol. 2018, 199, 298–303. [Google Scholar] [CrossRef]
- Crreon, M.A.; Li, S.; Falconer, J.L.; Noble, R.D. Alumina-supported SAPO-34 membranes for CO2/CH4 separation. J. Am. Chem. Soc. 2008, 130, 5412–5413. [Google Scholar] [CrossRef] [PubMed]
- Van den Bergh, J.; Zhu, W.; Gascon, J.; Moulijn, J.A.; Kapteijn, F. Separation and permeation characteristics of a DD3R zeolite membrane. J. Membr. Sci. 2008, 316, 35–45. [Google Scholar] [CrossRef]
- Suzuki, S.; Takaba, H.; Yamaguchi, T.; Nakao, S. Estimation of gas permeability of a zeolite membrane, based on a molecular simulation technique and permeation model. J. Phys. Chem. B 2000, 104, 1971–1976. [Google Scholar] [CrossRef]
- Talu, O.; Myers, A.L. Reference potentials for adsorption of helium, argon, methane, and krypton in high-silica zeolites. Colloids Surf. A 2001, 187–188, 83–93. [Google Scholar] [CrossRef]
- Makrodimitris, K.; Papadopoulos, G.K.; Theodorou, D.N. Prediction and permeation properties of CO2 and N2 through silicalite via molecular simulations. J. Phys. Chem. B 2001, 105, 777–788. [Google Scholar] [CrossRef]
- Bai, P.; Tsapatsis, M.; Siepmann, J.I. TraPPE-zeo: Transferable potentials for phase equilibria force field for all-silica zeolites. J. Phys. Chem. C 2013, 117, 24375–24387. [Google Scholar] [CrossRef]
- Rahmati, M.; Modarress, H. Selectivity of new siliceous zeolites for separation of methane and carbon dioxide by Monte Carlo simulation. Micropor. Mesopor. Mater. 2013, 176, 168–177. [Google Scholar] [CrossRef]
- Pham, T.D.; Xiong, R.; Dandler, S.I.; Lobo, R.F. Experimental and computational studies on the adsorption of CO2 and N2 on pure silica zeolites. Micropor. Mesopor. Mater. 2014, 185, 157–166. [Google Scholar] [CrossRef]
- Vujic, B.; Lyubartsev, A.P. Transferable forcefield for modelling of CO2, N2, O2, and Ar in all-silica and Na+ exchanged zeolites. Model. Simul. Mater. Sci. Eng. 2016, 24, 045002. [Google Scholar] [CrossRef]
- Sun, H. COMPASS: An ab initio force-field optimized for condensed phase application-overview with details on alkane and benzene compounds. J. Phys. Chem. B 1998, 102, 7338–7364. [Google Scholar] [CrossRef]
- Yang, J.; Ren, Y.; Tian, A.; Sun, H. COMPASS force field for 14 inorganic molecules, He, N2, Ar, Kr, Xe, H2, O2, N2, NO, CO, CO2, NO2, CS2, and SO2 in liquid phase. J. Phys. Chem. B 2000, 104, 4951–4957. [Google Scholar] [CrossRef]
- Martin, M.G.; Siepmann, J.I. Transferable potentials for phase equilibria. 1. United-atom description of n-alkanes. J. Phys. Chem. B 1998, 102, 2569–2577. [Google Scholar] [CrossRef]
- Martin, M.G.; Siepmann, J.I. Novel configurational Monte Carlo method for branched molecules. Transferable potentials for phase equilibria. 2. United-atom description of branched alkanes. J. Phys. Chem. B 1999, 103, 4508–4517. [Google Scholar] [CrossRef]
- Wick, C.D.; Martin, M.G.; Siepmann, J.I. Transferable potentials for phase equilibria. 4. United-atom description of linear and branched alkanes and alkylbenzenes. J. Phys. Chem. B 2000, 104, 8008–8016. [Google Scholar] [CrossRef] [Green Version]
- Potoff, J.J.; Siepmann, J.I. Vapor-liquid equilibria of mixtures containing alkanes, carbon dioxide, and nitrogen. AIChE J. 2001, 47, 1676–1682. [Google Scholar] [CrossRef]
- Camblor, M.A.; Diaz-Cabanas, M.J. A Synthesis, MAS NMR, Synchrotron X-ray PowderDiffraction, and Computational Study of Zeolite SSZ-23. Chem. Mater. 1999, 11, 2878–2885. [Google Scholar] [CrossRef]
- Ikeda, T.; Akiyama, Y.; Oumi, Y.; Kawai, A.; Mizukami, F. The toporactic conversion of a novel layered silicate into a new framework zeolites. Angew. Chem. Int. Ed. 2004, 43, 4892–4896. [Google Scholar] [CrossRef]
- den Exter, M.J.; Jansen, J.C.; van Bekkum, H. Separation of Permanent Gases on the All-Silica 8-Ring Clathrasil DD3R. Stud. Surf. Sci. Catal. 1994, 84, 1159–1166. [Google Scholar]
- Ban, T.; Takahashi, Y.; Ohya, Y.; Ozeki, Y.; Yoshikawa, M. JP2003-238147.
- Harris, J.G.; Yung, K.H. Carbon dioxide’s liquid-vapor coexistence curve and critical properties as predicted by a simple molecular model. J. Phys. Chem. 1995, 99, 12021–12024. [Google Scholar] [CrossRef]
- Treacy, M.M.J.; Higgins, J.B. Collection of Simulated XRD Powder Patterns for Zeolites, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Sun, M.S.; Shah, D.B.; Xu, H.H.; Talu, O. Adsorption Equilibria of C1 to C4 Alkanes, CO2, and SF6 on Silicalite. J. Phys. Chem. B 1998, 102, 1466–1473. [Google Scholar] [CrossRef]
- Abdul-Rehman, H.B.; Hasanain, M.A.; Loughlin, K.F. Quaternary, Ternary, Binary, and Pure Component Sorption on Zeolites. 1. Light Alkanes on Linde S-115 Silicalite at Moderate to High Pressures. Ind. Eng. Chem. Res. 1990, 29, 1525–1535. [Google Scholar] [CrossRef]
- Himeno, S.; Tomita, T.; Suzuki, K.; Yoshida, S. Characterization and selectivity for methane and carbon dioxide adsorption on the all-silica DD3R zeolite. Micropor. Mesopor. Mater. 2007, 98, 62–69. [Google Scholar] [CrossRef]
- Batsanov, S.S. Van der Waals Radii of Elements. Inorg. Mater. 2001, 37, 871–885. [Google Scholar] [CrossRef]
- Rege, S.U.; Yang, R.T. Corrected Horvath-Kawazoe equations for pore-size distribution. AIChE J. 2000, 46, 734–750. [Google Scholar] [CrossRef] [Green Version]
- Raymonda, J.W.; Muenter, J.S.; Klemperer, W.A. Electric Dipole Moment of SiO and GeO. J. Chem. Phys. 1970, 52, 3458–3461. [Google Scholar] [CrossRef]
- Caro, J.; Bulow, M.; Schirmer, W. Microdynamics of methane, ethane and propane in ZSM-5 type zeolites. J. Chem. Soc. Faraday Trans. 1985, 81, 2541–2550. [Google Scholar] [CrossRef]
- Karger, J.; Pfeifer, H.; Stallmach, F. 129Xe and 13C PFG n.m.r. study of the intracrystalline self-diffusion of Xe, CO2 and CO. Zeolites 1993, 13, 50–55. [Google Scholar] [CrossRef]
- Millot, B.; Methivier, A.; Jobic, H.; Moueddeb, H.; Dalmon, J.A. Permeation of linear and branched alkanes in ZSM-5 supported membranes. Micropor. Mesopor. Mater. 2000, 38, 85–95. [Google Scholar] [CrossRef]
- Talu, O.; Sun, M.S.; Shah, S.B. Diffusivities of n-alkanes in silicalite by steady-state single-crystal membrane technique. AIChE J. 1998, 44, 681–694. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Zeolite | Space Group | a (nm) | b (nm) | c (nm) | α (°) | β (°) | γ (°) | Composition |
|---|---|---|---|---|---|---|---|---|
| BEA | P4122 (No.91) | 1.266 | 1.266 | 2.641 | 90.00 | 90.00 | 90.00 | Si64O128 |
| CDO | Pnma (No.62) | 1.836 | 1.378 | 7.367 | 90.00 | 90.00 | 90.00 | Si36O72 |
| CHA | R3m (No.166) | 0.942 | 0.942 | 0.942 | 94.07 | 94.07 | 94.07 | Si12O24 |
| DDR | R3m (No.166) | 1.386 | 1.386 | 4.089 | 90.00 | 90.00 | 120.00 | Si120O240 |
| MFI | Pnma (No.62) | 2.002 | 1.990 | 1.338 | 90.00 | 90.00 | 90.00 | Si96O192 |
| STT | P21/N (No.14) | 1.309 | 2.167 | 1.373 | 90.00 | 102.58 | 90.00 | Si64O128 |
| Molecule | Element | σ (nm) | ε/k (K) | q (e) | Ref. |
|---|---|---|---|---|---|
| CH4 | C | 0.3730 | 148.0 | 0 | [17] |
| H | --- | --- | 0 | ||
| CO2 | C | 0.2757 | 28.1 | 0.6512 | [25] |
| O | 0.3033 | 80.5 | −0.3256 |
| Elements | σ (nm) | ε (kJ mol−1) | q (e) |
|---|---|---|---|
| Si | 0.421 | 0.954 | 1.10 |
| O | --- | --- | −0.55 |
| Element | Parameter | Bai et al. [11] | Pham et al. [13] | Vujic et al. [14] | This Work |
|---|---|---|---|---|---|
| Si | σ (nm) | 0.231 | --- | 0.297 | 0.421 |
| ε (kJ mol−1) | 0.185 | --- | 0.266 | 0.954 | |
| q (e) | 1.5 | 2.0 | 1.413 | 1.10 | |
| O | σ (nm) | 0.3304 | 0.2806 | 0.3011 | --- |
| ε (kJ mol−1) | 0.442 | 0.744 | 0.432 | --- | |
| q (e) | −0.75 | −1.0 | −0.7065 | −0.55 | |
| Average of difference (mol kg−1) | 0.09 | 0.13 | 0.09 | 0.06 | |
| Standard deviation (mol kg−1) | 0.09 | 0.18 | 0.10 | 0.06 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasegawa, Y.; Abe, C. Prediction of Adsorption and Diffusion Behaviors of CO2 and CH4 in All-Silica Zeolites Using Molecular Simulation. Membranes 2021, 11, 392. https://doi.org/10.3390/membranes11060392
Hasegawa Y, Abe C. Prediction of Adsorption and Diffusion Behaviors of CO2 and CH4 in All-Silica Zeolites Using Molecular Simulation. Membranes. 2021; 11(6):392. https://doi.org/10.3390/membranes11060392
Chicago/Turabian StyleHasegawa, Yasuhisa, and Chie Abe. 2021. "Prediction of Adsorption and Diffusion Behaviors of CO2 and CH4 in All-Silica Zeolites Using Molecular Simulation" Membranes 11, no. 6: 392. https://doi.org/10.3390/membranes11060392
APA StyleHasegawa, Y., & Abe, C. (2021). Prediction of Adsorption and Diffusion Behaviors of CO2 and CH4 in All-Silica Zeolites Using Molecular Simulation. Membranes, 11(6), 392. https://doi.org/10.3390/membranes11060392