
Mechanism Study of Proteins under Membrane Environment

 , , ,
, , , 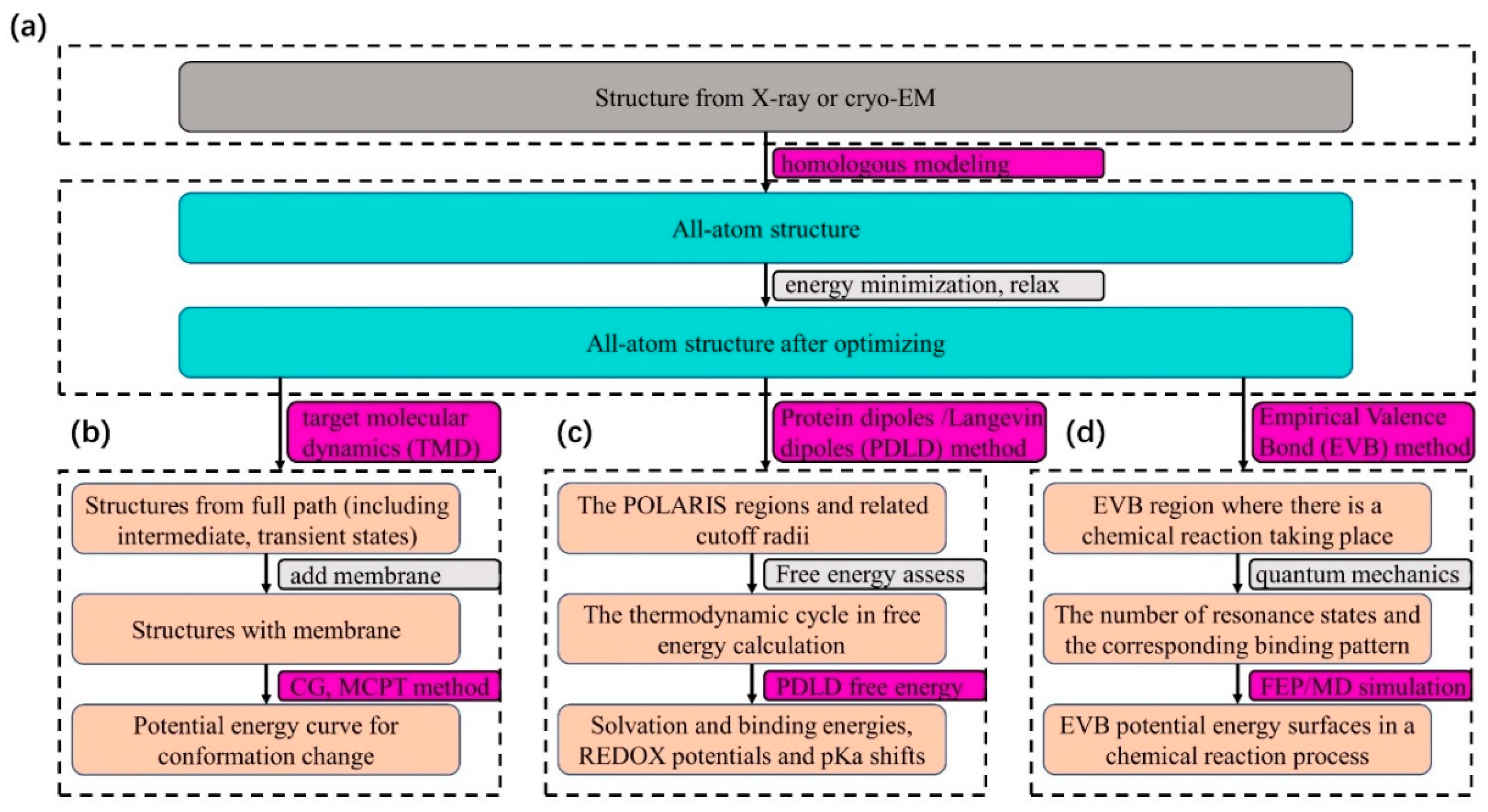{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Model Assembling
2.2. Coarse-Grained (CG) Model, Monte Carlo Proton Transfer (MCPT) Algorithm, and the Calculation Process of Folding Free Energy
2.3. Protein Dipoles/Langevin Dipoles (PDLD) Method and PDLD Energy Calculation Process
2.4. Empirical Valence Bond (EVB) Method and EVB Energy Calculation Process
3. The Gating Mechanism of TMEM16A Ion Channel
4. The Transduction Mechanism of mGlu2 Receptor
5. The Transport Cycle of P4-ATPase Flippase
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Gromiha, M.M.; Ou, Y.Y. Bioinformatics approaches for functional annotation of membrane proteins. Brief. Bioinform. 2014, 15, 155–168. [Google Scholar] [CrossRef]
- Wallin, E.; Heijne, G.V. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998, 7, 1029–1038. [Google Scholar] [CrossRef]
- Yıldırım, M.A.; Goh, K.-I.; Cusick, M.E.; Barabási, A.-L.; Vidal, M. Drug-target network. Nat. Biotechnol. 2007, 25, 1119–1126. [Google Scholar] [CrossRef]
- Yin, H.; Flynn, A.D. Drugging Membrane Protein Interactions. Annu. Rev. Biomed. Eng. 2016, 18, 51–76. [Google Scholar] [CrossRef]
- Errasti-Murugarren, E.; Bartoccioni, P.; Palacín, M. Membrane Protein Stabilization Strategies for Structural and Functional Studies. Membranes 2021, 11, 155. [Google Scholar] [CrossRef]
- Oprea, T.I.; Bologa, C.G.; Brunak, S.; Campbell, A.; Gan, G.N.; Gaulton, A.; Gomez, S.M.; Guha, R.; Hersey, A.; Holmes, J.; et al. Unexplored therapeutic opportunities in the human genome. Nat. Rev. Drug Discov. 2018, 17, 317–332. [Google Scholar] [CrossRef]
- Arinaminpathy, Y.; Khurana, E.; Engelman, D.M.; Gerstein, M.B. Computational analysis of membrane proteins: The largest class of drug targets. Drug Discov. Today 2009, 14, 1130–1135. [Google Scholar] [CrossRef]
- Sachs, J.N.; Engelman, D.M. Introduction to the membrane protein reviews: The interplay of structure, dynamics, and environment in membrane protein function. Annu. Rev. Biochem. 2006, 75, 707–712. [Google Scholar] [CrossRef]
- Bernaudat, F.; Frelet-Barrand, A.; Pochon, N.; Dementin, S.; Hivin, P.; Boutigny, S.; Rioux, J.B.; Salvi, D.; Seigneurin-Berny, D.; Richaud, P.; et al. Heterologous expression of membrane proteins: Choosing the appropriate host. PLoS ONE 2011, 6, e29191. [Google Scholar] [CrossRef]
- Zuo, X.; Li, S.; Hall, J.; Mattern, M.R.; Tran, H.; Shoo, J.; Tan, R.; Weiss, S.R.; Butt, T.R. Enhanced Expression and Purification of Membrane Proteins by SUMO Fusion in Escherichia coli. J. Struct. Funct. Genom. 2005, 6, 103–111. [Google Scholar] [CrossRef]
- Liao, M.; Cao, E.; Julius, D.; Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 2013, 504, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Kolev, V.; Warshel, A. Validating a Coarse-Grained Voltage Activation Model by Comparing Its Performance to the Results of Monte Carlo Simulations. J. Phys. Chem. B 2017, 121, 11284–11291. [Google Scholar] [CrossRef] [PubMed]
- Vorobyov, I.; Kim, I.; Chu, Z.T.; Warshel, A. Refining the treatment of membrane proteins by coarse-grained models. Proteins Struct. Funct. Bioinform. 2016, 84, 92–117. [Google Scholar] [CrossRef] [PubMed]
- Vicatos, S.; Rychkova, A.; Mukherjee, S.; Warshel, A. An effective Coarse-grained model for biological simulations: Recent refinements and validations. Proteins Struct. Funct. Bioinform. 2014, 82, 1168–1185. [Google Scholar] [CrossRef]
- Lin, Y.-L.; Aleksandrov, A.; Simonson, T.; Roux, B. An Overview of Electrostatic Free Energy Computations for Solutions and Proteins. J. Chem. Theory Comput. 2014, 10, 2690–2709. [Google Scholar] [CrossRef]
- Dong, F.; Zhou, H.-X. Electrostatic contribution to the binding stability of protein–protein complexes. Proteins Struct. Funct. Bioinform. 2006, 65, 87–102. [Google Scholar] [CrossRef]
- Alhadeff, R.; Warshel, A. A free-energy landscape for the glucagon-like peptide 1 receptor GLP1R. Proteins 2020, 88, 127–134. [Google Scholar] [CrossRef]
- Bai, C.; Wang, J.; Mondal, D.; Du, Y.; Ye, R.D.; Warshel, A. Exploring the Activation Process of the β2AR-Gs Complex. J. Am. Chem. Soc. 2021, 143, 11044–11051. [Google Scholar] [CrossRef]
- Alhadeff, R.; Vorobyov, I.; Yoon, H.W.; Warshel, A. Exploring the free-energy landscape of GPCR activation. Proc. Natl. Acad. Sci. USA 2018, 115, 10327–10332. [Google Scholar] [CrossRef]
- Bai, C.; Wang, J.; Chen, G.; Zhang, H.; An, K.; Xu, P.; Du, Y.; Ye, R.D.; Saha, A.; Zhang, A.; et al. Predicting Mutational Effects on Receptor Binding of the Spike Protein of SARS-CoV-2 Variants. J. Am. Chem. Soc. 2021, 143, 17646–17654. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Bai, C.; Feliks, M.; Alhadeff, R.; Warshel, A. On the control of the proton current in the voltage-gated proton channel Hv1. Proc. Natl. Acad. Sci. USA 2018, 115, 10321–10326. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [PubMed]
- Martí-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sánchez, R.; Melo, F.; Šali, A. Comparative Protein Structure Modeling of Genes and Genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef]
- Lee, F.S.; Chu, Z.T.; Warshel, A. Microscopic and semimicroscopic calculations of electrostatic energies in proteins by the POLARIS and ENZYMIX programs. J. Comput. Chem. 1993, 14, 161–185. [Google Scholar] [CrossRef]
- Kamerlin, S.C.L.; Vicatos, S.; Dryga, A.; Warshel, A. Coarse-Grained (Multiscale) Simulations in Studies of Biophysical and Chemical Systems. Annu. Rev. Phys. Chem. 2011, 62, 41–64. [Google Scholar] [CrossRef]
- Bai, C.; Asadi, M.; Warshel, A. The catalytic dwell in ATPases is not crucial for movement against applied torque. Nat. Chem. 2020, 12, 1187–1192. [Google Scholar] [CrossRef]
- Shi, D.; An, K.; Zhang, H.; Xu, P.; Bai, C. Application of Coarse-Grained (CG) Models to Explore Conformational Pathway of Large-Scale Protein Machines. Entropy 2022, 24, 620. [Google Scholar] [CrossRef]
- Messer, B.M.; Roca, M.; Chu, Z.T.; Vicatos, S.; Kilshtain, A.V.; Warshel, A. Multiscale simulations of protein landscapes: Using coarse-grained models as reference potentials to full explicit models. Proteins 2010, 78, 1212–1227. [Google Scholar] [CrossRef]
- Beroza, P.; Fredkin, D.R.; Okamura, M.Y.; Feher, G. Protonation of interacting residues in a protein by a Monte Carlo method Application to lysozyme and the photosynthetic reaction center of Rhodobact. Proc. Natl. Acad. Sci. USA 1991, 88, 5804–5808. [Google Scholar] [CrossRef]
- Sham, Y.Y.; Chu, Z.T.; Warshel, A. Consistent calculations of pKa’s of ionizable residues in proteins semi-microscopic and microscopic approaches. J. Phys. Chem. B 1997, 101, 4458–4472. [Google Scholar] [CrossRef]
- Lee, F.S.; Chu, Z.T.; Bolger, M.B.; Warshel, A. Calculations of antibody-antigen interactions microscopic and semi-microscopic evaluation of the free energies of binding of phosphorylcholine analogs to McPC603. Protein Eng. 1992, 5, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Warshel, A.; Weiss, R.M. Empirical valence bond calculations of enzyme catalysis. Ann. N. Y. Acad. Sci. 1981, 367, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Åqvist, J.; Warshel, A. Simulation of Enzyme Reactions Using Valence Bond Force Fields and Other Hybrid Quantum/Classical Approaches. Chem. Rev. 1993, 93, 2523–2544. [Google Scholar] [CrossRef]
- Schroeder, B.C.; Cheng, T.; Jan, Y.N.; Jan, L.Y. Expression Cloning of TMEM16A as a Calcium-Activated Chloride Channel Subunit. Cell 2008, 134, 1019–1029. [Google Scholar] [CrossRef]
- Caputo, A.; Caci, E.; Ferrera, L.; Pedemonte, N.; Barsanti, C.; Sondo, E.; Pfeffer, U.; Ravazzolo, R.; Zegarra-Moran, O.; Galietta, L.J.V. TMEM16A, A Membrane Protein Associated with Calcium-Dependent Chloride Channel Activity. Science 2008, 322, 590–594. [Google Scholar] [CrossRef]
- Yang, Y.D.; Cho, H.; Koo, J.Y.; Tak, M.H.; Cho, Y.; Shim, W.-S.; Park, S.P.; Lee, J.; Lee, B.; Kim, B.-M.; et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 2008, 455, 1210–1215. [Google Scholar] [CrossRef] [PubMed]
- Paulino, C.; Kalienkova, V.; Lam, A.K.M.; Neldner, Y.; Dutzler, R. Activation mechanism of the calcium-activated chloride channel TMEM16A revealed by cryo-EM. Nature 2017, 552, 421–425. [Google Scholar] [CrossRef]
- Lam, A.K.M.; Rheinberger, J.; Paulino, C.; Dutzler, R. Gating the pore of the calcium-activated chloride channel TMEM16A. Nat. Commun. 2021, 12, 785. [Google Scholar] [CrossRef]
- Paulino, C.; Neldner, Y.; Lam, A.K.M.; Kalienkova, V.; Brunner, J.D.; Schenck, S.; Dutzler, R. Structural basis for anion conduction in the calcium-activated chloride channel TMEM16A. eLife 2017, 6, e26232. [Google Scholar] [CrossRef]
- Lam, A.K.M.; Dutzler, R. Mechanism of pore opening in the calcium-activated chloride channel TMEM16A. Nat. Commun. 2021, 12, 786. [Google Scholar] [CrossRef] [PubMed]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.M.; Glukhova, A.; Sexton, P.M.; Christopoulos, A. Structural insights into G-protein-coupled receptor allostery. Nature 2018, 559, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Niswender, C.M.; Conn, P.J. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef] [PubMed]
- Monn, J.A.; Prieto, L.; Taboada, L.; Hao, J.; Reinhard, M.R.; Henry, S.S.; Beadle, C.D.; Walton, L.; Man, T.; Rudyk, H. Synthesis and Pharmacological Characterization of C 4-(Thiotriazolyl)-substituted-2-aminobicyclo [3.1. 0] hexane-2, 6-dicarboxylates. Identification of (1 R, 2 S, 4 R, 5 R, 6 R)-2-Amino-4-(1 H-1, 2, 4-triazol-3-ylsulfanyl) bicyclo [3.1. 0] hexane-2, 6-dicarboxylic Acid (LY2812223), a Highly Potent, Functionally Selective mGlu2 Receptor Agonist. J. Med. Chem. 2015, 58, 7526–7548. [Google Scholar]
- Chappell, M.D.; Li, R.; Smith, S.C.; Dressman, B.A.; Tromiczak, E.G.; Tripp, A.E.; Blanco, M.-J.; Vetman, T.; Quimby, S.J.; Matt, J. Discovery of (1 S, 2 R, 3 S, 4 S, 5 R, 6 R)-2-Amino-3-[(3, 4-difluorophenyl) sulfanylmethyl]-4-hydroxy-bicyclo [3.1. 0] hexane-2, 6-dicarboxylic Acid Hydrochloride (LY3020371 HCl): A Potent, Metabotropic Glutamate 2/3 Receptor Antagonist with Antidepressant-Like Activity. J. Med. Chem. 2016, 59, 10974–10993. [Google Scholar]
- Koehl, A.; Hu, H.; Feng, D.; Sun, B.; Zhang, Y.; Robertson, M.J.; Chu, M.; Kobilka, T.S.; Laeremans, T.; Steyaert, J.; et al. Structural insights into the activation of metabotropic glutamate receptors. Nature 2019, 566, 79–84. [Google Scholar] [CrossRef]
- Wu, H.; Wang, C.; Gregory, K.J.; Han, G.W.; Cho, H.P.; Xia, Y.; Niswender, C.M.; Katritch, V.; Meiler, J.; Cherezov, V.; et al. Structure of a Class C GPCR Metabotropic Glutamate Receptor 1 Bound to an Allosteric Modulator. Science 2014, 344, 58–64. [Google Scholar] [CrossRef]
- Du, J.; Wang, D.; Fan, H.; Xu, C.; Tai, L.; Lin, S.; Han, S.; Tan, Q.; Wang, X.; Xu, T.; et al. Structures of human mGlu2 and mGlu7 homo- and heterodimers. Nature 2021, 594, 589–593. [Google Scholar] [CrossRef]
- Liauw, B.W.; Afsari, H.S.; Vafabakhsh, R. Conformational rearrangement during activation of a metabotropic glutamate receptor. Nat. Chem. Biol. 2021, 17, 291–297. [Google Scholar] [CrossRef]
- Lin, S.; Han, S.; Cai, X.; Tan, Q.; Zhou, K.; Wang, D.; Wang, X.; Du, J.; Yi, C.; Chu, X.; et al. Structures of Gi-bound metabotropic glutamate receptors mGlu2 and mGlu4. Nature 2021, 594, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Seven, A.B.; Barros-Alvarez, X.; de Lapeyriere, M.; Papasergi-Scott, M.M.; Robertson, M.J.; Zhang, C.; Nwokonko, R.M.; Gao, Y.; Meyerowitz, J.G.; Rocher, J.P.; et al. G-protein activation by a metabotropic glutamate receptor. Nature 2021, 595, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Sun, Q. Cyro-EM structure of human mGlus: Leading therapeutic potential to neurological diseases. Signal Transduct. Target. Ther. 2021, 6, 302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Qu, L.; Wu, L.; Tang, X.; Luo, F.; Xu, W.; Xu, Y.; Liu, Z.J.; Hua, T. Structural insights into the activation initiation of full-length mGlu1. Protein Cell 2021, 12, 662–667. [Google Scholar] [CrossRef]
- Patil, S.T.; Zhang, L.; Martenyi, F.; Lowe, S.L.; Jackson, K.A.; Andreev, B.V.; Avedisova, A.S.; Bardenstein, L.M.; Gurovich, I.Y.; Morozova, M.A.; et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: A randomized Phase 2 clinical trial. Nat. Med. 2007, 13, 1102–1107. [Google Scholar] [CrossRef]
- Mantas, I.; Saarinen, M.; Xu, Z.D.; Svenningsson, P. Update on GPCR-based targets for the development of novel antidepressants. Mol. Psychiatry 2021, 27, 534–558. [Google Scholar] [CrossRef]
- Bretscher, M.S. Asymmetrical lipid bilayer structure for biological membranes. Nat. New Biol. 1972, 236, 11–12. [Google Scholar] [CrossRef]
- Op den Kamp, J.A.F. Lipid Asymmetry in Membranes. Annu. Rev. Biochem. 1979, 48, 47–71. [Google Scholar] [CrossRef]
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Doktorova, M.; Symons, J.L.; Levental, I. Structural and functional consequences of reversible lipid asymmetry in living membranes. Nat. Chem. Biol. 2020, 16, 1321–1330. [Google Scholar] [CrossRef]
- Gurtovenko, A.A.; Vattulainen, I. Molecular Mechanism for Lipid Flip-Flops. J. Phys. Chem. B 2007, 111, 13554–13559. [Google Scholar] [CrossRef] [PubMed]
- Timcenko, M.; Dieudonné, T.; Montigny, C.; Boesen, T.; Lyons, J.; Lenoir, G.; Nissen, P. Structural Basis of Substrate-Independent Phosphorylation in a P4-ATPase Lipid Flippase. J. Mol. Biol. 2021, 443, 167062. [Google Scholar] [CrossRef] [PubMed]
- Nintemann, S.J.; Palmgren, M.; López-Marqués, R.L. Catch You on the Flip Side: A Critical Review of Flippase Mutant Phenotypes. Trends Plant Sci. 2019, 24, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.P.; Vestergaard, A.L.; Mikkelsen, S.A.; Mogensen, L.S.; Chalat, M.; Molday, R.S. P4-ATPases as Phospholipid Flippases-Structure, Function, and Enigmas. Front. Physiol. 2016, 7, 275. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Marques, R.L.; Theorin, L.; Palmgren, M.G.; Pomorski, T.G. P4-ATPases: Lipid flippases in cell membranes. Pflügers Arch. Eur. J. Physiol. 2014, 466, 1227–1240. [Google Scholar] [CrossRef]
- Bryde, S.; Hennrich, H.; Verhulst, P.M.; Devaux, P.F.; Lenoir, G.; Holthuis, J.C. CDC50 proteins are critical components of the human class-1 P4-ATPase transport machinery. J. Biol. Chem. 2010, 285, 40562–40572. [Google Scholar] [CrossRef]
- Hiraizumi, M.; Yamashita, K.; Nishizawa, T.; Nureki, O. Cryo-EM structures capture the transport cycle of the P4-ATPase flippase. Science 2019, 365, 1149–1155. [Google Scholar] [CrossRef]
- Fan, Z.Z.; Hwang, J.K.; Warshel, A. Using simplified protein representation as a reference potential for all-atom calculations of folding free energy. Theor. Chem. Acc. 1999, 103, 77–80. [Google Scholar] [CrossRef]
- Stone, A.; Williamson, P. Outside of the box: Recent news about phospholipid translocation by P4 ATPases. J. Chem. Biol. 2012, 5, 131–136. [Google Scholar] [CrossRef]
- Lenoir, G.; Williamson, P.; Holthuis, J.C.M. On the origin of lipid asymmetry: The flip side of ion transport. Curr. Opin. Chem. Biol. 2007, 11, 654–661. [Google Scholar] [CrossRef]
- Bublitz, M.; Morth, J.P.; Nissen, P. P-type ATPases at a glance. J. Cell Sci. 2011, 124, 2515–2519. [Google Scholar] [CrossRef] [PubMed]
- Dyla, M.; Terry, D.S.; Kjaergaard, M.; Sørensen, T.L.M.; Lauwring Andersen, J.; Andersen, J.P.; Rohde Knudsen, C.; Altman, R.B.; Nissen, P.; Blanchard, S.C. Dynamics of P-type ATPase transport revealed by single-molecule FRET. Nature 2017, 551, 346–351. [Google Scholar] [CrossRef]
- Nakanishi, H.; Nishizawa, T.; Segawa, K.; Nureki, O.; Fujiyoshi, Y.; Nagata, S.; Abe, K. Transport Cycle of Plasma Membrane Flippase ATP11C by Cryo-EM. Cell Rep. 2020, 32, 108208. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, H.; Irie, K.; Segawa, K.; Hasegawa, K.; Fujiyoshi, Y.; Nagata, S.; Abe, K. Crystal structure of a human plasma membrane phospholipid flippase. J. Biol. Chem. 2020, 295, 10180–10194. [Google Scholar] [CrossRef] [PubMed]
- Zuttion, F.; Redondo-Morata, L.; Marchesi, A.; Casuso, I. High-Resolution and High-Speed Atomic Force Microscope Imaging. In Nanoscale Imaging: Methods and Protocols; Lyubchenko, Y.L., Ed.; Springer: New York, NY, USA, 2018; pp. 181–200. [Google Scholar]
- Marchesi, A.; Gao, X.; Adaixo, R.; Rheinberger, J.; Stahlberg, H.; Nimigean, C.; Scheuring, S. An iris diaphragm mechanism to gate a cyclic nucleotide-gated ion channel. Nat. Commun. 2018, 9, 3978. [Google Scholar] [CrossRef]
- Ruan, Y.; Miyagi, A.; Wang, X.; Chami, M.; Boudker, O.; Scheuring, S. Direct visualization of glutamate transporter elevator mechanism by high-speed AFM. Proc. Natl. Acad. Sci. USA 2017, 114, 1584–1588. [Google Scholar] [CrossRef]
- Napolitano, L.M.R.; Torre, V.; Marchesi, A. CNG channel structure, function, and gating: A tale of conformational flexibility. Pflügers Arch. Eur. J. Physiol. 2021, 473, 1423–1435. [Google Scholar] [CrossRef]
- Arcangeletti, M.; Marchesi, A.; Mazzolini, M.; Torre, V. Multiple mechanisms underlying rectification in retinal cyclic nucleotide-gated (CNGA1) channels. Physiol. Rep. 2013, 1, e00148. [Google Scholar] [CrossRef]
- Aguilella, V.M.; Bezrukov, S.M. Alamethicin channel conductance modified by lipid charge. Eur. Biophys. J. 2001, 30, 233–241. [Google Scholar]
- Cantor, R.S. The influence of membrane lateral pressures on simple geometric models of protein conformational equilibria. Chem. Phys. Lipids 1999, 101, 45–56. [Google Scholar] [CrossRef]
- Lee, A.G. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta 2004, 1612, 62–87. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Zhu, X.; Zhang, H.; Yan, J.; Xu, P.; Wu, P.; Wu, S.; Bai, C. Mechanism Study of Proteins under Membrane Environment. Membranes 2022, 12, 694. https://doi.org/10.3390/membranes12070694
Zhang Y, Zhu X, Zhang H, Yan J, Xu P, Wu P, Wu S, Bai C. Mechanism Study of Proteins under Membrane Environment. Membranes. 2022; 12(7):694. https://doi.org/10.3390/membranes12070694
Chicago/Turabian StyleZhang, Yue, Xiaohong Zhu, Honghui Zhang, Junfang Yan, Peiyi Xu, Peng Wu, Song Wu, and Chen Bai. 2022. "Mechanism Study of Proteins under Membrane Environment" Membranes 12, no. 7: 694. https://doi.org/10.3390/membranes12070694
APA StyleZhang, Y., Zhu, X., Zhang, H., Yan, J., Xu, P., Wu, P., Wu, S., & Bai, C. (2022). Mechanism Study of Proteins under Membrane Environment. Membranes, 12(7), 694. https://doi.org/10.3390/membranes12070694