
Self-Assembly of Lipid Mixtures in Solutions: Structures, Dynamics Processes and Mechanical Properties


Abstract
:1. Introduction
2. Model and Methodology
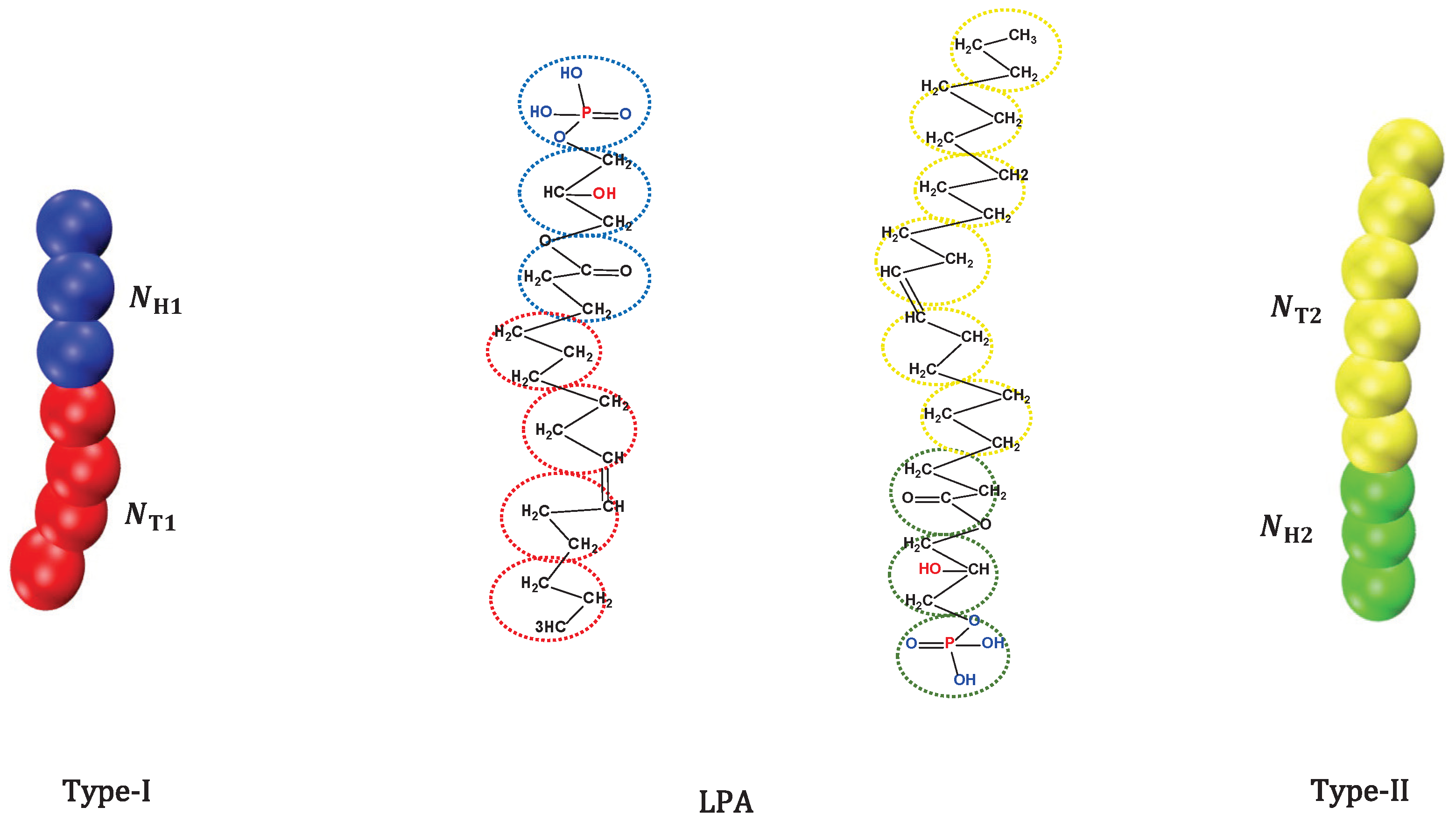
2.1. CG Lipid Model
2.2. Dissipative Particle Dynamics
2.3. Simulation Parameters
3. Results and Discussion
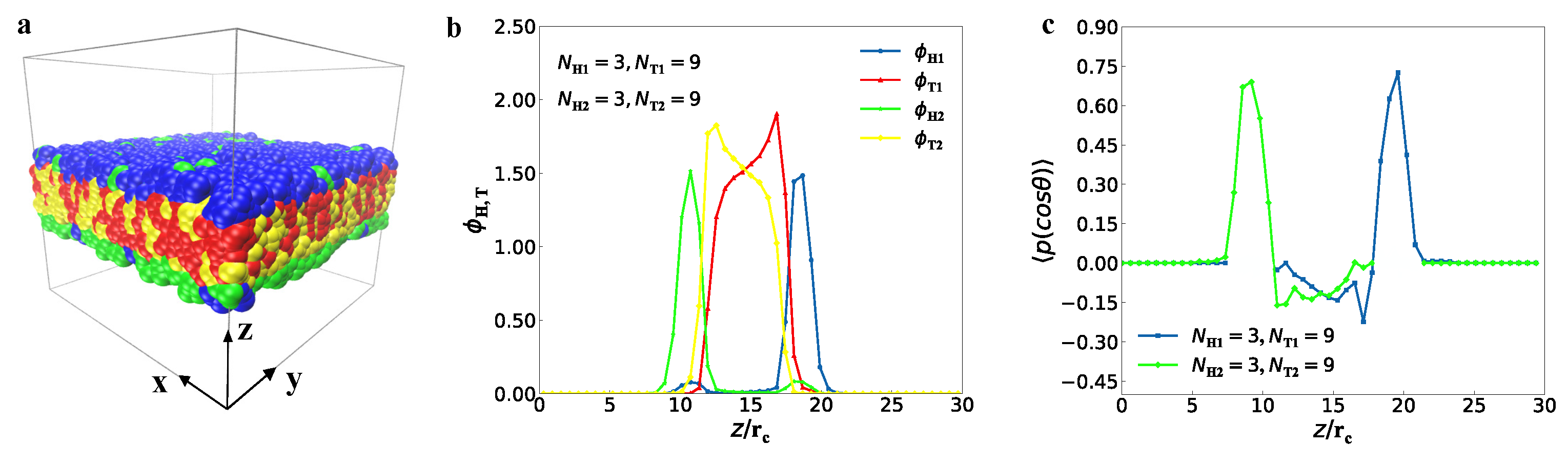
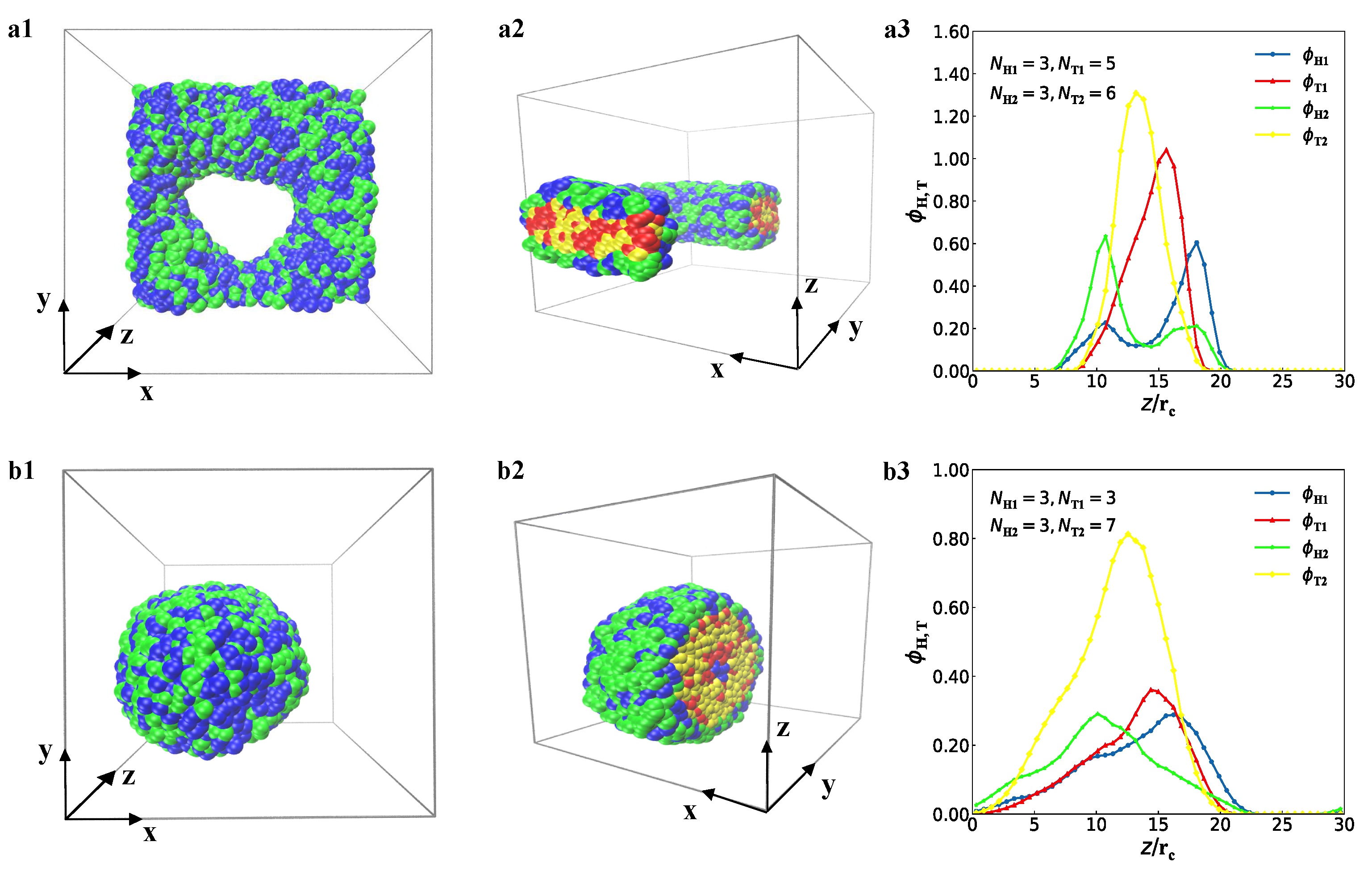
3.1. Typical Structures
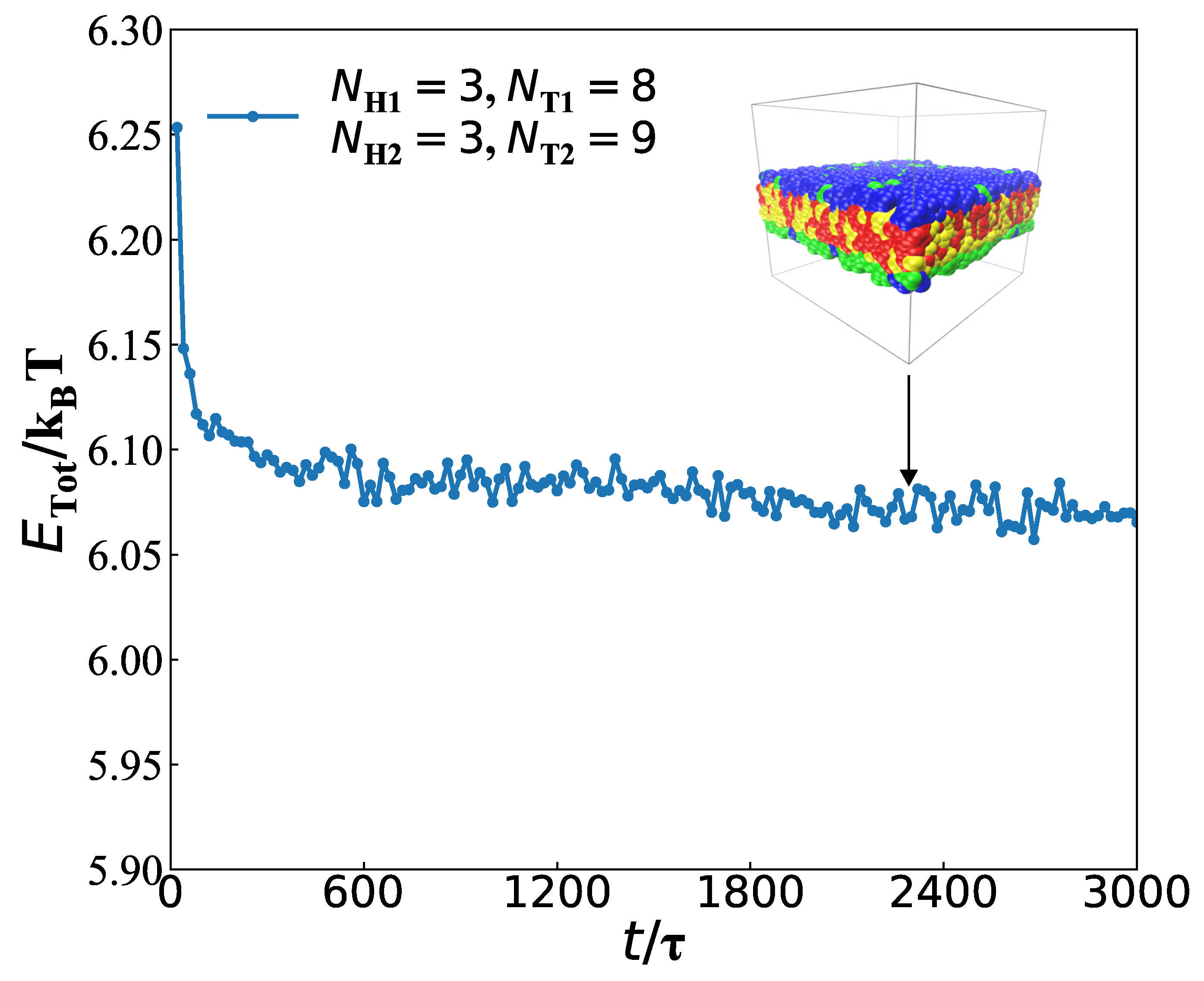
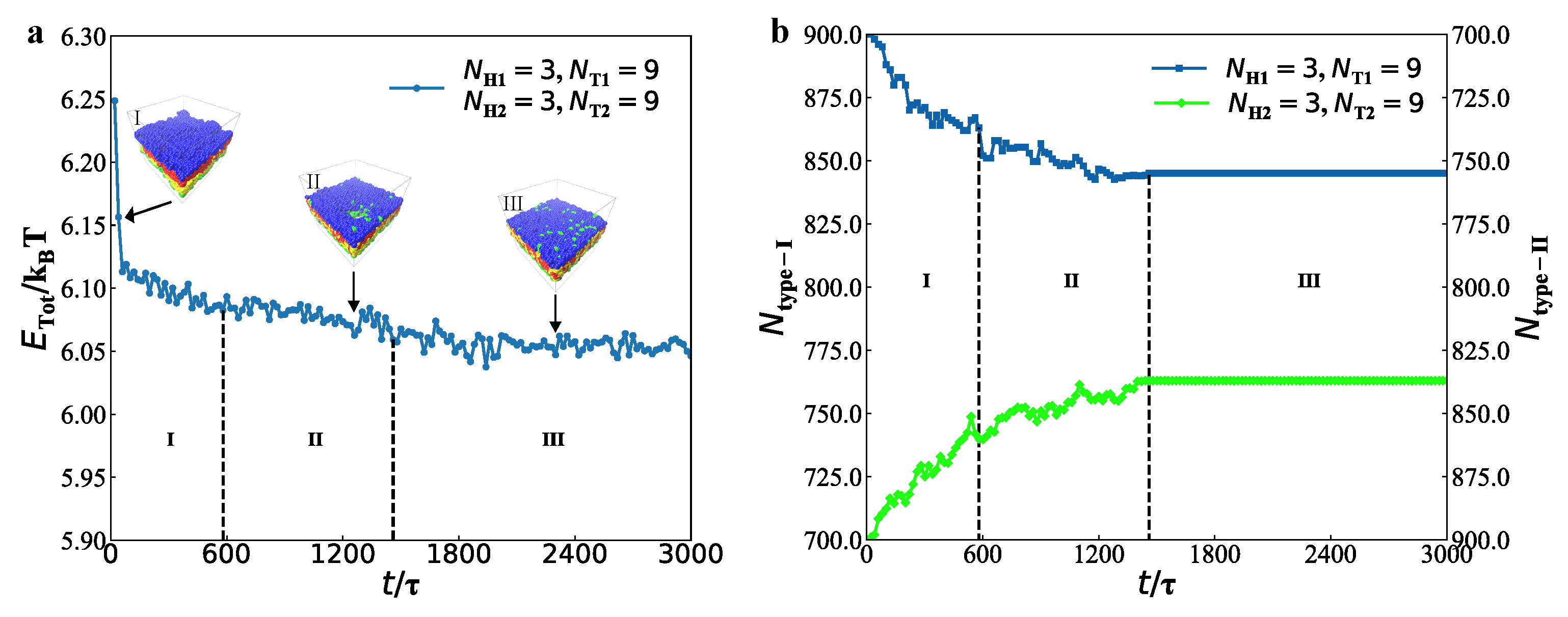
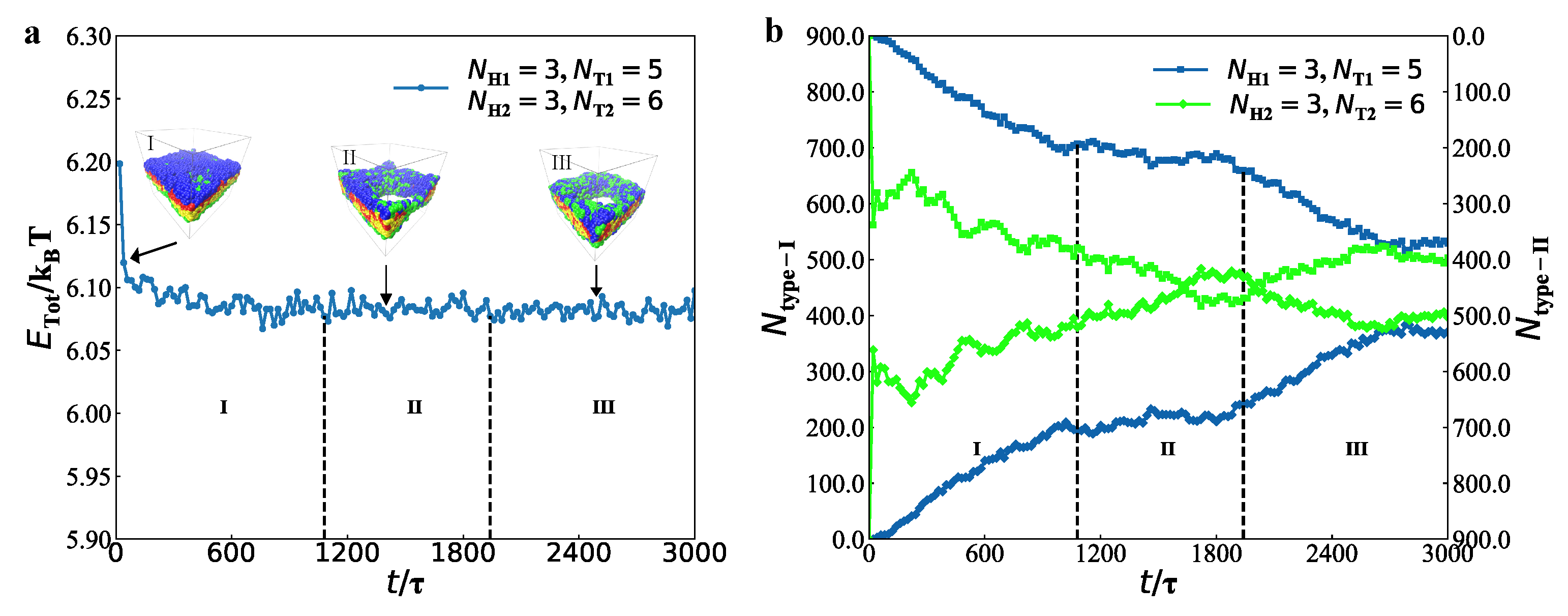
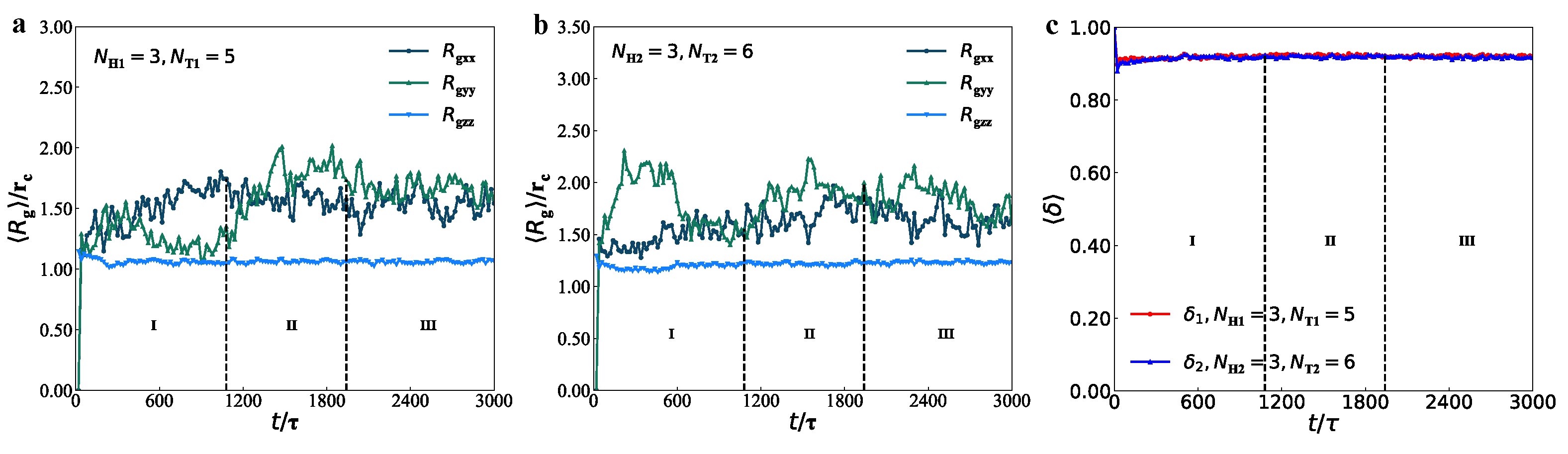
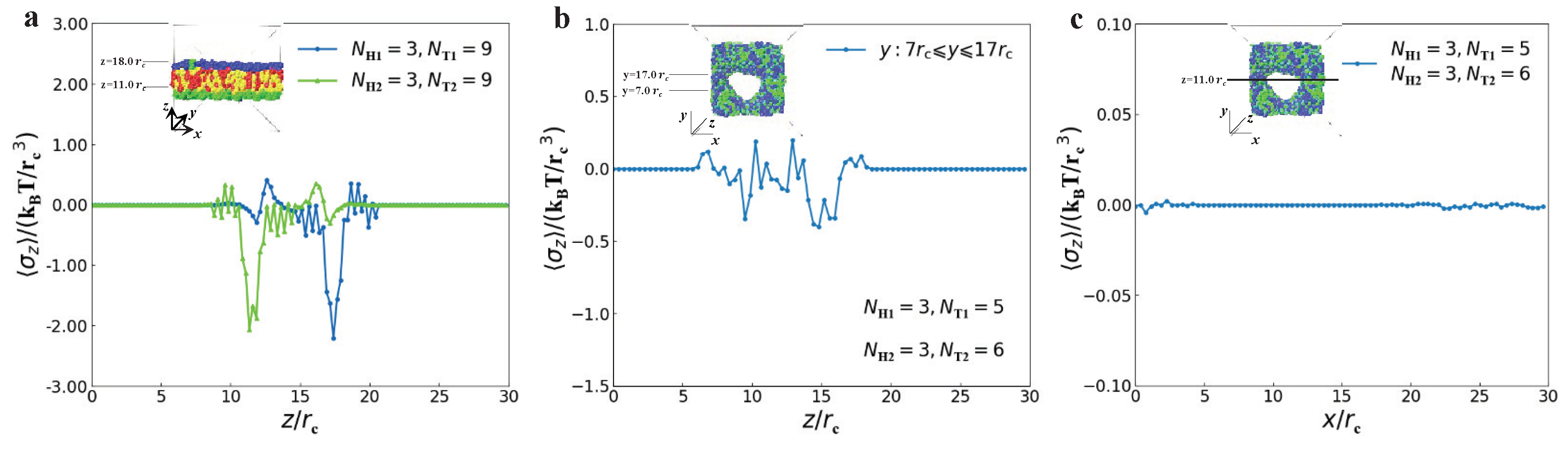
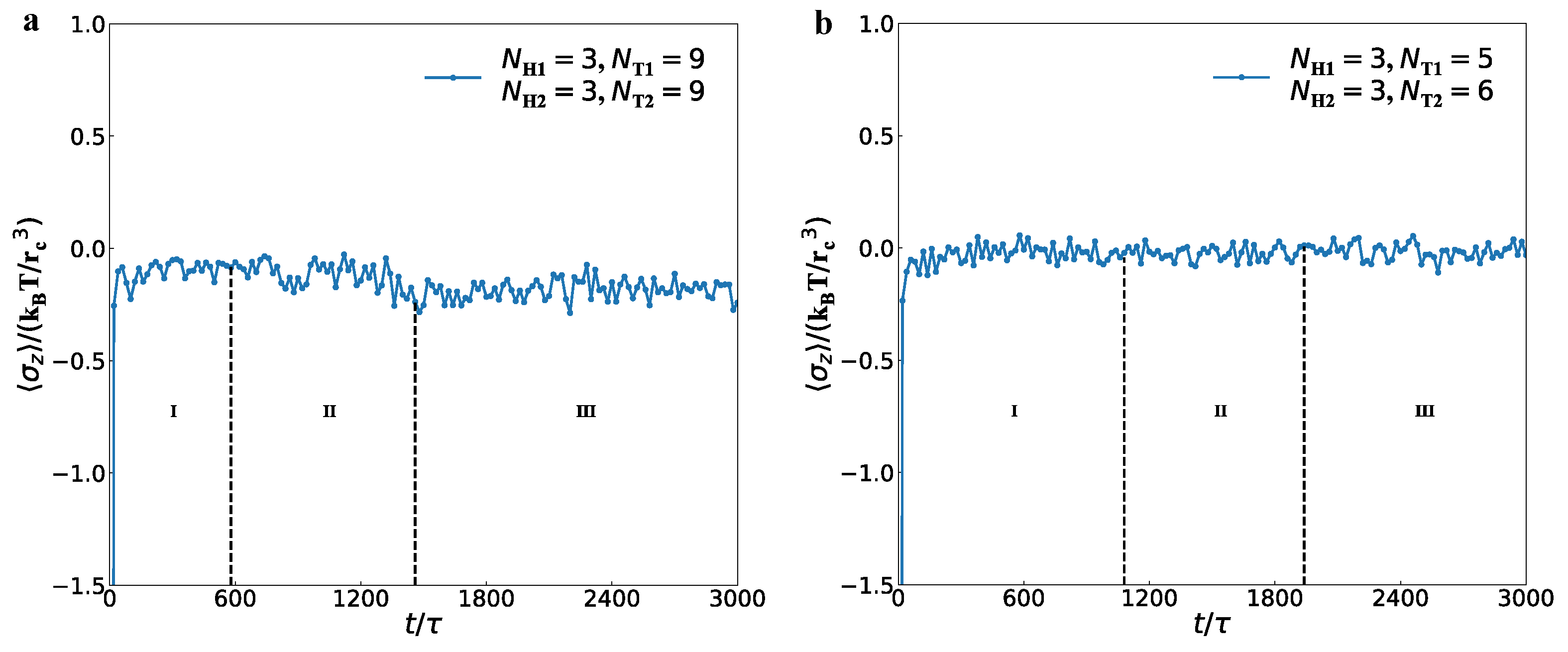
3.2. Dynamics Processes
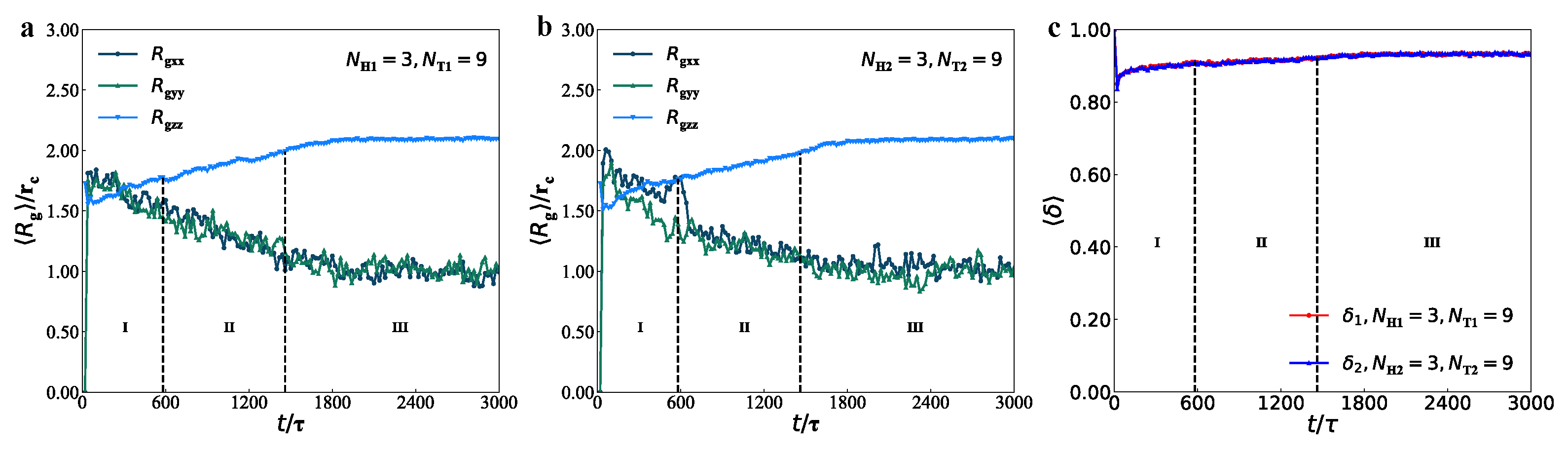
3.3. Mechanical Properties
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kulkarni, C.V. Lipid crystallization: From self-assembly to hierarchical and biological ordering. Nanoscale 2012, 4, 5779–5791. [Google Scholar] [CrossRef]
- Milak, S.; Zimmer, A. Glycerol monooleate liquid crystalline phases used in drug delivery systems. Int. J. Pharm. 2015, 478, 569–587. [Google Scholar] [CrossRef] [PubMed]
- Shi, A.; Claridge, S.A. Lipids: An atomic toolkit for the endless frontier. ACS Nano 2021, 15, 15429–15445. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.M.; Ma, Y.Q. Theoretical and computational investigations of nanoparticle-biomembrane interactions in cellular delivery. Small 2015, 11, 1055–1071. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Chen, P.Y.; Liang, J.S.; Guo, R.H.; Yan, L.T. Receptor-mediated endocytosis of two-dimensional nanomaterials undergoes flat vesiculation and occurs by revolution and self-rotation. ACS Nano 2016, 10, 1493–1502. [Google Scholar] [CrossRef]
- Kang, M.; Tuteja, M.; Centrone, A.; Topgaard, D.; Leal, C. Nanostructured lipid-based films for substrate mediated applications in biotechnology. Adv. Funct. Mater. 2018, 28, 1704356. [Google Scholar] [CrossRef]
- Sud, M.; Fahy, E.; Cotter, D.; Brown, A.; Dennis, E.A.; Glass, C.K.; Merrill, A.H.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.W.; et al. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2007, 35, D527–D532. [Google Scholar] [CrossRef] [Green Version]
- Manni, L.S.; Fong, W.-K.; Mezzenga, R. Lipid-based mesophases as matrices for nanoscale reactions. Nanoscale Horiz. 2020, 5, 914–927. [Google Scholar] [CrossRef]
- Ostroverkhova, O. Organic optoelectronic materials: Mechanisms and applications. Chem. Rev. 2016, 116, 13279–13412. [Google Scholar] [CrossRef]
- Liu, J.H.; Wu, J.H.; Sun, J.J.; Wang, D.Y.; Wang, Z.W. Investigation of the phase behavior of food-grade microemulsions by mesoscopic simulation. Colloids Surf. A 2015, 487, 75–83. [Google Scholar] [CrossRef]
- Kim, S.; Li, C.H.; Farese, R.V.; Walther, T.C.; Voth, G.A. Key factors governing initial stages of lipid droplet formation. J. Phys. Chem. B 2022, 126, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.M.; Ma, Y.Q. Design maps for cellular uptake of gene nanovectors by computer simulation. Biomaterials 2013, 34, 8401–8407. [Google Scholar] [CrossRef] [PubMed]
- Bunker, A.; Magarkar, A.; Viitala, T. Rational design of liposomal drug delivery systems, a review: Combined experimental and computational studies of lipid membranes, liposomes and their PEGylation. Biochim. Biophys. Acta 2016, 1858, 2334–2352. [Google Scholar] [CrossRef] [PubMed]
- Hishida, M.; Seto, H.; Kaewsaiha, P.; Matsuoka, H.; Yoshikawa, K. Stacking structures of dry phospholipid films on a solid substrate. Colloids Surf. A 2006, 284, 444–447. [Google Scholar] [CrossRef] [Green Version]
- Deschamps, J.; Kantsler, V.; Steinberg, V. Phase diagram of single vesicle dynamical states in shear flow. Phys. Rev. Lett. 2009, 102, 118105. [Google Scholar] [CrossRef]
- Qiu, H.; Caffrey, M. The phase diagram of the monoolein/water system: Metastability and equilibrium aspects. Biomaterials 2000, 21, 223–234. [Google Scholar] [CrossRef]
- Schwarz, U.S.; Gompper, G. Stability of inverse bicontinuous cubic phases in lipid-water mixtures. Phys. Rev. Lett. 2000, 85, 1472–1475. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, C.V.; Tang, T.Y.; Seddon, A.M.; Seddon, J.M.; Ces, O.; Templer, R.H. Engineering bicontinuous cubic structures at the nanoscale-the role of chain splay. Soft Matter 2010, 6, 3191–3194. [Google Scholar] [CrossRef]
- Kulkarni, C.V. Nanostructural studies on monoelaidin-water systems at low temperatures. Langmuir 2011, 27, 11790–11800. [Google Scholar] [CrossRef]
- Barriga, H.M.G.; Tyler, A.I.I.; McCarthy, N.L.C.; Parsons, E.S.; Ces, O.; Law, R.V.; Seddon, J.M.; Brooks, N.J. Temperature and pressure tuneable swollen bicontinuous cubic phases approaching nature’s length scales. Soft Matter 2015, 11, 600–607. [Google Scholar] [CrossRef] [Green Version]
- Manni, L.S.; Zabara, A.; Osornio, Y.M.; Schöppe, J.; Batyuk, A.; Plückthun, A.; Siegel, J.S.; Mezzenga, R.; Landau, E.M. Phase behavior of a designed cyclopropyl analogue of monoolein: Implications for low-temperature membrane protein crystallization. Angew. Chem. Int. Ed. 2015, 54, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Oka, T. Transformation between inverse bicontinuous cubic phases of a lipid from diamond to primitive. Langmuir 2015, 31, 3180–3185. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Saiki, T.; Alam, J.M.; Yamazaki, M. Activation energy of the low-pH-induced lamellar to bicontinuous cubic phase transition in dioleoylphosphatidylserine/monoolein. Langmuir 2016, 32, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; Manesi, G.M.; Yang, C.Y.; Hung, Y.C.; Yang, K.C.; Chiu, P.T.; Avgeropoulos, A.; Ho, R.M. Mesoscale networks and corresponding transitions from self-assembly of block copolymers. Proc. Natl. Acad. Sci. USA 2021, 118, e2022275118. [Google Scholar] [CrossRef]
- Den Otter, W.K. Free energies of stable and metastable pores in lipid membranes under tension. J. Chem. Phys. 2009, 131, 205101. [Google Scholar] [CrossRef]
- Petelska, A.D. Interfacial tension of bilayer lipid membranes. Cent. Eur. J. Chem. 2012, 10, 16–26. [Google Scholar] [CrossRef]
- Levadny, V.; Tsuboi, T.-A.; Belaya, M.; Yamazaki, M. Rate constant of tension-induced pore formation in lipid membranes. Langmuir 2013, 29, 3848–3852. [Google Scholar] [CrossRef]
- Takei, T.; Yaguchi, T.; Fujii, T.; Nomoto, T.; Toyota, T.; Fujinami, M. Measurement of membrane tension of free standing lipid bilayers via laser-induced surface deformation spectroscopy. Soft Matter 2015, 11, 8641–8647. [Google Scholar] [CrossRef]
- Elani, Y.; Purushothaman, S.; Booth, P.J.; Seddon, J.M.; Brooks, N.J.; Law, R.V.; Ces, O. Measurements of the effect of membrane asymmetry on the mechanical properties of lipid bilayers. Chem. Commun. 2015, 51, 6976–6979. [Google Scholar] [CrossRef] [Green Version]
- Karal, M.A.; Yamazaki, M. Communication: Activation energy of tension-induced pore formation in lipid membranes. J. Chem. Phys. 2015, 143, 081103. [Google Scholar] [CrossRef] [Green Version]
- Chandran, S.; Baschnagel, J.; Cangialosi, D.; Fukao, K.; Glynos, E.; Janssen, L.M.C.; Müller, M.; Muthukumar, M.; Steiner, U.; Xu, J.; et al. Processing pathways decide polymer properties at the molecular level. Macromolecules 2019, 52, 7146–7156. [Google Scholar] [CrossRef] [Green Version]
- Kei, P.; Howell, M.T.; Chavez, C.A.; Mai, J.C.; Do, C.; Hong, K.; Nesterov, E.E. Kinetically controlled formation of semi-crystalline conjugated polymer nanostructures. Macromolecules 2021, 54, 2162–2177. [Google Scholar] [CrossRef]
- Tran, N.; Hawley, A.M.; Zhai, J.; Muir, B.W.; Fong, C.; Drummond, C.J.; Mulet, X. High-throughput screening of saturated fatty acid influence on nanostructure of lyotropic liquid crystalline lipid nanoparticles. Langmuir 2016, 32, 4509–4520. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Abberton, B.C.; Kröger, M.; Liu, W.K. Challenges in multiscale modeling of polymer dynamics. Polymers 2013, 5, 751–832. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Becton, M.; Wang, X. Designing nanoparticle translocation through cell membranes by varying amphiphilic polymer coatings. J. Phys. Chem. B 2015, 119, 3786–3794. [Google Scholar] [CrossRef]
- Różycki, B.; Lipowsky, R. Spontaneous curvature of bilayer membranes from molecular simulations: Asymmetric lipid densities and asymmetric adsorption. J. Chem. Phys. 2015, 142, 054101. [Google Scholar] [CrossRef]
- Venturoli, M.; Smit, B.; Sperotto, M.M. Simulation studies of protein-induced bilayer deformations, and lipid-induced protein tilting, on a mesoscopic model for lipid bilayers with embedded proteins. Biophys. J. 2005, 88, 1778–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groot, R.D.; Rabone, K.L. Mesoscopic simulation of cell membrane damage, morphology change and rupture by nonionic surfactants. Biophys. J. 2001, 81, 725–736. [Google Scholar] [CrossRef] [Green Version]
- Qiang, X.W.; Wang, X.H.; Ji, Y.Y.; Li, S.B.; He, L.L. Liquid-crystal self-assembly of lipid membranes on solutions: A dissipative particle dynamic simulation study. Polymer 2017, 115, 1–11. [Google Scholar] [CrossRef]
- Español, P.; Warren, P. Statistical mechanics of dissipative particle dynamics. EPL 1995, 30, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Flekkøy, E.G.; Coveney, P.V. From molecular dynamics to dissipative particle dynamics. Phys. Rev. Lett. 1999, 83, 1775–1778. [Google Scholar] [CrossRef] [Green Version]
- Sevink, G.J.; Fraaije, J.G. Efficient solvent-free dissipative particle dynamics for lipid bilayers. Soft Matter 2014, 10, 5129–5146. [Google Scholar] [CrossRef] [PubMed]
- Basan, M.; Prost, J.; Joanny, J.F.; Elgeti, J. Dissipative particle dynamics simulations for biological tissues: Rheology and competition. Phys. Biol. 2011, 8, 026014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Español, P.; Warren, P.B. Perspective: Dissipative particle dynamics. J. Chem. Phys. 2017, 146, 150901. [Google Scholar] [CrossRef]
- Liu, M.B.; Liu, G.R.; Zhou, L.W.; Chang, J.Z. Dissipative particle dynamics (DPD): An overview and recent developments. Arch. Comput. Methods Eng. 2014, 22, 529–556. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.H.; Han, Y.F.; Xu, Z.Y.; Yang, X.Z.; Ramakrishna, S.; Liu, Y. Dissipative particle dynamics simulation: A review on investigating mesoscale properties of polymer systems. Macromol. Mater. Eng. 2021, 306, 2000724. [Google Scholar] [CrossRef]
- Ding, H.M.; Ma, Y.Q. Interactions between Janus particles and membranes. Nanoscale 2012, 4, 1116–1122. [Google Scholar] [CrossRef]
- Kliesch, T.T.; Dietz, J.; Turco, L.; Halder, P.; Polo, E.; Tarantola, M.; Jahn, R.; Janshoff, A. Membrane tension increases fusion efficiency of model membranes in the presence of SNAREs. Sci. Rep. 2017, 7, 12070. [Google Scholar] [CrossRef] [Green Version]
- Li, X.J.; Liu, Y.; Wang, L.; Deng, M.G.; Liang, H.J. Fusion and fission pathways of vesicles from amphiphilic triblock copolymers: A dissipative particle dynamics simulation study. Phys. Chem. Chem. Phys. 2009, 11, 4051–4059. [Google Scholar] [CrossRef]
- Velázquez, M.E.; Gama-Goicochea, A.; González-Melchor, M.; Neria, M.; Alejandre, J. Finite-size effects in dissipative particle dynamics simulations. J. Chem. Phys. 2006, 124, 084104. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Kim, B.N.; Stansfeld, P.J.; Sansom, M.S.; Lindau, M. A coarse grained model for a lipid membrane with physiological composition and leaflet asymmetry. PLoS ONE 2015, 10, e0144814. [Google Scholar] [CrossRef] [Green Version]
- Leermakers, F.A.M.; Scheutjens, J.M.H.M. Statistical thermodynamics of association colloids. I. Lipid bilayer membranes. J. Chem. Phys. 1988, 89, 3264–3274. [Google Scholar] [CrossRef] [Green Version]
- Özen, A.S.; Sen, U.; Atilgan, C. Complete mapping of the morphologies of some linear and graft fluorinated co-oligomers in an aprotic solvent by dissipative particle dynamics. J. Chem. Phys. 2006, 124, 064905. [Google Scholar] [CrossRef] [PubMed]
- Maiti, A.; McGrother, S. Bead-bead interaction parameters in dissipative particle dynamics: Relation to bead-size, solubility parameter, and surface tension. J. Chem. Phys. 2004, 120, 1594–1601. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Wang, Z.G.; Ji, Y.Y.; He, L.L.; Wang, X.H.; Li, S.B. Asymmetric lipid membranes under shear flows: A dissipative particle dynamics study. Membranes 2021, 11, 655. [Google Scholar] [CrossRef]
- Seeger, H.; Marino, G.; Alessandrini, A.; Facci, P. Effect of physical parameters on the main phase transition of supported lipid bilayers. Biophys. J. 2009, 97, 1067–1076. [Google Scholar] [CrossRef] [Green Version]
- Goertz, M.P.; Goyal, N.; Bunker, B.C.; Montaño, G.A. Substrate effects on interactions of lipid bilayer assemblies with bound nanoparticles. J. Colloid Interface Sci. 2011, 358, 635–638. [Google Scholar] [CrossRef]
- Kamiński, D.M.; Matwijczuk, A.; Pociecha, D.; Górecka, E.; Niewiadomy, A.; Dmowska, M.; Gagoś, M. Effect of 2-(4-fluorophenylamino)-5-(2, 4-dihydroxyphenyl)-1, 3, 4-thiadiazole on the molecular organisation and structural properties of the DPPC lipid multibilayers. Biochimi. Biophys. Acta (BBA)-Biomembr. 2012, 1818, 2850–2859. [Google Scholar] [CrossRef] [Green Version]
- Kluczyk, D.; Matwijczuk, A.; Górecki, A.; Karpińska, M.M.; Szymanek, M.; Niewiadomy, A.; Gagoś, M. Molecular organization of dipalmitoylphosphatidylcholine bilayers containing bioactive compounds 4-(5-heptyl-1, 3, 4-thiadiazol-2-yl) benzene-1, 3-diol and 4-(5-methyl-1, 3, 4-thiadiazol-2-yl) benzene-1, 3-diols. J. Phys. Chem. B 2016, 120, 12047–12063. [Google Scholar] [CrossRef] [PubMed]
- Bennett, W.F.; Tieleman, D.P. Computer simulations of lipid membrane domains. Biochim. Biophys. Acta 2013, 1828, 1765–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.Y.; Li, X.; Li, H.; Karniadakis, G.E. MD/DPD multiscale framework for predicting morphology and stresses of red blood cells in health and disease. PLoS Comput. Biol. 2016, 12, e1005173. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, T.M.; Coreta-Gomes, F.; Ollila, O.H.; Moreno, M.J.; Vaz, W.L.; Topgaard, D. Cholesterol and POPC segmental order parameters in lipid membranes: Solid state 1H-13C NMR and MD simulation studies. Phys. Chem. Chem. Phys. 2013, 15, 1976–1989. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Z.; Xu, R.; Kan, D.; He, X.H. Molecular dynamics simulation of electric-field-induced self-assembly of diblock copolymers. J. Chem. Phys. 2016, 144, 234901. [Google Scholar] [CrossRef] [PubMed]
- Alexeev, A.; Uspal, W.E.; Balazs, A.C. Harnessing janus nanoparticles to create controllable pores in membranes. ACS Nano 2008, 2, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Canton, I.; Battaglia, G. Endocytosis at the nanoscale. Chem. Soc. Rev. 2012, 41, 2718–2739. [Google Scholar] [CrossRef]
- Lin, J.; Alexander-Katz, A. Cell membranes open “doors” for cationic nanoparticles/biomolecules: Insights into uptake kinetics. ACS Nano 2013, 7, 10799–10808. [Google Scholar] [CrossRef]
- Stansfeld, P.J.; Goose, J.E.; Caffrey, M.; Carpenter, E.P.; Parker, J.L.; Newstead, S.; Sansom, M.S. MemProtMD: Automated insertion of membrane protein structures into explicit lipid membranes. Structure 2015, 23, 1350–1361. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.H.; Tang, H.Y.; Andrikopoulos, N.; Javed, I.; Cecchetto, L.; Nandakumar, A.; Kakinen, A.; Davis, T.P.; Ding, F.; Ke, P.C. The membrane axis of Alzheimer’s nanomedicine. Adv. NanoBiomed Res. 2021, 1, 2000040. [Google Scholar] [CrossRef]
- Santos, D.E.S.; Pontes, F.J.S.; Lins, R.D.; Coutinho, K.; Soares, T.A. SuAVE: A tool for analyzing curvature-dependent properties in chemical interfaces. J. Chem. Inf. Model. 2020, 60, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Mark, A.E. Molecular dynamics simulation of the formation, structure, and dynamics of small phospholipid vesicles. J. Am. Chem. Soc. 2003, 125, 15233–15242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsen, M.W.; Schick, M. Stable and unstable phases of a diblock copolymer melt. Phys. Rev. Lett. 1994, 72, 2660–2663. [Google Scholar] [CrossRef] [PubMed]
- Li, S.B.; Chen, P.; Zhang, L.X.; Liang, H.J. Geometric frustration phases of diblock copolymers in nanoparticles. Langmuir 2011, 27, 5081–5089. [Google Scholar] [CrossRef] [PubMed]
- Grason, G.M.; DiDonna, B.A.; Kamien, R.D. Geometric theory of diblock copolymer phases. Phys. Rev. Lett. 2003, 91, 058304. [Google Scholar] [CrossRef] [Green Version]
- Shan, Y.; Ji, Y.Y.; Wang, X.H.; He, L.L.; Li, S.B. Predicting asymmetric phospholipid microstructures in solutions. RSC Adv. 2020, 10, 24521–24532. [Google Scholar] [CrossRef]
- Bakardzhiev, P.; Rangelov, S.; Trzebicka, B.; Momekova, D.; Lalev, G.; Garamus, V.M. Nanostructures by self-assembly of polyglycidol-derivatized lipids. RSC Adv. 2014, 4, 37208–37219. [Google Scholar] [CrossRef] [Green Version]
- MacCallum, J.L.; Tieleman, D.P. Computer simulation of the distribution of hexane in a lipid bilayer: Spatially resolved free energy, entropy, and enthalpy profiles. J. Am. Chem. Soc. 2006, 128, 125–130. [Google Scholar] [CrossRef]
- Choudhury, N.; Pettitt, B.M. Enthalpy-entropy contributions to the potential of mean force of nanoscopic hydrophobic solutes. J. Phys. Chem. B 2006, 110, 8459–8463. [Google Scholar] [CrossRef] [Green Version]
- García Daza, F.A.; Colville, A.J.; Mackie, A.D. Chain architecture and micellization: A mean-field coarse-grained model for poly(ethylene oxide) alkyl ether surfactants. J. Chem. Phys. 2015, 142, 114902. [Google Scholar] [CrossRef]
- Su, Z.Q.; Ravindhran, G.; Dias, C.L. Effects of trimethylamine-N-oxide (TMAO) on hydrophobic and charged interactions. J. Phys. Chem. B 2018, 122, 5557–5566. [Google Scholar] [CrossRef] [PubMed]
- Azman, N.; Bekale, L.; Nguyen, T.X.; Kah, J.C.Y. Polyelectrolyte stiffness on gold nanorods mediates cell membrane damage. Nanoscale 2020, 12, 14021–14036. [Google Scholar] [CrossRef] [PubMed]
- Li, W.H.; Liu, M.J.; Qiu, F.; Shi, A.C. Phase diagram of diblock copolymers confined in thin films. J. Phys. Chem. B 2013, 117, 5280–5288. [Google Scholar] [CrossRef]
- Verma, P.; Gandhi, S.; Lata, K.; Chattopadhyay, K. Pore-forming toxins in infection and immunity. Biochem. Soc. Trans. 2021, 49, 455–465. [Google Scholar] [CrossRef]
- Raghupathy, R.; Anilkumar, A.A.; Polley, A.; Singh, P.P.; Yadav, M.; Johnson, C.; Suryawanshi, S.; Saikam, V.; Sawant, S.D.; Panda, A.; et al. Transbilayer lipid interactions mediate nanoclustering of lipid-anchored proteins. Cell 2015, 161, 581–594. [Google Scholar] [CrossRef] [Green Version]
- Doktorova, M.; Symons, J.L.; Levental, I. Structural and functional consequences of reversible lipid asymmetry in living membranes. Nat. Chem. Biol. 2020, 16, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zaburdaev, V. The shape of pinned forced polymer loops. Soft Matter 2019, 15, 1785–1792. [Google Scholar] [CrossRef] [Green Version]
- Jalali, A.; Shahbikian, S.; Huneault, M.A.; Elkoun, S. Effect of molecular weight on the shear-induced crystallization of poly(lactic acid). Polymer 2017, 112, 393–401. [Google Scholar] [CrossRef]
- Rudnick, J.; Gaspari, G. The aspherity of random walks. J. Phys. A Math. Gen. 1986, 19, L191–L193. [Google Scholar] [CrossRef]
- Diehl, H.W.; Eisenriegler, E. Universal shape ratios for open and closed random walks: Exact results for all d. J. Phys. A Math. Gen. 1989, 22, L87–L91. [Google Scholar] [CrossRef]
- Zifferer, G.; Eggerstorfer, D. Monte Carlo simulation studies of the size and shape of linear and star-branched copolymers embedded in a tetrahedral lattice. Macromol. Theory Simul. 2010, 19, 458–482. [Google Scholar] [CrossRef]
- Irving, J.H.; Kirkwood, J.G. The statistical mechanical theory of transport processes. IV. the equations of hydrodynamics. J. Chem. Phys. 1950, 18, 817–829. [Google Scholar] [CrossRef]
- Jiang, F.Y.; Bouret, Y.; Kindt, J.T. Molecular dynamics simulations of the lipid bilayer edge. Biophys. J. 2004, 87, 182–192. [Google Scholar] [CrossRef] [Green Version]
- De Joannis, J.; Jiang, F.Y.; Kindt, J.T. Coarse-grained model simulations of mixed-lipid systems:composition and line tension of a stabilized bilayer edge. Langmuir 2006, 22, 998–1005. [Google Scholar] [CrossRef]
- Wohlert, J.; Otter, W.K.d.; Edholm, O.; Briels, W.J. Free energy of a trans-membrane pore calculated from atomistic molecular dynamics simulations. J. Chem. Phys. 2006, 124, 154905. [Google Scholar] [CrossRef]
- Yamamoto, T.; Safran, S.A. Line tension between domains in multicomponent membranes is sensitive to degree of unsaturation of hybrid lipids. Soft Matter 2011, 7, 7021–7033. [Google Scholar] [CrossRef]
- Li, J.F.; Pastor, K.A.; Shi, A.-C.; Schmid, F.; Zhou, J.J. Elastic properties and line tension of self-assembled bilayer membranes. Phys. Rev. E 2013, 88, 012718. [Google Scholar] [CrossRef] [Green Version]




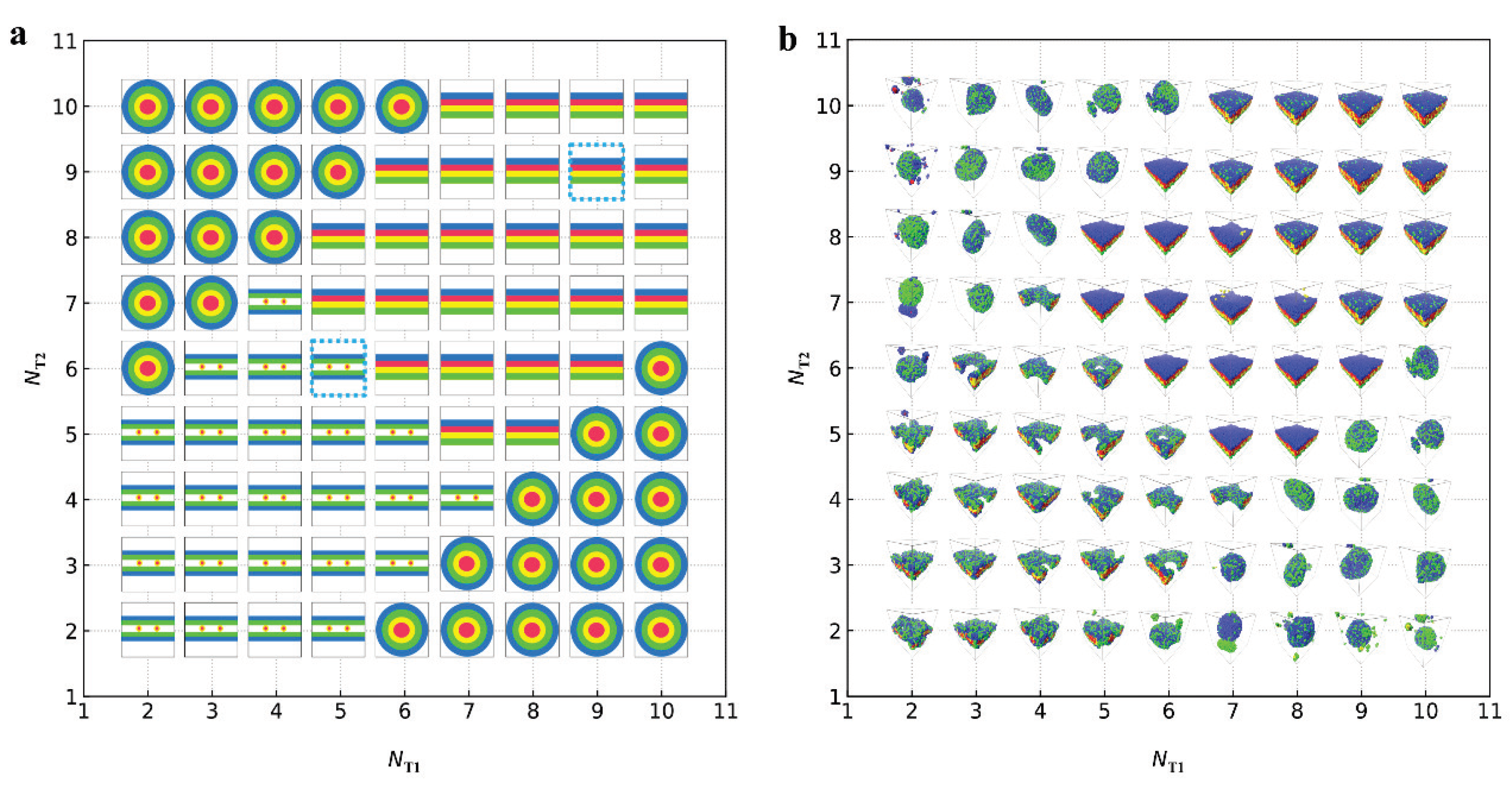
 represent the bilayer membranes, perforated bilayer membranes and spherical vesicles, respectively. (b) Phase diagram arranged with the real snapshots. All the phases are arranged by the numbers of tail beads for two types of lipid chains.
represent the bilayer membranes, perforated bilayer membranes and spherical vesicles, respectively. (b) Phase diagram arranged with the real snapshots. All the phases are arranged by the numbers of tail beads for two types of lipid chains.
represent the bilayer membranes, perforated bilayer membranes and spherical vesicles, respectively. (b) Phase diagram arranged with the real snapshots. All the phases are arranged by the numbers of tail beads for two types of lipid chains.
represent the bilayer membranes, perforated bilayer membranes and spherical vesicles, respectively. (b) Phase diagram arranged with the real snapshots. All the phases are arranged by the numbers of tail beads for two types of lipid chains.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Box size | |||||||
| DPD parameters | |||||||
| Beads | H1  | T1  | T1 | H2  | T2  | ||
| Beads | |||||||
| H1 | 25 | ||||||
| T1 | 100 | 25 | |||||
W  | 25 | 100 | 25 | ||||
| H2 | 25 | 100 | 25 | 25 | |||
| T2 | 100 | 100 | 100 | 100 | 25 | ||
 are the head and tail beads of type-I,
are the head and tail beads of type-I,  is water molecule,
is water molecule,  are the head and tail beads of type-II
respectively.
are the head and tail beads of type-II
respectively.Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, L.; Pan, F.; Li, S. Self-Assembly of Lipid Mixtures in Solutions: Structures, Dynamics Processes and Mechanical Properties. Membranes 2022, 12, 730. https://doi.org/10.3390/membranes12080730
Sun L, Pan F, Li S. Self-Assembly of Lipid Mixtures in Solutions: Structures, Dynamics Processes and Mechanical Properties. Membranes. 2022; 12(8):730. https://doi.org/10.3390/membranes12080730
Chicago/Turabian StyleSun, Lingling, Fan Pan, and Shiben Li. 2022. "Self-Assembly of Lipid Mixtures in Solutions: Structures, Dynamics Processes and Mechanical Properties" Membranes 12, no. 8: 730. https://doi.org/10.3390/membranes12080730
APA StyleSun, L., Pan, F., & Li, S. (2022). Self-Assembly of Lipid Mixtures in Solutions: Structures, Dynamics Processes and Mechanical Properties. Membranes, 12(8), 730. https://doi.org/10.3390/membranes12080730