
A Simple, Semi-Quantitative Acyl Biotin Exchange-Based Method to Detect Protein S-Palmitoylation Levels
 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials and Reagents
2.2. Extraction of Proteins from C2C12 Cells
2.3. Immunoprecipitation and Gel Electrophoresis
2.4. ABE onto PVDF Membranes and in Solution
2.5. Western Blotting
2.6. Statistical Analysis
3. Results and Discussion
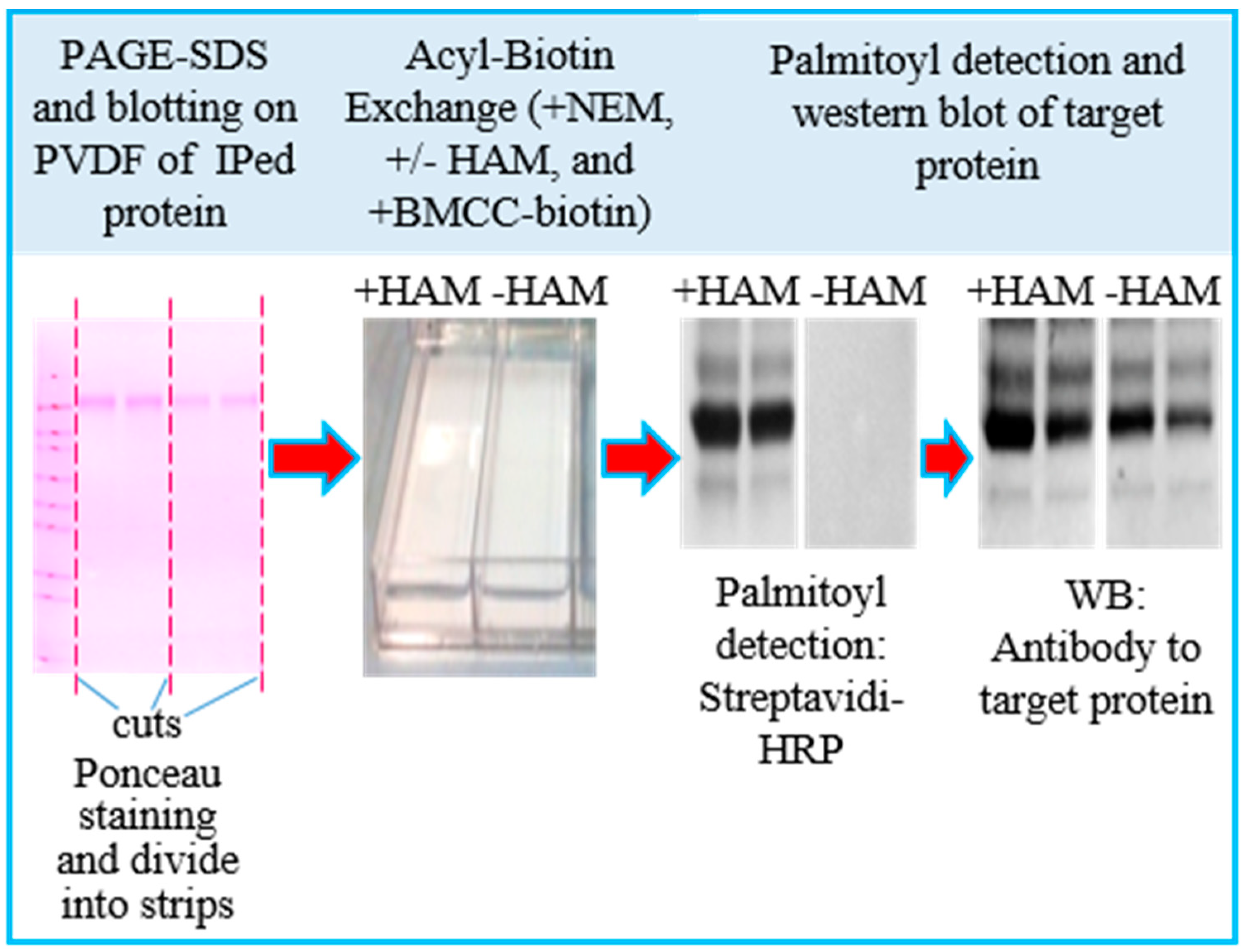
3.1. Assay Design
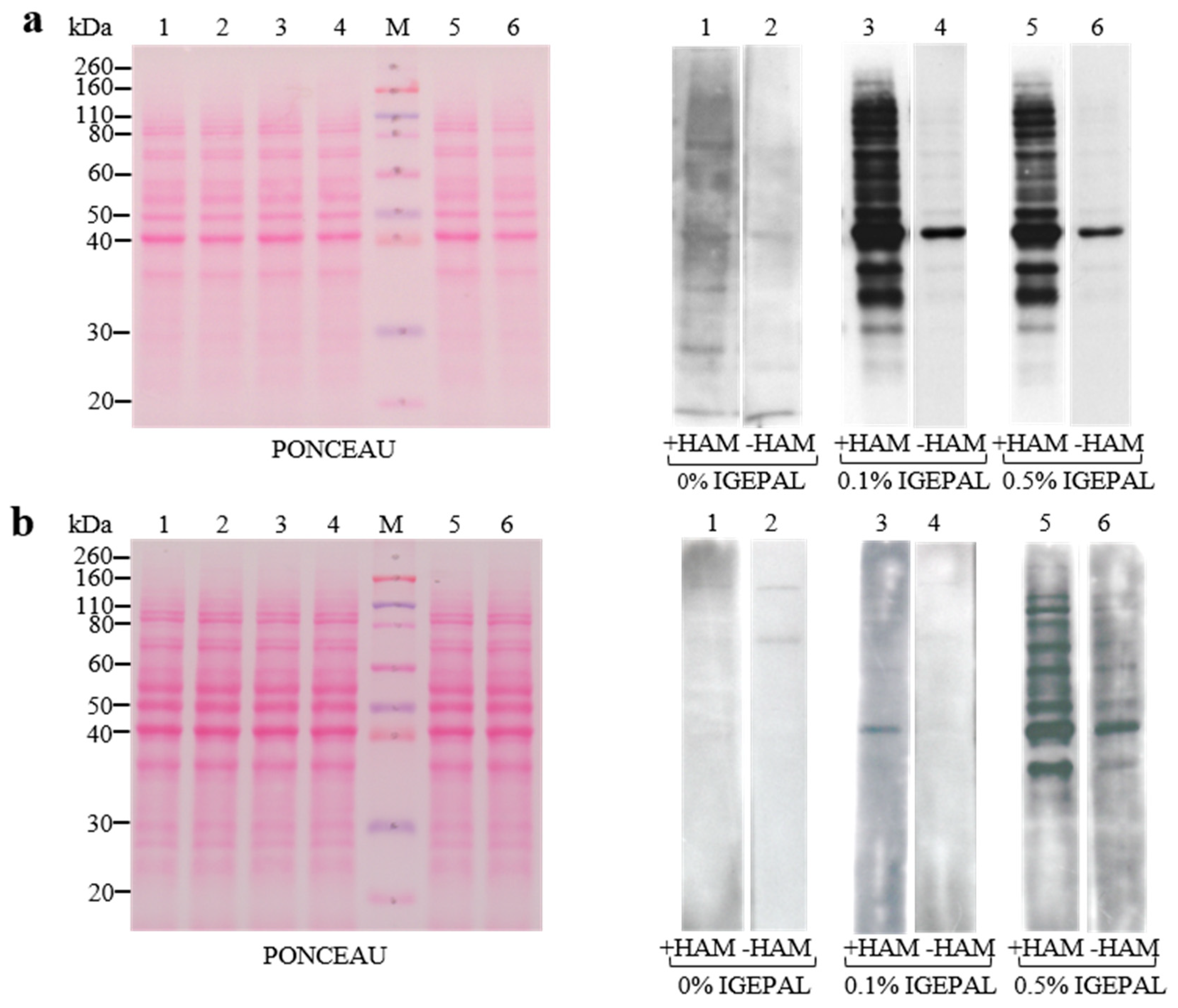
3.2. Setup of Detergent Concentrations in ABE Buffers and Membranes
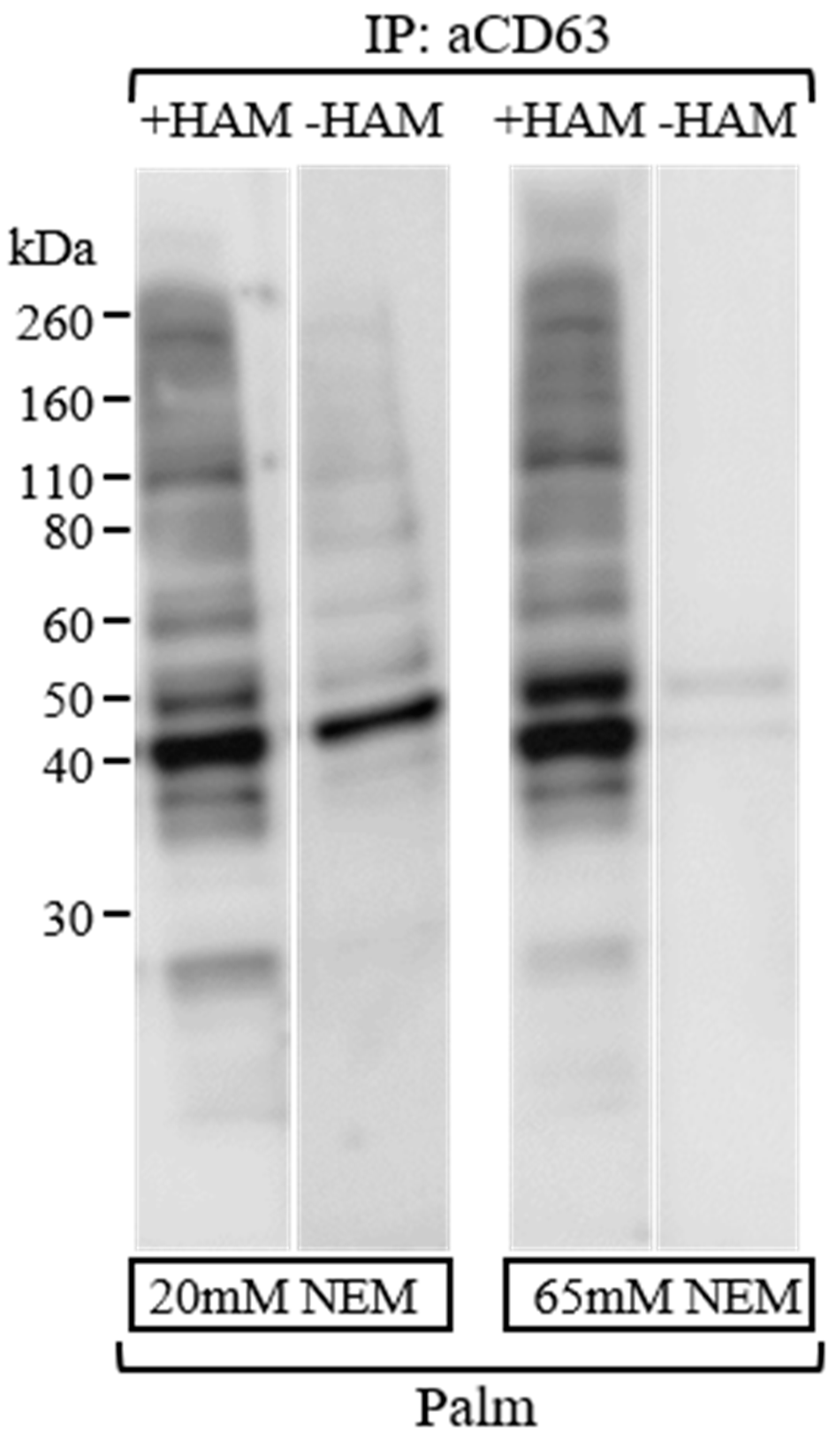
3.3. Efficient Blockage of Unspecific Free Sulfhydryl Groups Using NEM as an Alkylating Agent
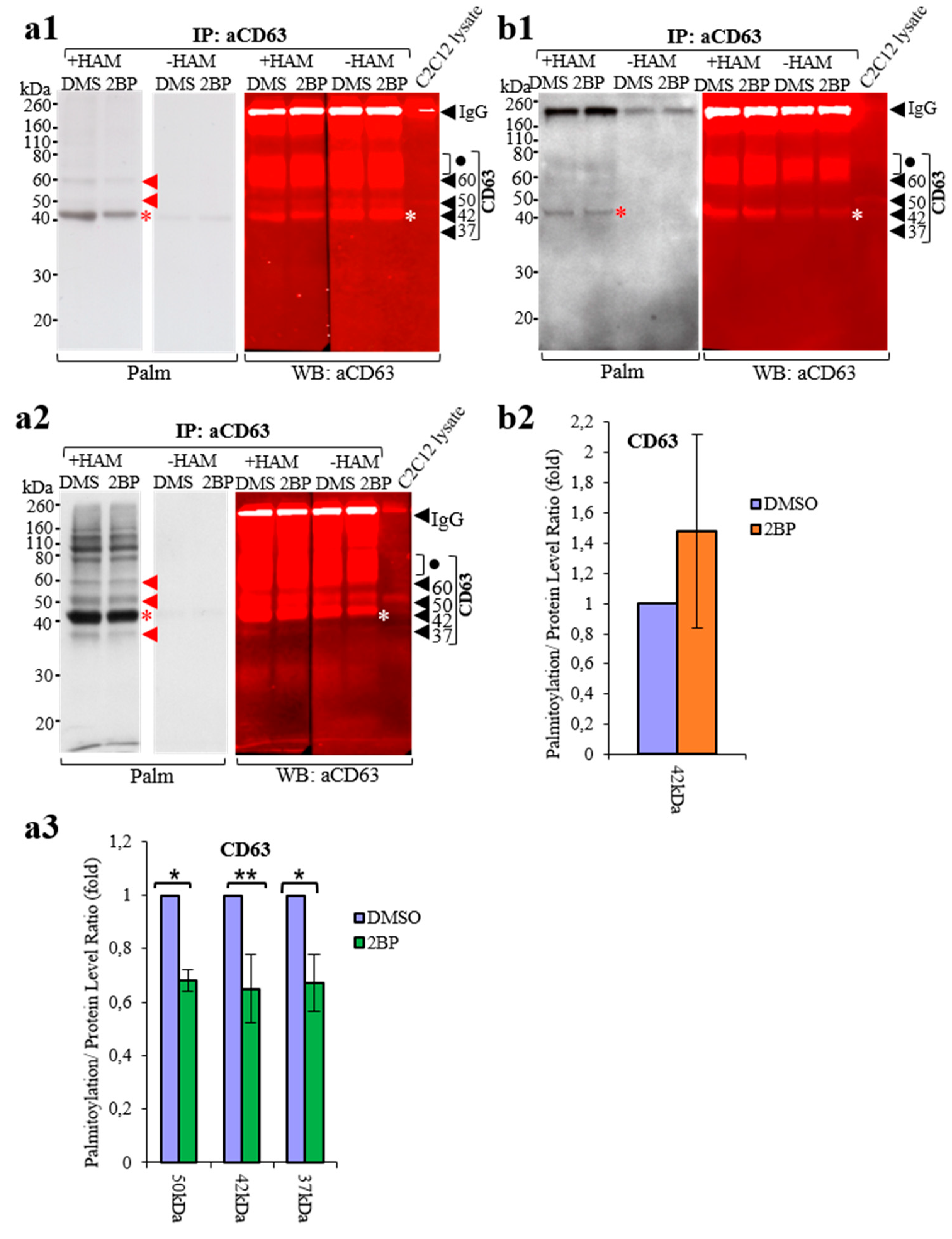
3.4. Comparison of ABE Protocols
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smotrys, J.E.; Linder, M.E. Palmitoylation of intracellular signaling proteins: Regulation and function. Annu. Rev. Biochem. 2004, 73, 559–587. [Google Scholar] [CrossRef]
- Sefton, B.M.; Buss, J.E. The covalent modification of eukaryotic proteins with lipid. J. Cell Biol. 1987, 104, 1449–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aicart-Ramos, C.; Valero, R.A.; Rodriguez-Crespo, I. Protein palmitoylation and subcellular trafficking. Biochim. Biophys. Acta (BBA) Biomembr. 2011, 1808, 2981–2994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaskovic, S.; Adibekian, A.; Blanc, M.; van der Goot, G.F. Mechanistic effects of protein palmitoylation and the cellular consequences thereof. Chem. Phys. Lipids 2014, 180, 44–52. [Google Scholar] [CrossRef]
- Drisdel, R.C.; Alexander, J.K.; Sayeed, A.; Green, W.N. Assays of protein palmitoylation. Methods 2006, 40, 127–134. [Google Scholar] [CrossRef]
- Roth, A.F.; Wan, J.; Bailey, A.O.; Sun, B.; Kuchar, J.A.; Green, W.N.; Phinney, B.S.; Yates, J.R., III; Davis, N.G. Global Analysis of Protein Palmitoylation in Yeast. Cell 2006, 125, 1003–1013. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Zhi, X.; Wang, X.; Meng, D. Protein palmitoylation and its pathophysiological relevance. J. Cell Physiol. 2021, 236, 3220–3233. [Google Scholar] [CrossRef]
- Buss, J.E.; Sefton, B.M. Direct identification of palmitic acid as the lipid attached to p21ras. Mol. Cell Biol. 1986, 6, 116–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancock, J.F.; Magee, A.I.; Childs, J.E.; Marshall, C.J. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 1989, 57, 1167–1177. [Google Scholar] [CrossRef]
- Koegl, M.; Zlatkine, P.; Ley, S.C.; Courtneidge, S.A.; Magee, A.I. Palmitoylation of multiple Src-family kinases at a homologous N-terminal motif. Biochem. J. 1994, 303, 749–753. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Claas, C.; Kraeft, S.-K.; Chen, L.B.; Wang, Z.; Kreidberg, J.A.; Hemler, M.E. Palmitoylation of Tetraspanin Proteins: Modulation of CD151 Lateral Interactions, Subcellular Distribution, and Integrin-dependent Cell Morphology. Mol. Biol. Cell 2002, 13, 767–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romancino, D.P.; Buffa, V.; Caruso, S.; Ferrara, I.; Raccosta, S.; Notaro, A.; Campos, Y.; Noto, R.; Martorana, V.; Cupane, A.; et al. Palmitoylation is a post-translational modification of Alix regulating the membrane organization of exosome-like small extracellular vesicles. Biochim. Biophys. Acta (BBA) Gen. Subj. 2018, 1862, 2879–2887. [Google Scholar] [CrossRef] [Green Version]
- Picciotto, S.; Romancino, D.P.; Buffa, V.; Cusimano, A.; Bongiovanni, A.; Adamo, G. Post-translational lipidation in extracellular vesicles: Chemical mechanisms, biological functions and applications. Adv. Biomembr. Lipid Self-Assem. 2020, 32, 1–29. [Google Scholar] [CrossRef]
- Mariscal, J.; Vagner, T.; Kim, M.; Zhou, B.; Chin, A.; Zandian, M.; Freeman, M.R.; You, S.; Zijlstra, A.; Yang, W.; et al. Comprehensive palmitoyl-proteomic analysis identifies distinct protein signatures for large and small cancer-derived extracellular vesicles. J. Extracell. Vesicles 2020, 9, 1764192. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Schmidt, M.F.G.; Rott, R. Chemical identification of cysteine as palmitoylation site in a transmembrane protein (Semliki Forest virus E1). J. Biol. Chem. 1988, 263, 18635–18639. [Google Scholar] [CrossRef]
- Yap, M.C.; Kostiuk, M.A.; Martin, D.D.O.; Perinpanayagam, M.A.; Hak, P.G.; Siddam, A.; Majjigapu, J.R.; Rajaiah, G.; Keller, B.O.; Prescher, J.A.; et al. Rapid and selective detection of fatty acylated proteins using ω-alkynyl-fatty acids and click chemistry. J. Lipid Res. 2010, 51, 1566–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linder, M.E.; Deschenes, R. Palmitoylation: Policing protein stability and traffic. Nat. Rev. Mol. Cell Biol. 2007, 8, 74–84. [Google Scholar] [CrossRef]
- Wan, J.; Roth, A.F.; Bailey, A.O.; Davis, N.G. Palmitoylated proteins: Purification and identification. Nat. Protoc. 2007, 2, 1573–1584. [Google Scholar] [CrossRef]
- Kostiuk, M.A.; Corvi, M.M.; Keller, B.O.; Plummer, G.; Prescher, J.A.; Hangauer, M.J.; Bertozzi, C.R.; Rajaiah, G.; Falck, J.R.; Berthiaume, L.G. Identification of palmitoylated mitochondrial proteins using a bio-orthogonal azido-palmitate analogue. FASEB J. 2008, 22, 721–732. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, J.L.; Majmudar, J.D.; Martin, B.R. Profiling and inhibiting reversible palmitoylation. Curr. Opin. Chem. Biol. 2013, 17, 20–26. [Google Scholar] [CrossRef] [Green Version]
- Drisdel, R.C.; Green, W.N. Labeling and quantifying sites of protein palmitoylation. Biotechniques 2004, 36, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Brigidi, G.S.; Bamji, S.X. Detection of protein palmitoylation in cultured hippocampal neurons by immunoprecipitation and acyl-biotin exchange (ABE). JoVE-J. Vis. Exp. 2013, 72, 50031–50036. [Google Scholar] [CrossRef] [Green Version]
- Martin, B.R.; Wang, C.; Adibekian, A.; Tully, S.E.; Cravatt, B.F. Global profiling of dynamic protein palmitoylation. Nat. Methods 2012, 9, 84–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Wan, J.; Arstikaitis, P.; Takahashi, H.; Huang, K.; Bailey, A.O.; Thompson, J.X.; Roth, A.F.; Drisdel, R.C.; Mastro, R.; et al. Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature 2008, 456, 904–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Wang, Y.; Yan, Y.; Mariscal, J.; Di Vizio, D.; Freeman, M.R.; Yang, W. Low-Background Acyl-Biotinyl Exchange Largely Eliminates the Coisolation of Non-S-Acylated Proteins and Enables Deep S-Acylproteomic Analysis. Anal. Chem. 2019, 91, 9858–9866. [Google Scholar] [CrossRef]
- Hurst, C.H.; Turnbull, D.; Plain, F.; Fuller, W.; Hemsley, P.A. Maleimide scavenging enhances determination of protein S-palmitoylation state in acyl-exchange methods. Biotechniques 2017, 62, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Tewari, R.; West, S.J.; Shayahati, B.; Akimzhanov, A.M. Detection of Protein S-Acylation using Acyl-Resin Assisted Capture. J. Vis. Exp. 2020, 158, e61016. [Google Scholar] [CrossRef]
- Edmonds, M.J.; Geary, B.; Doherty, M.K.; Morgan, A. Analysis of the brain palmitoyl-proteome using both acyl-biotin exchange and acyl-resin-assisted capture methods. Sci. Rep. 2017, 7, 3299. [Google Scholar] [CrossRef] [Green Version]
- Coleman, R.A.; Rao, P.; Fogelsong, R.J.; Bardes, E.S.-G. 2-Bromopalmitoyl-CoA and 2-bromopalmitate: Promiscuous inhibitors of membrane-bound enzymes. Biochim. Biophys. Acta (BBA) Lipids Lipid Metab. 1992, 1125, 203–209. [Google Scholar] [CrossRef]
- Pedro, M.D.P.; Vilcaes, A.A.; Tomatis, V.; Oliveira, R.G.; Gomez, G.; Daniotti, J.L. 2-Bromopalmitate Reduces Protein Deacylation by Inhibition of Acyl-Protein Thioesterase Enzymatic Activities. PLoS ONE 2013, 8, e75232. [Google Scholar] [CrossRef] [Green Version]
- Charrin, S.; Manié, S.; Oualid, M.; Billard, M.; Boucheix, C.; Rubinstein, E. Differential stability of tetraspanin/tetraspanin interactions: Role of palmitoylation. FEBS Lett. 2002, 516, 139–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tominaga, N.; Hagiwara, K.; Kosaka, N.; Honma, K.; Nakagama, H.; Ochiya, T. RPN2-mediated glycosylation of tetraspanin CD63 regulates breast cancer cell malignancy. Mol. Cancer 2014, 13, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buffa, V.; Adamo, G.; Picciotto, S.; Bongiovanni, A.; Romancino, D.P. A Simple, Semi-Quantitative Acyl Biotin Exchange-Based Method to Detect Protein S-Palmitoylation Levels. Membranes 2023, 13, 361. https://doi.org/10.3390/membranes13030361
Buffa V, Adamo G, Picciotto S, Bongiovanni A, Romancino DP. A Simple, Semi-Quantitative Acyl Biotin Exchange-Based Method to Detect Protein S-Palmitoylation Levels. Membranes. 2023; 13(3):361. https://doi.org/10.3390/membranes13030361
Chicago/Turabian StyleBuffa, Valentina, Giorgia Adamo, Sabrina Picciotto, Antonella Bongiovanni, and Daniele P. Romancino. 2023. "A Simple, Semi-Quantitative Acyl Biotin Exchange-Based Method to Detect Protein S-Palmitoylation Levels" Membranes 13, no. 3: 361. https://doi.org/10.3390/membranes13030361
APA StyleBuffa, V., Adamo, G., Picciotto, S., Bongiovanni, A., & Romancino, D. P. (2023). A Simple, Semi-Quantitative Acyl Biotin Exchange-Based Method to Detect Protein S-Palmitoylation Levels. Membranes, 13(3), 361. https://doi.org/10.3390/membranes13030361