

Specific Features of Mitochondrial Dysfunction under Conditions of Ferroptosis Induced by t-Butylhydroperoxide and Iron: Protective Role of the Inhibitors of Lipid Peroxidation and Mitochondrial Permeability Transition Pore Opening


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
2.2. Preparation of Rat Liver Mitochondria
2.3. Estimation of Swelling of Mitochondria
2.4. Estimation of Oxidation of Succinate and NAD-Dependent Substrates by the Methyl Thiazolyl Tetrazolium (MTT) Assay
2.5. Determination of the Redox State of Pyridine Nucleotides and Oxidative Phosphorylation in Mitochondria
2.6. Statistical Analysis
3. Results
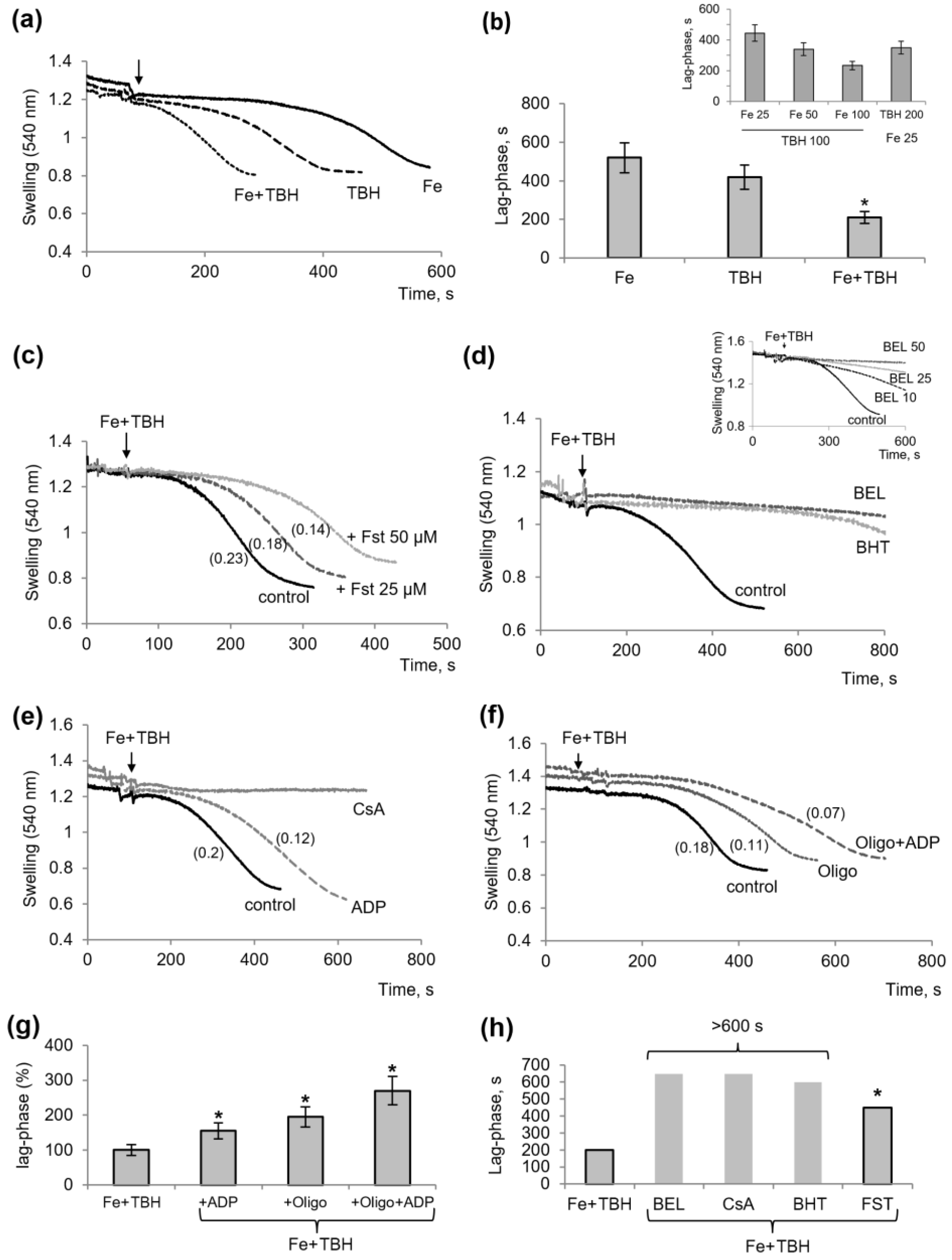
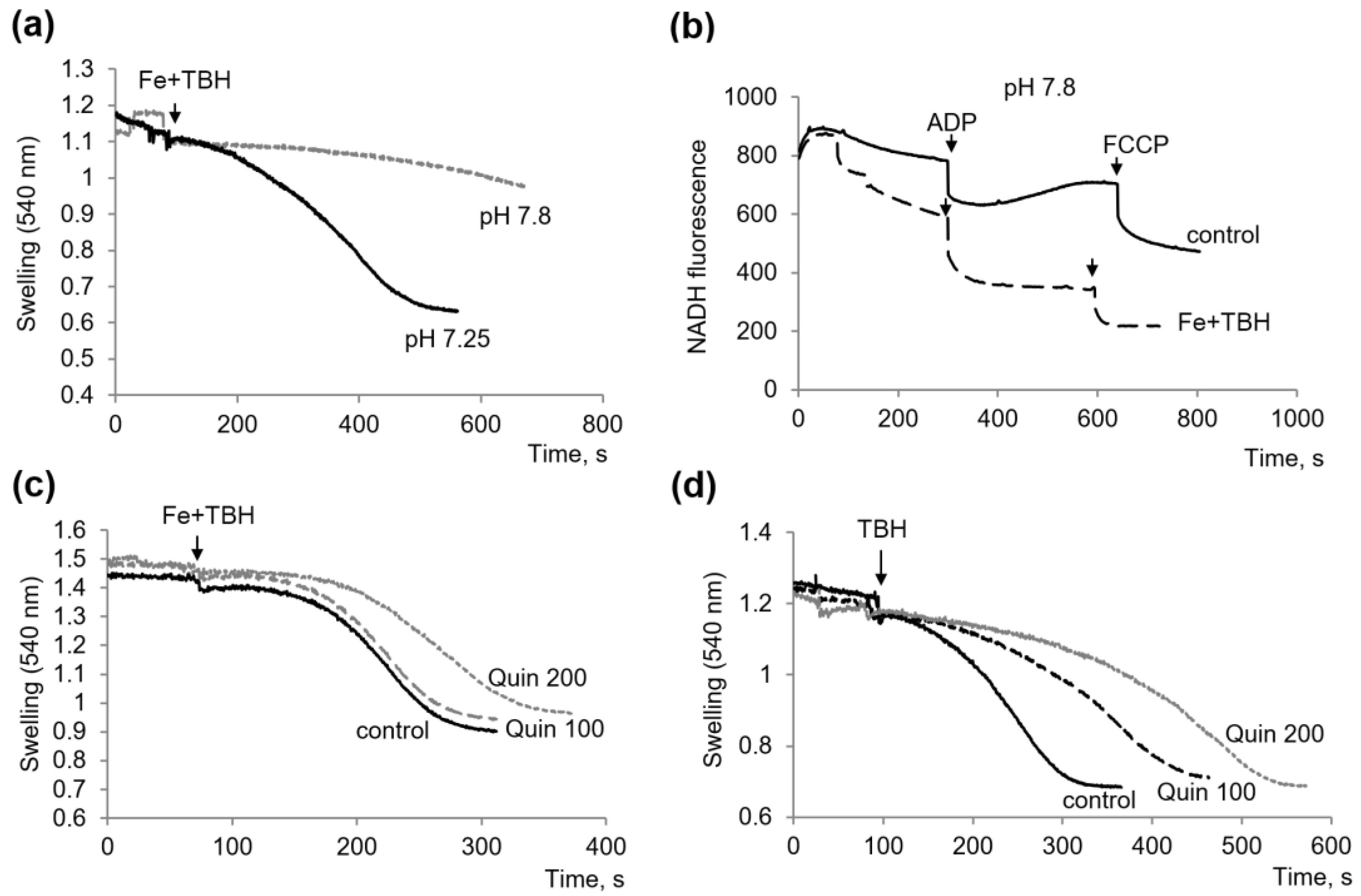
3.1. Influence of TBH and Ferrous Ions on the Swelling of Mitochondria
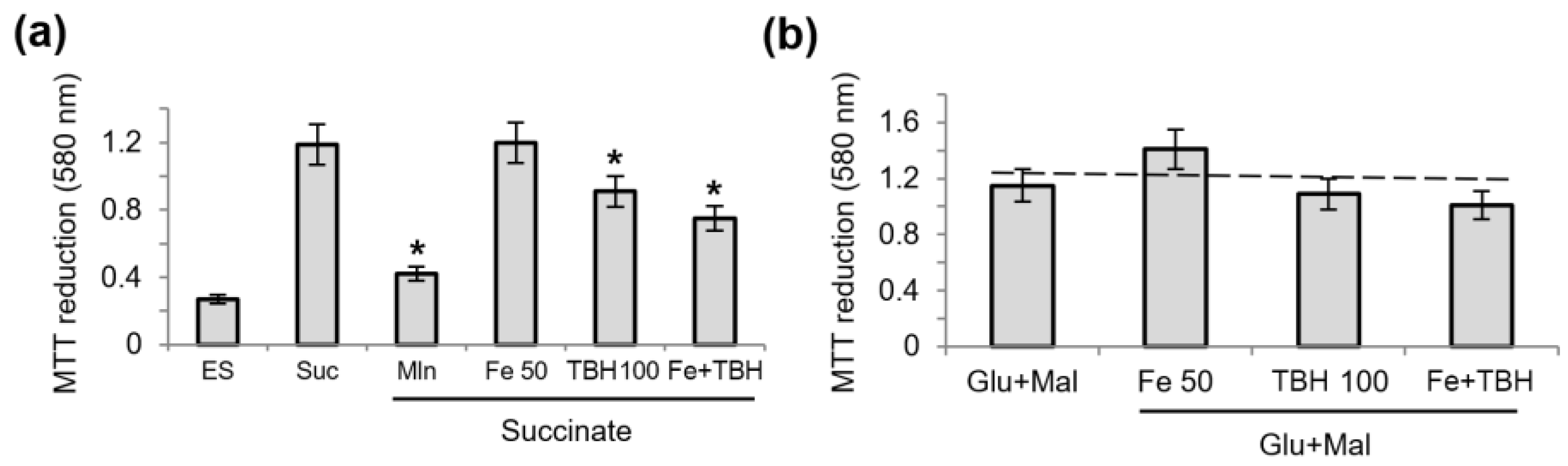
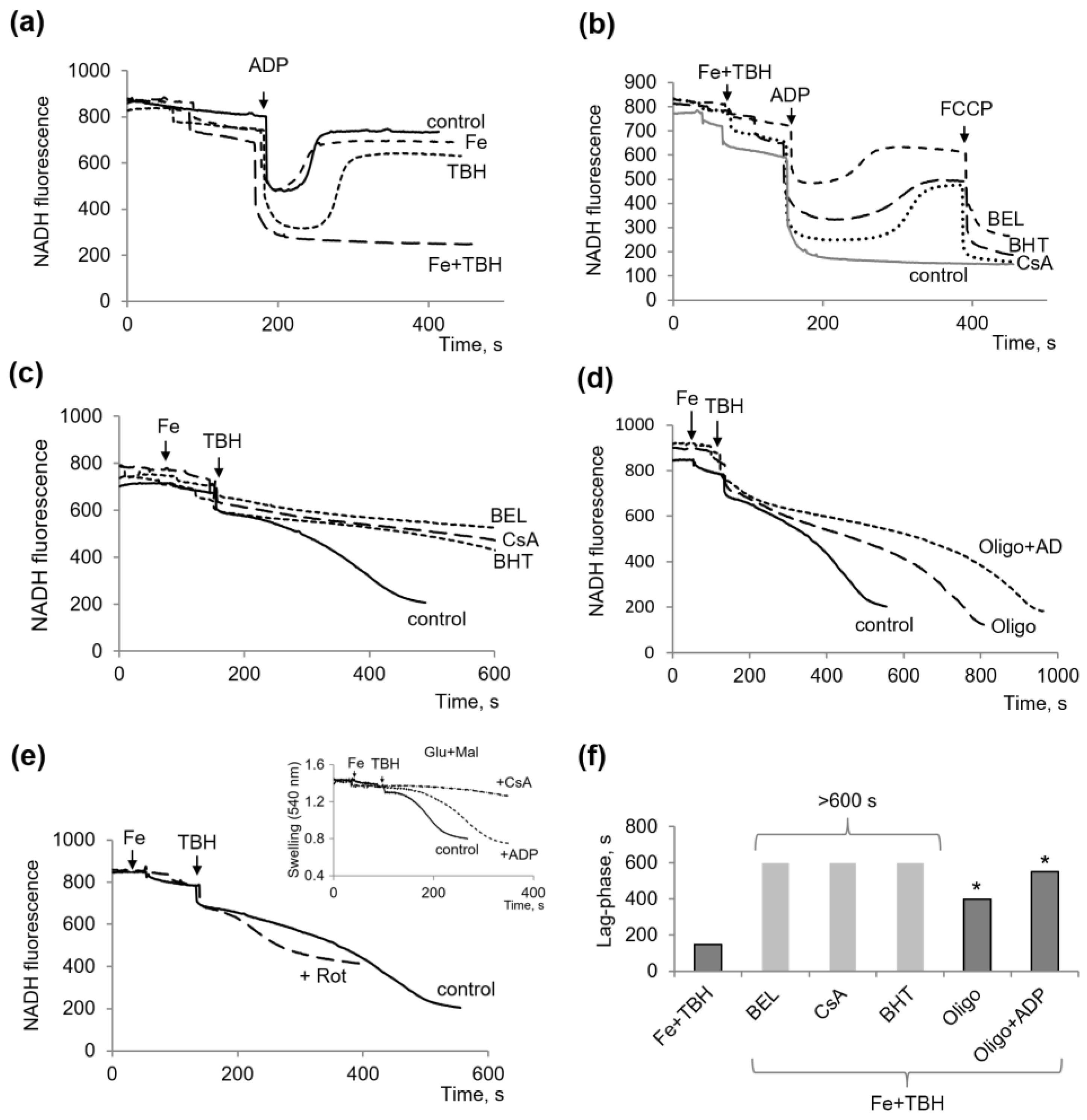
3.2. Influence of TBH and Ferrous Ions on the Oxidation of Succinate and NAD-Dependent Substrates and Oxidative Phosphorylation
3.3. Other Protectors against the Mitochondrial Dysfunction Induced by TBH and Ferrous Ions
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Lv, J.; Hou, B.; Song, J.; Xu, Y.; Xie, S.J. The Relationship Between Ferroptosis and Diseases. Multidiscip. Healthc. 2022, 15, 2261–2275. [Google Scholar] [CrossRef] [PubMed]
- Ajoolabady, A.; Tang, D.; Kroemer, G.; Ren, J. Ferroptosis in hepatocellular carcinoma: Mechanisms and targeted therapy. Br. J. Cancer 2023, 128, 190–205. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, X.Z.; Wang, Y.H.; Cheng, X.L.; Zhao, Y.; Zhou, L.Y.; Wang, K. Emerging roles of ferroptosis in cardiovascular diseases. Cell Death Discov. 2022, 8, 394. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.H.; Fefelova, N.; Pamarthi, S.H.; Gwathmey, J.K. Molecular Mechanisms of Ferroptosis and Relevance to Cardiovascular Disease. Cells 2022, 11, 2726. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Conrad, M.; Derek, A.; Pratt, D.A. The chemical basis of ferroptosis. Nat. Chem. Biol. 2019, 15, 1137–1147. [Google Scholar] [CrossRef]
- Gan, B. Mitochondrial regulation of ferroptosis. J. Cell Biol. 2021, 220, e202105043. [Google Scholar] [CrossRef]
- Javadov, S. Mitochondria and ferroptosis. Curr. Opin. Physiol. 2022, 25, 100483. [Google Scholar] [CrossRef]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.; Lee, J.; You, J.H.; Kim, D.; Roh, J.L. Dihydrolipoamide dehydrogenase regulates cystine deprivation-induced ferroptosis in head and neck cancer. Redox Biol. 2020, 30, 101418. [Google Scholar] [CrossRef]
- Li, C.; Liu, J.; Hou, W.; Kang, R.; Tang, D. STING1 Promotes Ferroptosis Through MFN1/2-Dependent Mitochondrial Fusion. Front. Cell Dev. Biol. 2021, 9, 698679. [Google Scholar] [CrossRef] [PubMed]
- Ballard, A.; Zeng, R.; Zarei, A.; Shao, C.; Cox, L.; Yan, H.; Franco, A.; Dorn, G.W., 2nd; Faccio, R.; Veis, D.J. The tethering function of mitofusin2 controls osteoclast differentiation by modulating the Ca2+-NFATc1 axis. J. Biol. Chem. 2020, 295, 6629–6640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, A.; Walton, C.E.; Dang, X. Mitochondria Clumping vs. Mitochondria Fusion in CMT2A Diseases. Life 2022, 12, 2110. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; He, S.; Guo, N.; Tian, W.; Zhang, W.; Luo, L. Molecular mechanisms of ferroptosis and relevance to inflammation. Inflamm. Res. 2023, 72, 281–299. [Google Scholar] [CrossRef]
- Tadokoro, T.; Ikeda, M.; Ide, T.; Deguchi, H.; Ikeda, S.; Okabe, K.; Ishikita, A.; Matsushima, S.; Koumura, T.; Yamada, K.I.; et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 2020, 5, e132747. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Naga Prasad, S.V.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Lemasters, J.J. Translocation of iron from lysosomes to mitochondria during ischemia predisposes to injury after reperfusion in rat hepatocytes. Free Radic. Biol. Med. 2013, 63, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Zhao, W.; Yu, J.; Li, S.; Lin, L.; Chen, X. Induction of ferroptosis and mitochondrial dysfunction by oxidative stress in PC12 cells. Sci. Rep. 2018, 8, 574. [Google Scholar] [CrossRef] [Green Version]
- Wenz, C.; Faust, D.; Linz, B.; Turmann, C.; Nikolova, T.; Bertin, J.; Gough, P.; Wipf, P.; Schröder, A.S.; Krautwald, S.; et al. t-BuOOH induces ferroptosis in human and murine cell lines. Arch. Toxicol. 2018, 92, 759–775. [Google Scholar] [CrossRef]
- Han, L.; Wang, Y.L.; Sun, Y.C.; Hu, Z.Y.; Hu, K.; Du, L.B. tert-Butylhydroperoxide induces apoptosis in RAW264.7 macrophages via a mitochondria-mediated signaling pathway. Toxicol. Res. 2018, 7, 970–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedotcheva, N.I.; Mokhova, E.N. Mechanism of induction of oxidative stress in liver mitochondria by low concentrations of tert-butyl hydroperoxide. Biochemistry 2013, 78, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Fedotcheva, N.; Olenin, A.; Beloborodova, N. Influence of Microbial Metabolites on the Nonspecific Permeability of Mitochondrial Membranes under Conditions of Acidosis and Loading with Calcium and Iron Ions. Biomedicines 2021, 9, 558. [Google Scholar] [CrossRef] [PubMed]
- Fedotcheva, T.A.; Fedotcheva, N.I. Protectors of the Mitochondrial Permeability Transition Pore Activated by Iron and Doxorubicin. Curr. Cancer Drug Targets 2021, 21, 514–525. [Google Scholar] [CrossRef]
- Fedotcheva, T.; Shimanovsky, N.; Fedotcheva, N. Involvement of multidrug resistance modulators in the regulation of the mitochondrial permeability transition pore. Membranes 2022, 12, 890. [Google Scholar] [CrossRef]
- Fedotcheva, N.; Beloborodova, N. Influence of microbial metabolites and itaconic acid involved in bacterial inflammation on the activity of mitochondrial enzymes and the protective role of alkalization. Int. J. Mol. Sci. 2022, 23, 9069. [Google Scholar] [CrossRef]
- Jang, S.; Javadov, S. Association between ROS production, swelling and the respirasome integrity in cardiac mitochondria. Arch. Biochem. Biophys. 2017, 630, 1–8. [Google Scholar] [CrossRef]
- Endlicher, R.; Drahota, Z.; Červinková, Z. Modification of calcium retention capacity of rat liver mitochondria by phosphate and tert-butyl hydroperoxide. Physiol. Res. 2019, 68, 59–65. [Google Scholar] [CrossRef]
- Sokolova, N.; Pan, S.; Provazza, S.; Beutner, G.; Vendelin, M.; Birkedal, R.; Sheu, S.S. ADP protects cardiac mitochondria under severe oxidative stress. PLoS ONE 2013, 8, e83214. [Google Scholar] [CrossRef] [Green Version]
- Gogvadze, V.; Walter, P.B.; Ames, B.N. The role of Fe2+-induced lipid peroxidation in the initiation of the mitochondrial permeability transition. Arch. Biochem. Biophys. 2003, 414, 255–260. [Google Scholar] [CrossRef]
- Rauckhorst, A.J.; Pfeiffer, D.R.; Broekemeier, K.M. The iPLA(2)γ is identified as the membrane potential sensitive phospholipase in liver mitochondria. FEBS Lett. 2015, 589, 2367–2371. [Google Scholar] [CrossRef] [Green Version]
- Hara, S.; Yoda, E.; Sasaki, Y.; Nakatani, Y.; Kuwata, H. Calcium-independent phospholipase A2γ (iPLA2γ) and its roles in cellular functions and diseases. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 861–868. [Google Scholar] [CrossRef]
- Rauckhorst, A.J.; Broekemeier, K.M.; Pfeiffer, D.R. Regulation of the Ca2+-independent phospholipase A2 in liver mitochondria by changes in the energetic state. J. Lipid Res. 2014, 55, 826–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garlid, A.O.; Jaburek, M.; Jacobs, J.P.; Garlid, K.D. Mitochondrial reactive oxygen species: Which ROS signals cardioprotection? Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H960–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol. 2021, 23, 266–285. [Google Scholar] [CrossRef] [PubMed]
- Carraro, M.; Carrer, A.; Urbani, A.; Bernardi, P. Molecular nature and regulation of the mitochondrial permeability transition pore(s), drug target(s) in cardioprotection. J. Mol. Cell. Cardiol. 2020, 144, 76–86. [Google Scholar] [CrossRef]
- Mnatsakanyan, N.; Llaguno, M.C.; Yang, Y.; Yan, Y.; Weber, J.; Sigworth, F.J.; Jonas, E.A. A mitochondrial megachannel resides in monomeric F1FO ATP synthase. Nat. Commun. 2019, 10, 5823. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, A.; Biswas, S.; Bandyopadhyay, D.; Sarkar, C.; Datta, A.G. Effect of isoproterenol on lipid peroxidation and antioxidant enzymes of myocardial tissue of mice and protection by quinidine. Mol. Cell. Biochem. 2003, 245, 43–49. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fedotcheva, T.; Shimanovsky, N.; Fedotcheva, N. Specific Features of Mitochondrial Dysfunction under Conditions of Ferroptosis Induced by t-Butylhydroperoxide and Iron: Protective Role of the Inhibitors of Lipid Peroxidation and Mitochondrial Permeability Transition Pore Opening. Membranes 2023, 13, 372. https://doi.org/10.3390/membranes13040372
Fedotcheva T, Shimanovsky N, Fedotcheva N. Specific Features of Mitochondrial Dysfunction under Conditions of Ferroptosis Induced by t-Butylhydroperoxide and Iron: Protective Role of the Inhibitors of Lipid Peroxidation and Mitochondrial Permeability Transition Pore Opening. Membranes. 2023; 13(4):372. https://doi.org/10.3390/membranes13040372
Chicago/Turabian StyleFedotcheva, Tatiana, Nikolai Shimanovsky, and Nadezhda Fedotcheva. 2023. "Specific Features of Mitochondrial Dysfunction under Conditions of Ferroptosis Induced by t-Butylhydroperoxide and Iron: Protective Role of the Inhibitors of Lipid Peroxidation and Mitochondrial Permeability Transition Pore Opening" Membranes 13, no. 4: 372. https://doi.org/10.3390/membranes13040372
APA StyleFedotcheva, T., Shimanovsky, N., & Fedotcheva, N. (2023). Specific Features of Mitochondrial Dysfunction under Conditions of Ferroptosis Induced by t-Butylhydroperoxide and Iron: Protective Role of the Inhibitors of Lipid Peroxidation and Mitochondrial Permeability Transition Pore Opening. Membranes, 13(4), 372. https://doi.org/10.3390/membranes13040372