

Dimerization of Transmembrane Proteins in Cancer Immunotherapy

Abstract
:1. Introduction
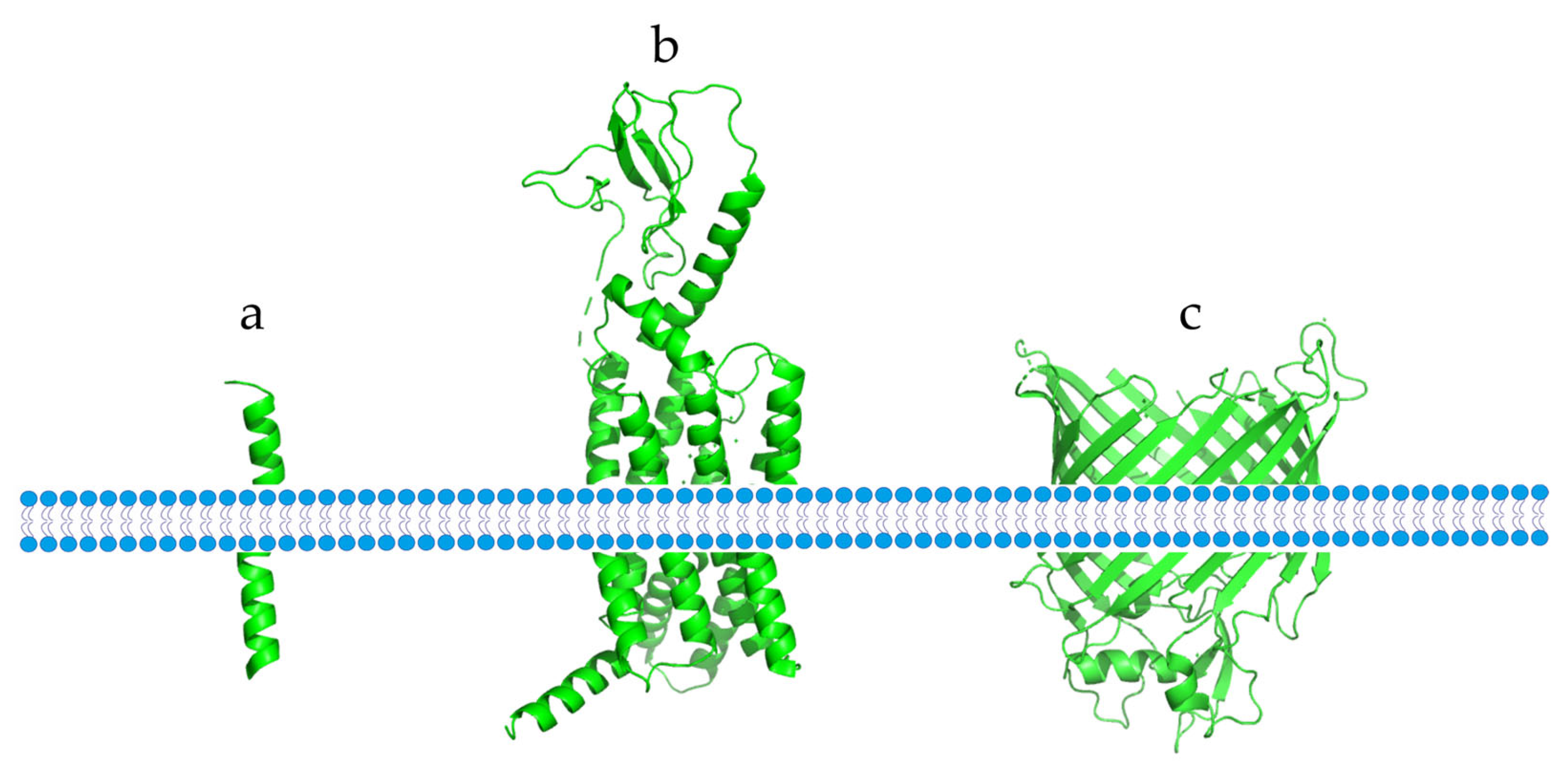
2. Structures and Functions of Immune Checkpoint Proteins in Cancer Immunotherapy
2.1. PD-L1
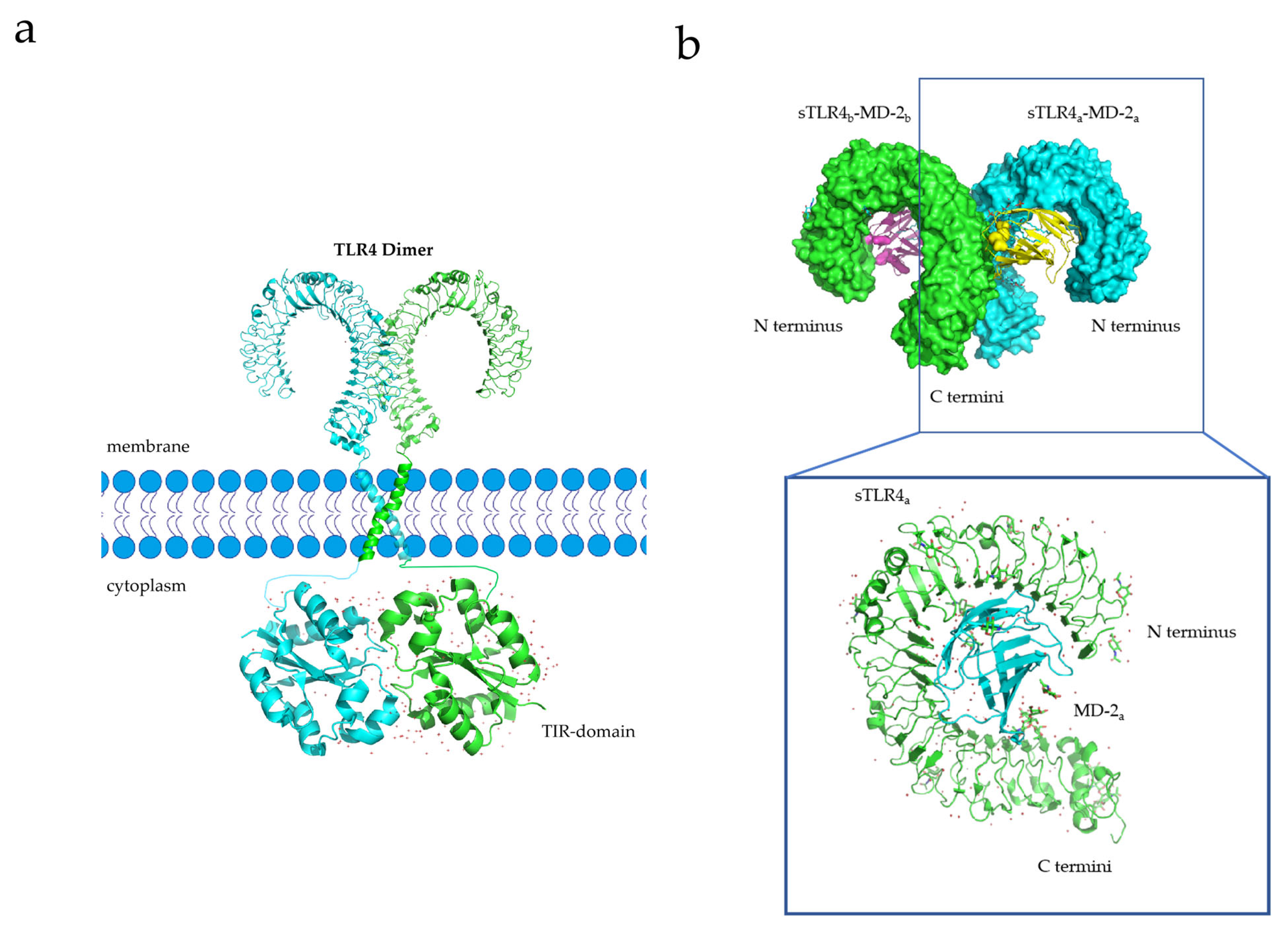
2.2. TLR-4
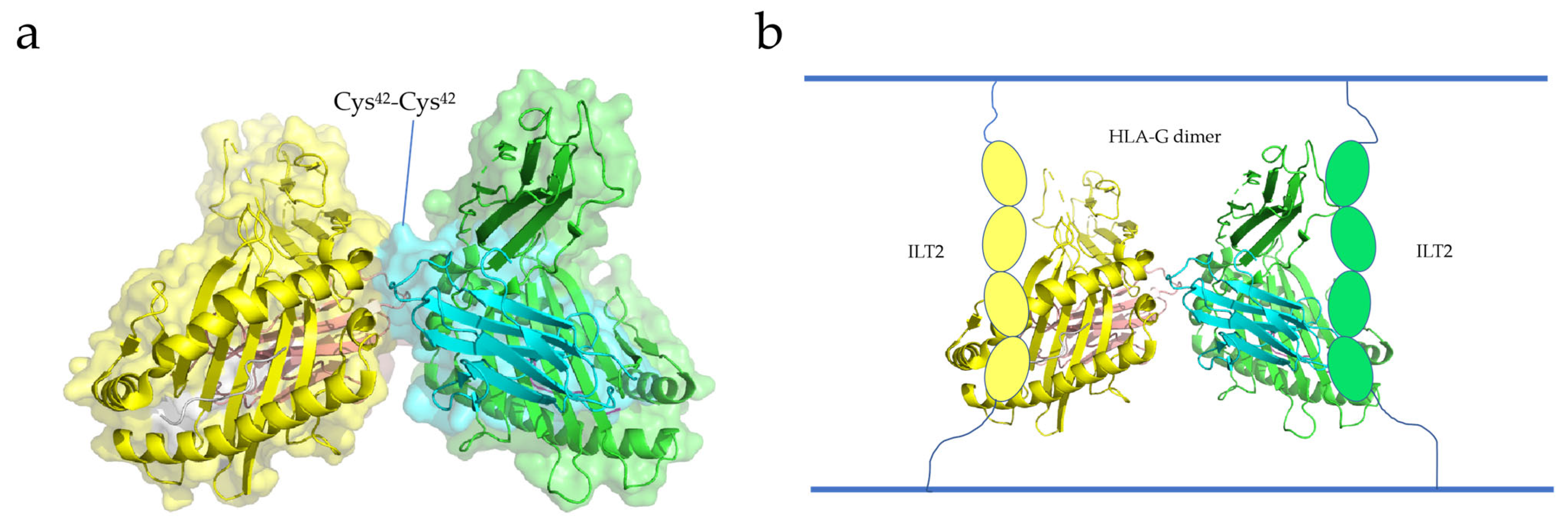
2.3. HLA-G
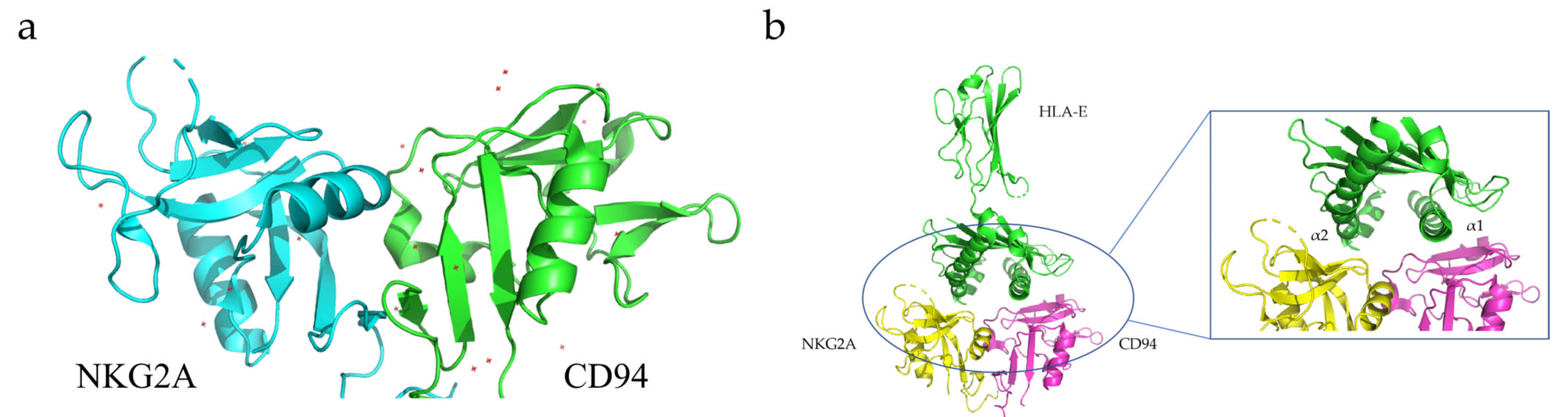
2.4. NKG2A
3. Binding Characteristics and Function of Transmembrane Protein-Receptor Dimers
3.1. PD-L1 Dimerization

3.2. TLR-4 Dimerization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3.3. HLA-G Dimerization
3.4. NKG2A Dimerization
4. Application of Regulation of TMEM Dimerization in Anti-Tumor Immunity
5. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Smith, S.M. Strategies for the purification of membrane proteins. In Protein Chromatography; Springer: Berlin/Heidelberg, Germany, 2011; pp. 485–496. [Google Scholar] [CrossRef] [Green Version]
- Schmit, K.; Michiels, C. TMEM proteins in cancer: A review. Front. Pharmacol. 2018, 9, 1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinothkumar, K.R.; Henderson, R. Structures of membrane proteins. Q. Rev. Biophys. 2010, 43, 65–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Liang, D.; Liu, J.; Zeng, J.; Zeng, Y. The Breakthroughs in Cancer Immune Checkpoint Based Therapy: A Review of Development in Immune Checkpoint Study and its Application. Comb. Chem. High Throughput Screen. 2017, 20, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Zhang, C.; Jin, S.; Gao, Z.; Cao, J.; Wang, A.; Li, D.; Wang, Q.; Sun, X.; Bai, D. Progress of immune checkpoint therapy in the clinic (Review). Oncol. Rep. 2019, 41, 3–14. [Google Scholar] [CrossRef]
- Litak, J.; Grochowski, C.; Litak, J.; Osuchowska, I.; Gosik, K.; Radzikowska, E.; Kamieniak, P.; Rolinski, J. TLR-4 Signaling vs. Immune Checkpoints, miRNAs Molecules, Cancer Stem Cells, and Wingless-Signaling Interplay in Glioblastoma Multiforme-Future Perspectives. Int. J. Mol. Sci. 2020, 21, 3114. [Google Scholar] [CrossRef]
- Ying, H.; Zhang, X.; Duan, Y.; Lao, M.; Xu, J.; Yang, H.; Liang, T.; Bai, X. Non-cytomembrane PD-L1: An atypical target for cancer. Pharmacol. Res. 2021, 170, 105741. [Google Scholar] [CrossRef]
- Sugiura, D.; Maruhashi, T.; Okazaki, I.M.; Shimizu, K.; Maeda, T.K.; Takemoto, T.; Okazaki, T. Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science 2019, 364, 558–566. [Google Scholar] [CrossRef]
- Park, S.N.; Noh, K.T.; Jeong, Y.I.; Jung, I.D.; Kang, H.K.; Cha, G.S.; Lee, S.J.; Seo, J.K.; Kang, D.H.; Hwang, T.H.; et al. Rhamnogalacturonan II is a Toll-like receptor 4 agonist that inhibits tumor growth by activating dendritic cell-mediated CD8+ T cells. Exp. Mol. Med. 2013, 45, e8. [Google Scholar] [CrossRef]
- Khan, M.; Arooj, S.; Wang, H. NK Cell-Based Immune Checkpoint Inhibition. Front. Immunol. 2020, 11, 167. [Google Scholar] [CrossRef]
- Godal, R.; Bachanova, V.; Gleason, M.; McCullar, V.; Yun, G.H.; Cooley, S.; Verneris, M.R.; McGlave, P.B.; Miller, J.S. Natural killer cell killing of acute myelogenous leukemia and acute lymphoblastic leukemia blasts by killer cell immunoglobulin-like receptor-negative natural killer cells after NKG2A and LIR-1 blockade. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 2010, 16, 612–621. [Google Scholar] [CrossRef] [Green Version]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Biassoni, R.; Mingari, M.C.; Moretta, L. Receptors for HLA class-I molecules in human natural killer cells. Annu. Rev. Immunol. 1996, 14, 619–648. [Google Scholar] [CrossRef] [PubMed]
- Moretta, L.; Mingari, M.C.; Pende, D.; Bottino, C.; Biassoni, R.; Moretta, A. The molecular basis of natural killer (NK) cell recognition and function. J. Clin. Immunol. 1996, 16, 243–253. [Google Scholar] [CrossRef]
- Wagtmann, N.; Rajagopalan, S.; Winter, C.C.; Peruzzi, M.; Long, E.O. Killer cell inhibitory receptors specific for HLA-C and HLA-B identified by direct binding and by functional transfer. Immunity 1995, 3, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Clements, C.S.; Kjer-Nielsen, L.; McCluskey, J.; Rossjohn, J. Structural studies on HLA-G: Implications for ligand and receptor binding. Hum. Immunol. 2007, 68, 220–226. [Google Scholar] [CrossRef]
- Hò, G.T.; Celik, A.A.; Huyton, T.; Hiemisch, W.; Blasczyk, R.; Simper, G.S.; Bade-Doeding, C. NKG2A/CD94 Is a New Immune Receptor for HLA-G and Distinguishes Amino Acid Differences in the HLA-G Heavy Chain. Int. J. Mol. Sci. 2020, 21, 4362. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xiong, H.; Ning, Z. Implications of NKG2A in immunity and immune-mediated diseases. Front. Immunol. 2022, 13, 960852. [Google Scholar] [CrossRef]
- Brown, J.A.; Dorfman, D.M.; Ma, F.R.; Sullivan, E.L.; Munoz, O.; Wood, C.R.; Greenfield, E.A.; Freeman, G.J. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J. Immunol. 2003, 170, 1257–1266. [Google Scholar] [CrossRef] [Green Version]
- Greenwald, R.J.; Freeman, G.J.; Sharpe, A.H. The B7 family revisited. Annu. Rev. Immunol. 2005, 23, 515. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Honjo, T. PD-1: An inhibitory immunoreceptor involved in peripheral tolerance. Trends Immunol. 2001, 22, 265–268. [Google Scholar] [CrossRef]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef]
- Lin, D.Y.-W.; Tanaka, Y.; Iwasaki, M.; Gittis, A.G.; Su, H.-P.; Mikami, B.; Okazaki, T.; Honjo, T.; Minato, N.; Garboczi, D.N. The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 3011–3016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, D.; Zhou, S.; Liu, X.; Zhao, C.; Liu, H.; Yao, X. Understanding the structural and energetic basis of PD-1 and monoclonal antibodies bound to PD-L1: A molecular modeling perspective. Biochim. et Biophys. Acta (BBA)-Gen. Subj. 2018, 1862, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Zak, K.M.; Kitel, R.; Przetocka, S.; Golik, P.; Guzik, K.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A.J.S. Structure of the complex of human programmed death 1, PD-1, and its ligand PD-L1. Structure 2015, 23, 2341–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, M.; Simonetta, F.; Baker, J.; Morrison, A.R.; Wenokur, A.S.; Pierini, A.; Berraondo, P.; Negrin, R.S. Indirect impact of PD-1/PD-L1 blockade on a murine model of NK cell exhaustion. Front. Immunol. 2020, 11, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J.; Kurachi, M.J.N.R.I. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Swaika, A.; Hammond, W.A.; Joseph, R.W. Current state of anti-PD-L1 and anti-PD-1 agents in cancer therapy. Mol. Immunol. 2015, 67, 4–17. [Google Scholar] [CrossRef]
- Xing, Y.; Hogquist, K. T-cell tolerance: Central and peripheral. Cold Spring Harb. Perspect. Biol. 2012, 4, a006957. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Casals, M.; Brahmer, J.R.; Callahan, M.K.; Flores-Chávez, A.; Keegan, N.; Khamashta, M.A.; Lambotte, O.; Mariette, X.; Prat, A.; Suárez-Almazor, M.E. Immune-related adverse events of checkpoint inhibitors. Nat. Rev. Dis. Prim. 2020, 6, 38. [Google Scholar] [CrossRef]
- Sibaud, V. Dermatologic reactions to immune checkpoint inhibitors. Am. J. Clin. Dermatol. 2018, 19, 345–361. [Google Scholar] [CrossRef]
- Hua, Z.; Hou, B. TLR signaling in B-cell development and activation. Cell. Mol. Immunol. 2013, 10, 103–106. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.-J.; Gong, H.-F.; Zhao, Q.-Q.; Liu, X.-S.; Liu, C.; Wang, H.J.T.L. Critical role of toll-like receptor 4 (TLR4) in dextran sulfate sodium (DSS)-Induced intestinal injury and repair. Toxicol. Lett. 2019, 315, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Pahwa, R.; Devaraj, S.; Jialal, I. The effect of the accessory proteins, soluble CD14 and lipopolysaccharide-binding protein on Toll-like receptor 4 activity in human monocytes and adipocytes. Int. J. Obes. 2016, 40, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Mishra, V.; Pathak, C. Human Toll-Like Receptor 4 (hTLR4): Structural and functional dynamics in cancer. Int. J. Biol. Macromol. 2019, 122, 425–451. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Park, B.S.; Kim, J.-I.; Kim, S.E.; Lee, J.; Oh, S.C.; Enkhbayar, P.; Matsushima, N.; Lee, H.; Yoo, O.J.J.C. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 2007, 130, 906–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Zhang, L.; Li, Z.; Zhang, Q. The roles of toll-like receptors in carcinogenesis and cancer immunotherapy. Chin.-Ger. J. Clin. Oncol. 2010, 9, 118–120. [Google Scholar] [CrossRef]
- Yirmiya, R.; Goshen, I.J.B. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav. Immun. 2011, 25, 181–213. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.J.N. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef]
- Kluwe, J.; Mencin, A.; Schwabe, R.F. Toll-like receptors, wound healing, and carcinogenesis. J. Mol. Med. 2009, 87, 125–138. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, K.; Selmani, Z.; Ishii, H.; Yamaguchi, K. Innate immunity and cancer therapy. Int. Immunopharmacol. 2011, 11, 350–357. [Google Scholar] [CrossRef]
- Shetab Boushehri, M.A.; Lamprecht, A. TLR4-based immunotherapeutics in cancer: A review of the achievements and shortcomings. Mol. Pharm. 2018, 15, 4777–4800. [Google Scholar] [CrossRef]
- Foroni, I.; Couto, A.R.; Bettencourt, B.F.; Santos, M.; Lima, M.; Bruges-Armas, J.J.H.; Diseases, A.I. HLA-E, HLA-F and HLA-G—The non-classical side of the MHC cluster. In HLA and Associated Important Diseases; IntechOpen: London, UK, 2014; Volume 3, pp. 61–109. [Google Scholar] [CrossRef] [Green Version]
- Carosella, E.D.; Rouas-Freiss, N.; Tronik-Le Roux, D.; Moreau, P.; LeMaoult, J. HLA-G: An immune checkpoint molecule. Adv. Immunol. 2015, 127, 33–144. [Google Scholar] [CrossRef]
- de Kruijf, E.M.; Sajet, A.; van Nes, J.G.; Natanov, R.; Putter, H.; Smit, V.T.; Liefers, G.J.; van den Elsen, P.J.; van de Velde, C.J.; Kuppen, P.J.K. HLA-E and HLA-G expression in classical HLA class I-negative tumors is of prognostic value for clinical outcome of early breast cancer patients. J. Immunol. 2010, 185, 7452–7459. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Wang, P.; Dai, P.; Jin, B.; Tong, Y.; Lin, H.; Shi, G. Correlation between human leukocyte antigen-G expression and clinical parameters in oral squamous cell carcinoma. Indian J. Cancer 2018, 55, 340. [Google Scholar] [CrossRef] [PubMed]
- Zeestraten, E.; Reimers, M.; Saadatmand, S.; Dekker, J.T.; Liefers, G.; Van Den Elsen, P.; Van De Velde, C.; Kuppen, P.J.K. Combined analysis of HLA class I, HLA-E and HLA-G predicts prognosis in colon cancer patients. Br. J. Cancer 2014, 110, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Arnaiz-Villena, A.; Juarez, I.; Suarez-Trujillo, F.; López-Nares, A.; Vaquero, C.; Palacio-Gruber, J.; Martin-Villa, J.M. HLA-G: Function, polymorphisms and pathology. Int. J. Immunogenet. 2021, 48, 172–192. [Google Scholar] [CrossRef] [PubMed]
- Diehl, M.; Münz, C.; Keilholz, W.; Stevanović, S.; Holmes, N.; Loke, Y.W.; Rammensee, H.-G.J.C.B. Nonclassical HLA-G molecules are classical peptide presenters. Curr. Biol. 1996, 6, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Paul, P.; Cabestre, F.A.; Lefebvre, S.; Khalil-Daher, I.; Vazeux, G.; Quiles, R.M.M.; Bermond, F.; Dausset, J.; Carosella, E.D. Identification of HLA-G7 as a new splice variant of the HLA-G mRNA and expression of soluble HLA-G5,-G6, and-G7 transcripts in human transfected cells. Hum. Immunol. 2000, 61, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Kuroki, K.; Okabe, Y.; Kasai, Y.; Matsumoto, N.; Yamada, C.; Takai, T.; Ose, T.; Kon, S.; Matsuda, T. The immunosuppressive effect of domain-deleted dimer of HLA-G2 isoform in collagen-induced arthritis mice. Hum. Immunol. 2016, 77, 754–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.-X.; Xie, Y.-M.; Zhao, S.-J.; Liu, C.-Y.; Mor, G.; Liao, A.-H. Human leukocyte antigens: The unique expression in trophoblasts and their crosstalk with local immune cells. Int. J. Biol. Sci. 2022, 18, 4043–4052. [Google Scholar] [CrossRef] [PubMed]
- Moffett, A.; Colucci, F. Co-evolution of NK receptors and HLA ligands in humans is driven by reproduction. Immunol. Rev. 2015, 267, 283–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Zhou, Y.; Wei, H. Roles of HLA-G in the maternal-fetal immune microenvironment. Front. Immunol. 2020, 11, 592010. [Google Scholar] [CrossRef] [PubMed]
- Saurabh, A.; Chakraborty, S.; Kumar, P.; Mohan, A.; Bhatnagar, A.K.; Rishi, N.; Mitra, D.K.J.T. Inhibiting HLA-G restores IFN-γ and TNF-α producing T cell in pleural tuberculosis. Tuberculosis 2018, 109, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Martín-Villa, J.M.; Vaquero-Yuste, C.; Molina-Alejandre, M.; Juarez, I.; Suárez-Trujillo, F.; López-Nares, A.; Palacio-Gruber, J.; Barrera-Gutiérrez, L.; Fernández-Cruz, E.; Rodríguez-Sainz, C. HLA-G: Too Much or Too Little? Role in Cancer and Autoimmune Disease. Front. Immunol. 2022, 67, 796054. [Google Scholar] [CrossRef] [PubMed]
- Ristich, V.; Liang, S.; Zhang, W.; Wu, J.; Horuzsko, A. Tolerization of dendritic cells by HLA-G. Eur. J. Immunol. 2005, 35, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Attia, J.V.; Dessens, C.E.; van de Water, R.; Houvast, R.D.; Kuppen, P.J.; Krijgsman, D. The molecular and functional characteristics of HLA-G and the interaction with its receptors: Where to intervene for cancer immunotherapy? Int. J. Mol. Sci. 2020, 21, 8678. [Google Scholar] [CrossRef] [PubMed]
- Muntasell, A.; Ochoa, M.C.; Cordeiro, L.; Berraondo, P.; de Cerio, A.L.-D.; Cabo, M.; López-Botet, M.; Melero, I. Targeting NK-cell checkpoints for cancer immunotherapy. Curr. Opin. Immunol. 2017, 45, 73–81. [Google Scholar] [CrossRef]
- Li, F.; Wei, H.; Wei, H.; Gao, Y.; Xu, L.; Yin, W.; Sun, R.; Tian, Z. Blocking the Natural Killer Cell Inhibitory Receptor NKG2A Increases Activity of Human Natural Killer Cells and Clears Hepatitis B Virus Infection in Mice. Gastroenterology 2013, 144, 392–401. [Google Scholar] [CrossRef]
- Rapaport, A.S.; Schriewer, J.; Gilfillan, S.; Hembrador, E.; Crump, R.; Plougastel, B.F.; Wang, Y.; Le Friec, G.; Gao, J.; Cella, M.; et al. The Inhibitory Receptor NKG2A Sustains Virus-Specific CD8+ T Cells in Response to a Lethal Poxvirus Infection. Immunity 2015, 43, 1112–1124. [Google Scholar] [CrossRef] [Green Version]
- Plougastel, B.; Jones, T.; Trowsdale, J. Genomic structure, chromosome location, and alternative splicing of the humanNKG2A gene. Immunogenetics 1996, 44, 286–291. [Google Scholar] [CrossRef]
- Iwaszko, M.; Bogunia-Kubik, K. Clinical Significance of the HLA-E and CD94/NKG2 Interaction. Arch. Immunol. et Ther. Exp. 2011, 59, 353–367. [Google Scholar] [CrossRef]
- Mingari, M.C.; Pietra, G.; Moretta, L. Immune Checkpoint Inhibitors: Anti-NKG2A Antibodies on Board. Trends Immunol. 2019, 40, 83–85. [Google Scholar] [CrossRef]
- Tang, L.; Bai, J.; Chung, C.-S.; Lomas-Neira, J.; Chen, Y.; Huang, X.; Ayala, A. Programmed Cell Death Receptor Ligand 1 Modulates the Regulatory T Cells’ Capacity to Repress Shock/Sepsis–Induced Indirect Acute Lung Injury by Recruiting Phosphatase Src Homology Region 2 Domain-Containing Phosphatase 1. Shock 2015, 43, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Wen, L.-Z.; Ding, K.; Wang, Z.-R.; Ding, C.-H.; Lei, S.-J.; Liu, J.-P.; Yin, C.; Hu, P.-F.; Ding, J.; Chen, W.-S.; et al. SHP-1 Acts as a Tumor Suppressor in Hepatocarcinogenesis and HCC Progression. Cancer Res. 2018, 78, 4680–4691. [Google Scholar] [CrossRef] [Green Version]
- Mahapatra, S.; Mace, E.M.; Minard, C.G.; Forbes, L.R.; Vargas-Hernandez, A.; Duryea, T.K.; Makedonas, G.; Banerjee, P.P.; Shearer, W.; Orange, J.S. High-resolution phenotyping identifies NK cell subsets that distinguish healthy children from adults. PLoS ONE 2017, 12, e0181134. [Google Scholar] [CrossRef]
- Kamiya, T.; Seow, S.V.; Wong, D.; Robinson, M.; Campana, D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J. Clin. Investig. 2019, 129, 2094–2106. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Cui, Y.; Luo, G.; Wang, Q.; Hu, J.; He, W.; Yuan, J.; Zhou, J.; Wu, Y.; Sun, X.; et al. Activated mouse CD4+Foxp3− T cells facilitate melanoma metastasis via Qa-1-dependent suppression of NK-cell cytotoxicity. Cell Res. 2012, 22, 1696–1706. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, X.-M.; Li, S.-R.; Twelkmeyer, T.; Wang, W.-H.; Zhang, S.-Y.; Wang, S.-F.; Chen, J.-Z.; Jin, X.; Wu, Y.-Z.; et al. NKG2A is a NK cell exhaustion checkpoint for HCV persistence. Nat. Commun. 2019, 10, 1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borst, L.; van der Burg, S.H.; van Hall, T. The NKG2A–HLA-E Axis as a Novel Checkpoint in the Tumor Microenvironment. Clin. Cancer Res. 2020, 26, 5549–5556. [Google Scholar] [CrossRef] [PubMed]
- Van Hall, T.; André, P.; Horowitz, A.; Ruan, D.F.; Borst, L.; Zerbib, R.; Narni-Mancinelli, E.; Van Der Burg, S.H.; Vivier, E. Monalizumab: Inhibiting the novel immune checkpoint NKG2A. J. Immunother. Cancer 2019, 7, 263. [Google Scholar] [CrossRef]
- Li, Q.; Cai, S.; Li, M.; Zhou, X.; Wu, G.; Kang, K.; Yuan, J.; Wang, R.; Huyan, T.; Zhang, W. Natural killer cell exhaustion in lung cancer. Int. Immunopharmacol. 2021, 96, 107764. [Google Scholar] [CrossRef]
- van Montfoort, N.; Borst, L.; Korrer, M.J.; Sluijter, M.; Marijt, K.A.; Santegoets, S.J.; van Ham, V.J.; Ehsan, I.; Charoentong, P.; André, P.; et al. NKG2A Blockade Potentiates CD8 T Cell Immunity Induced by Cancer Vaccines. Cell 2018, 175, 1744–1755.e15. [Google Scholar] [CrossRef] [Green Version]
- Sheu, B.-C.; Chiou, S.-H.; Lin, H.-H.; Chow, S.-N.; Huang, S.-C.; Ho, H.-N.; Hsu, S.-M. Up-regulation of Inhibitory Natural Killer Receptors CD94/NKG2A with Suppressed Intracellular Perforin Expression of Tumor-Infiltrating CD8+ T Lymphocytes in Human Cervical Carcinoma. Cancer Res. 2005, 65, 2921–2929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McWilliams, E.M.; Mele, J.M.; Cheney, C.; Timmerman, E.A.; Fiazuddin, F.; Strattan, E.J.; Mo, X.; Byrd, J.C.; Muthusamy, N.; Awan, F.T. Therapeutic CD94/NKG2A blockade improves natural killer cell dysfunction in chronic lymphocytic leukemia. OncoImmunology 2016, 5, e1226720. [Google Scholar] [CrossRef] [Green Version]
- Zak, K.M.; Grudnik, P.; Guzik, K.; Zieba, B.J.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget 2016, 7, 30323–30335. [Google Scholar] [CrossRef] [Green Version]
- Bailly, C.; Vergoten, G. Protein homodimer sequestration with small molecules: Focus on PD-L1. Biochem. Pharmacol. 2020, 174, 113821. [Google Scholar] [CrossRef] [PubMed]
- Zak, K.M.; Grudnik, P.; Magiera-Mularz, K.; Dömling, A.; Dubin, G.; Holak, T.A. Structural Biology of the Immune Checkpoint Receptor PD-1 and Its Ligands PD-L1/PD-L2. Structure 2017, 25, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Perry, E.; Mills, J.J.; Zhao, B.; Wang, F.; Sun, Q.; Christov, P.P.; Tarr, J.C.; Rietz, T.A.; Olejniczak, E.T.; Lee, T.; et al. Fragment-based screening of programmed death ligand 1 (PD-L1). Bioorganic Med. Chem. Lett. 2019, 29, 786–790. [Google Scholar] [CrossRef]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef] [Green Version]
- Schöniger, S.; Jasani, B. The PD-1/PD-L1 Pathway: A Perspective on Comparative Immuno-Oncology. Animals 2022, 12, 2661. [Google Scholar] [CrossRef]
- Wang, Q.; Bardhan, K.; Boussiotis, V.A.; Patsoukis, N. The PD-1 Interactome. Adv. Biol. 2021, 5, e2100758. [Google Scholar] [CrossRef]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, K.-A.; Fitz, L.J.; Lee, J.M.; Benander, C.; George, J.A.; Wooters, J.; Qiu, Y.; Jussif, J.M.; Carter, L.L.; Wood, C.R.; et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3ζ signalosome and downstream signaling to PKCθ. FEBS Lett. 2004, 574, 37–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.Y.-C.; Wang, Y.; Jhatakia, A.D.; Deng, A.X.; Bee, C.; Deshpande, S.; Rangan, V.S.; Bezman, N.; Gudmundsson, O.; Chen, G. Higher-Order Structure Characterization of NKG2A/CD94 Protein Complex and Anti-NKG2A Antibody Binding Epitopes by Mass Spectrometry-Based Protein Footprinting Strategies. J. Am. Soc. Mass Spectrom. 2021, 32, 1567–1574. [Google Scholar] [CrossRef]
- Kang-Pettinger, T.; Walker, K.; Brown, R.; Cowan, R.; Wright, H.; Baravalle, R.; Waters, L.C.; Muskett, F.W.; Bowler, M.W.; Sawmynaden, K.; et al. Identification, binding, and structural characterization of single domain anti-PD-L1 antibodies inhibitory of immune regulatory proteins PD-1 and CD80. J. Biol. Chem. 2022, 299, 102769. [Google Scholar] [CrossRef]
- Zhao, Y.; Lee, C.K.; Lin, C.-H.; Gassen, R.B.; Xu, X.; Huang, Z.; Xiao, C.; Bonorino, C.; Lu, L.-F.; Bui, J.D.; et al. PD-L1:CD80 Cis-Heterodimer Triggers the Co-stimulatory Receptor CD28 While Repressing the Inhibitory PD-1 and CTLA-4 Pathways. Immunity 2019, 51, 1059–1073.e9. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cai, S.; Cheng, Y.; Zhang, W.; Wang, M.; Sun, H.; Guo, B.; Li, Z.; Xiao, Y.; Jiang, S. Discovery of Small-Molecule Inhibitors of the PD-1/PD-L1 Axis That Promote PD-L1 Internalization and Degradation. J. Med. Chem. 2022, 65, 3879–3893. [Google Scholar] [CrossRef]
- Mineev, K.S.; Goncharuk, S.A.; Goncharuk, M.V.; Volynsky, P.E.; Novikova, E.V.; Aresinev, A.S. Spatial structure of TLR4 transmembrane domain in bicelles provides the insight into the receptor activation mechanism. Sci. Rep. 2017, 7, 6864. [Google Scholar] [CrossRef] [Green Version]
- Toshchakov, V.Y.; Szmacinski, H.; Couture, L.A.; Lakowicz, J.R.; Vogel, S.N. Targeting TLR4 Signaling by TLR4 Toll/IL-1 Receptor Domain-Derived Decoy Peptides: Identification of the TLR4 Toll/IL-1 Receptor Domain Dimerization Interface. J. Immunol. 2011, 186, 4819–4827. [Google Scholar] [CrossRef] [Green Version]
- Reuven, E.M.; Fink, A.; Shai, Y. Regulation of innate immune responses by transmembrane interactions: Lessons from the TLR family. Biochim. et Biophys. Acta (BBA)-Biomembr. 2014, 1838, 1586–1593. [Google Scholar] [CrossRef] [Green Version]
- Kargas, V.; Marzinek, J.K.; Holdbrook, D.A.; Yin, H.; Ford, R.C.; Bond, P.J. A polar SxxS motif drives assembly of the transmembrane domains of Toll-like receptor 4. Biochim. et Biophys. Acta (BBA)-Biomembr. 2017, 1859, 2086–2095. [Google Scholar] [CrossRef] [PubMed]
- Kashani, B.; Zandi, Z.; Pourbagheri-Sigaroodi, A.; Bashash, D.; Ghaffari, S.H. The role of toll-like receptor 4 (TLR4) in cancer progression: A possible therapeutic target? J. Cell. Physiol. 2020, 236, 4121–4137. [Google Scholar] [CrossRef] [PubMed]
- Krüger, C.L.; Zeuner, M.-T.; Cottrell, G.S.; Widera, D.; Heilemann, M. Quantitative single-molecule imaging of TLR4 reveals ligand-specific receptor dimerization. Sci. Signal. 2017, 10, eaan1308. [Google Scholar] [CrossRef] [Green Version]
- Soares, J.-B.; Pimentel-Nunes, P.; Jr, R.R.-A.; Leite-Moreira, A. The role of lipopolysaccharide/toll-like receptor 4 signaling in chronic liver diseases. Hepatol. Int. 2010, 4, 659–672. [Google Scholar] [CrossRef] [Green Version]
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.D.; et al. Tumor-infiltrating DCs suppress nucleic acid–mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 2012, 13, 832–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.-J.; Cheng, J.; Feng, X.; Yu, Y.; Tian, L.; Huang, Q. The dual role and therapeutic potential of high-mobility group box 1 in cancer. Oncotarget 2017, 8, 64534–64550. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Zhang, Q.; Zeh, H.J., 3rd; Lotze, M.T.; Tang, D. HMGB1 in Cancer: Good, Bad, or Both? Clin. Cancer Res. 2013, 19, 4046–4057. [Google Scholar] [CrossRef] [Green Version]
- Krysko, O.; Løve Aaes, T.; Bachert, C.; Vandenabeele, P.; Krysko, D.V. Many faces of DAMPs in cancer therapy. Cell Death Dis. 2013, 4, e631. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Cui, X.; Liu, Q. Emerging role of HMGB1 in lung diseases: Friend or foe. J. Cell. Mol. Med. 2016, 21, 1046–1057. [Google Scholar] [CrossRef]
- Lee, C.-H.; Yoon, S.-J.; Lee, S.-M. Chlorogenic Acid Attenuates High Mobility Group Box 1 (HMGB1) and Enhances Host Defense Mechanisms in Murine Sepsis. Mol. Med. 2012, 18, 1437–1448. [Google Scholar] [CrossRef]
- Tadie, J.-M.; Bae, H.-B.; Deshane, J.S.; Bell, C.P.; Lazarowski, E.R.; Chaplin, D.D.; Thannickal, V.J.; Abraham, E.; Zmijewski, J.W. Toll-Like Receptor 4 Engagement Inhibits Adenosine 5′-Monophosphate–Activated Protein Kinase Activation through a High Mobility Group Box 1 Protein–Dependent Mechanism. Mol. Med. 2012, 18, 659–668. [Google Scholar] [CrossRef]
- Poulain, L.; Richard, V.; Lévy, P.; Dematteis, M.; Arnaud, C. Toll-Like Receptor-4 Mediated Inflammation Is Involved in the Cardiometabolic Alterations Induced by Intermittent Hypoxia. Mediat. Inflamm. 2015, 2015, 620258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Insua-Rodríguez, J.; Oskarsson, T. The extracellular matrix in breast cancer. Adv. Drug Deliv. Rev. 2016, 97, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Tivari, S.; Lu, H.; Dasgupta, T.; De Lorenzo, M.S.; Wieder, R. Reawakening of dormant estrogen-dependent human breast cancer cells by bone marrow stroma secretory senescence. Cell Commun. Signal. 2018, 16, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankov, R.; Yamada, K.M. Fibronectin at a glance. J. Cell Sci. 2002, 115, 3861–3863. [Google Scholar] [CrossRef] [Green Version]
- Wu, C. Roles of integrins in fibronectin matrix assembly. Histol. Histopathol. 1997, 12, 233–240. [Google Scholar] [PubMed]
- Julier, Z.; Martino, M.M.; de Titta, A.; Jeanbart, L.; Hubbell, J.A. The TLR4 Agonist Fibronectin Extra Domain A is Cryptic, Exposed by Elastase-2; use in a fibrin matrix cancer vaccine. Sci. Rep. 2015, 5, 8569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.-I.; Hong, M.; Wilson, I.A. An unusual dimeric structure and assembly for TLR4 regulator RP105–MD-1. Nat. Struct. Mol. Biol. 2011, 18, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Shiroishi, M.; Kuroki, K.; Ose, T.; Rasubala, L.; Shiratori, I.; Arase, H.; Tsumoto, K.; Kumagai, I.; Kohda, D.; Maenaka, K. Efficient Leukocyte Ig-like Receptor Signaling and Crystal Structure of Disulfide-linked HLA-G Dimer. J. Biol. Chem. 2006, 281, 10439–10447. [Google Scholar] [CrossRef] [Green Version]
- Boyson, J.E.; Erskine, R.; Whitman, M.C.; Chiu, M.; Lau, J.M.; Koopman, L.A.; Valter, M.M.; Angelisova, P.; Horejsí, V.; Strominger, J.L. Disulfide bond-mediated dimerization of HLA-G on the cell surface. Proc. Natl. Acad. Sci. USA 2002, 99, 16180–16185. [Google Scholar] [CrossRef] [Green Version]
- Santos, S.; Powis, S.; Arosa, F.A. Misfolding of Major Histocompatibility Complex Class I Molecules in Activated T Cells Allows cis-Interactions with Receptors and Signaling Molecules and Is Associated with Tyrosine Phosphorylation. J. Biol. Chem. 2004, 279, 53062–53070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, X.; Kim, J.; Deng, M.; John, S.; Chen, H.; Wu, G.; Phan, H.; Zhang, C.C. Inhibitory leukocyte immunoglobulin-like receptors: Immune checkpoint proteins and tumor sustaining factors. Cell Cycle 2016, 15, 25–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krijgsman, D.; Roelands, J.; Hendrickx, W.; Bedognetti, D.; Kuppen, P.J.K. HLA-G: A New Immune Checkpoint in Cancer? Int. J. Mol. Sci. 2020, 21, 4528. [Google Scholar] [CrossRef] [PubMed]
- Hirayasu, K.; Arase, H. Functional and genetic diversity of leukocyte immunoglobulin-like receptor and implication for disease associations. J. Hum. Genet. 2015, 60, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, K.; Matsubara, H.; Kanda, R.; Miyashita, N.; Shiroishi, M.; Fukunaga, Y.; Kamishikiryo, J.; Fukunaga, A.; Fukuhara, H.; Hirose, K.; et al. Structural and Functional Basis for LILRB Immune Checkpoint Receptor Recognition of HLA-G Isoforms. J. Immunol. 2019, 203, 3386–3394. [Google Scholar] [CrossRef] [PubMed]
- Shiroishi, M.; Kuroki, K.; Rasubala, L.; Tsumoto, K.; Kumagai, I.; Kurimoto, E.; Kato, K.; Kohda, D.; Maenaka, K. Structural basis for recognition of the nonclassical MHC molecule HLA-G by the leukocyte Ig-like receptor B2 (LILRB2/LIR2/ILT4/CD85d). Proc. Natl. Acad. Sci. USA 2006, 103, 16412–16417. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Song, H.; Cheng, H.; Qi, J.; Nam, G.; Tan, S.; Wang, J.; Fang, M.; Shi, Y.; Tian, Z.; et al. Structures of the four Ig-like domain LILRB2 and the four-domain LILRB1 and HLA-G1 complex. Cell. Mol. Immunol. 2019, 17, 966–975. [Google Scholar] [CrossRef]
- Mandel, I.; Ziv, D.H.; Goldshtein, I.; Peretz, T.; Alishekevitz, D.; Dror, A.F.; Hakim, M.; Hashmueli, S.; Friedman, I.; Sapir, Y.; et al. BND-22, a first-in-class humanized ILT2-blocking antibody, promotes antitumor immunity and tumor regression. J. Immunother. Cancer 2022, 10, e004859. [Google Scholar] [CrossRef]
- Lin, A.; Yan, W.-H. Heterogeneity of HLA-G Expression in Cancers: Facing the Challenges. Front. Immunol. 2018, 9, 2164. [Google Scholar] [CrossRef]
- Zheng, G.; Jia, L.; Yang, A.-G. Roles of HLA-G/KIR2DL4 in Breast Cancer Immune Microenvironment. Front. Immunol. 2022, 13, 791975. [Google Scholar] [CrossRef]
- Van Beneden, K.; De Creus, A.; Stevenaert, F.; Debacker, V.; Plum, J.; Leclercq, G. Expression of Inhibitory Receptors Ly49E and CD94/NKG2 on Fetal Thymic and Adult Epidermal TCR Vγ3 Lymphocytes. J. Immunol. 2002, 168, 3295–3302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.S.; McCullar, V. Human natural killer cells with polyclonal lectin and immunoglobulinlike receptors develop from single hematopoietic stem cells with preferential expression of NKG2A and KIR2DL2/L3/S2. Blood 2001, 98, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Eugène, J.; Jouand, N.; Ducoin, K.; Dansette, D.; Oger, R.; Deleine, C.; Leveque, E.; Meurette, G.; Podevin, J.; Matysiak, T.; et al. The inhibitory receptor CD94/NKG2A on CD8+ tumor-infiltrating lymphocytes in colorectal cancer: A promising new druggable immune checkpoint in the context of HLAE/β2m overexpression. Mod. Pathol. 2020, 33, 468–482. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, B.K.; Pizarro, J.C.; Kerns, J.; Strong, R.K. Structural basis for NKG2A/CD94 recognition of HLA-E. Proc. Natl. Acad. Sci. USA 2008, 105, 6696–6701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veuillen, C.; Aurran-Schleinitz, T.; Castellano, R.; Rey, J.; Mallet, F.; Orlanducci, F.; Pouyet, L.; Just-Landi, S.; Coso, D.; Ivanov, V.; et al. Primary B-CLL Resistance to NK Cell Cytotoxicity can be Overcome In Vitro and In Vivo by Priming NK Cells and Monoclonal Antibody Therapy. J. Clin. Immunol. 2012, 32, 632–646. [Google Scholar] [CrossRef]
- Prašnikar, E.; Perdih, A.; Borišek, J. What a Difference an Amino Acid Makes: An All-Atom Simulation Study of Nonameric Peptides in Inhibitory HLA-E/NKG2A/CD94 Immune Complexes. Front. Pharmacol. 2022, 13, 925427. [Google Scholar] [CrossRef]
- Ruibal, P.; Franken, K.L.M.C.; van Meijgaarden, K.E.; van Loon, J.J.F.; van der Steen, D.; Heemskerk, M.H.M.; Ottenhoff, T.H.M.; Joosten, S.A. Peptide Binding to HLA-E Molecules in Humans, Nonhuman Primates, and Mice Reveals Unique Binding Peptides but Remarkably Conserved Anchor Residues. J. Immunol. 2020, 205, 2861–2872. [Google Scholar] [CrossRef]
- Chai, I.; Kornyeyev, D.; Hsieh, E.; Magombedze, G.; Stapleton, L.; Hung, M.; Kwon, H.J.; Stefanutti, E.; Belzile, J.; Czerwieniec, G.; et al. Effects of small molecule-induced dimerization on the programmed death ligand 1 protein life cycle. Sci. Rep. 2022, 12, 21286. [Google Scholar] [CrossRef]
- Guzik, K.; Zak, K.M.; Grudnik, P.; Magiera, K.; Musielak, B.; Törner, R.; Skalniak, L.; Dömling, A.; Dubin, G.; Holak, T.A. Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Death-Ligand 1 (PD-1/PD-L1) Interaction via Transiently Induced Protein States and Dimerization of PD-L1. J. Med. Chem. 2017, 60, 5857–5867. [Google Scholar] [CrossRef]
- Liu, C.; Seeram, N.P.; Ma, H. Small molecule inhibitors against PD-1/PD-L1 immune checkpoints and current methodologies for their development: A review. Cancer Cell Int. 2021, 21, 239. [Google Scholar] [CrossRef]
- Guo, Y.; Jin, Y.; Wang, B.; Liu, B. Molecular Mechanism of Small-Molecule Inhibitors in Blocking the PD-1/PD-L1 Pathway through PD-L1 Dimerization. Int. J. Mol. Sci. 2021, 22, 4766. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ye, W.; Wang, S.; He, Y.; Zhong, H.; Wang, Y.; Zhu, Y.; Han, J.; Bing, Z.; Ji, S.; et al. Discovery of a new inhibitor targeting PD-L1 for cancer immunotherapy. Neoplasia 2021, 23, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Ghochikyan, A.; Pichugin, A.; Bagaev, A.; Davtyan, A.; Hovakimyan, A.; Tukhvatulin, A.; Davtyan, H.; Shcheblyakov, D.; Logunov, D.; Chulkina, M.; et al. Targeting TLR-4 with a novel pharmaceutical grade plant derived agonist, Immunomax®, as a therapeutic strategy for metastatic breast cancer. J. Transl. Med. 2014, 12, 322. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Yang, Y.; Jiang, Y.; Shao, J.; Sun, X.; Chen, J.; Dong, L.; Zhang, J. Anti-tumor immune responses of tumor-associated macrophages via toll-like receptor 4 triggered by cationic polymers. Biomaterials 2013, 34, 746–755. [Google Scholar] [CrossRef]
- Ajith, A.; Portik-Dobos, V.; Nguyen-Lefebvre, A.T.; Callaway, C.; Horuzsko, D.D.; Kapoor, R.; Zayas, C.; Maenaka, K.; Mulloy, L.L.; Horuzsko, A. HLA-G dimer targets Granzyme B pathway to prolong human renal allograft survival. FASEB J. 2019, 33, 5220–5236. [Google Scholar] [CrossRef]
- Zilberman, S.; Schenowitz, C.; Agaugué, S.; Favier, B.; Riteau, B.; Rouzier, R.; Carosella, E.D.; Rouas-Freiss, N.; Menier, C. HLA-G1 and HLA-G5 active dimers are present in malignant cells and effusions: The influence of the tumor microenvironment. Eur. J. Immunol. 2012, 42, 1599–1608. [Google Scholar] [CrossRef] [Green Version]
- Rouas-Freiss, N.; Bruel, S.; Menier, C.; Marcou, C.; Moreau, P.; Carosella, E.D. Switch ofHLA-G alternative splicing in a melanoma cell line causes loss of HLA-G1 expression and sensitivity to NK lysis. Int. J. Cancer 2005, 117, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.; Elstrand, M.B.; McMaster, M.T.; Berner, A.; Kurman, R.J.; Risberg, B.; Trope, C.G.; Shih, I.-M. HLA-G expression in effusions is a possible marker of tumor susceptibility to chemotherapy in ovarian carcinoma. Gynecol. Oncol. 2005, 96, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Singer, G.; Rebmann, V.; Chen, Y.-C.; Liu, H.-T.; Ali, S.Z.; Reinsberg, J.; McMaster, M.T.; Pfeiffer, K.; Chan, D.W.; Wardelmann, E.; et al. HLA-G is a potential tumor marker in malignant ascites. Clin. Cancer Res. 2003, 9, 4460–4464. [Google Scholar]
- Schwich, E.; Rebmann, V.; Michita, R.T.; Rohn, H.; Voncken, J.W.; Horn, P.A.; Kimmig, R.; Kasimir-Bauer, S.; Buderath, P. HLA-G 3′ untranslated region variants +3187G/G, +3196G/G and +3035T define diametrical clinical status and disease outcome in epithelial ovarian cancer. Sci. Rep. 2019, 9, 5407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reches, A.; Nachmani, D.; Berhani, O.; Duev-Cohen, A.; Shreibman, D.; Ophir, Y.; Seliger, B.; Mandelboim, O. HNRNPR Regulates the Expression of Classical and Nonclassical MHC Class I Proteins. J. Immunol. 2016, 196, 4967–4976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducoin, K.; Oger, R.; Mutala, L.B.; Deleine, C.; Jouand, N.; Desfrançois, J.; Podevin, J.; Duchalais, E.; Cruard, J.; Benlalam, H.; et al. Targeting NKG2A to boost anti-tumor CD8 T-cell responses in human colorectal cancer. Oncoimmunology 2022, 11, 2046931. [Google Scholar] [CrossRef] [PubMed]
- Ram, D.R.; Lucar, O.; Hueber, B.; Reeves, R.K. Simian Immunodeficiency Virus Infection Modulates CD94 + (KLRD1+) NK Cells in Rhesus Macaques. J. Virol. 2019, 93, e00731-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, M.T.; Wu, J.; Fang, M.; Sigal, L.J.; Spee, P.; Egebjerg, T.; Dissen, E.; Fossum, S.; Phillips, J.H.; Lanier, L.L. Development and Function of CD94-Deficient Natural Killer Cells. PLoS ONE 2010, 5, e15184. [Google Scholar] [CrossRef] [Green Version]
- Vance, R.E.; Jamieson, A.M.; Cado, D.; Raulet, D.H. Implications of CD94 deficiency and monoallelic NKG2A expression for natural killer cell development and repertoire formation. Proc. Natl. Acad. Sci. USA 2002, 99, 868–873. [Google Scholar] [CrossRef] [Green Version]
- Mingari, M.C.; Ponte, M.; Bertone, S.; Schiavetti, F.; Vitale, C.; Bellomo, R.; Moretta, A.; Moretta, L. HLA class I-specific inhibitory receptors in human T lymphocytes: Interleukin 15-induced expression of CD94/NKG2A in superantigen- or alloantigen-activated CD8 + T cells. Proc. Natl. Acad. Sci. USA 1998, 95, 1172–1177. [Google Scholar] [CrossRef] [Green Version]
- Starzer, A.M.; Preusser, M.; Berghoff, A.S. Immune escape mechanisms and therapeutic approaches in cancer: The cancer-immunity cycle. Ther. Adv. Med Oncol. 2022, 14, 17588359221096219. [Google Scholar] [CrossRef]
- Wang, L.; Liang, H.; Sun, J.; Liu, Y.; Li, J.; Li, J.; Li, J.; Yang, H. Bispecific Aptamer Induced Artificial Protein-Pairing: A Strategy for Selective Inhibition of Receptor Function. J. Am. Chem. Soc. 2019, 141, 12673–12681. [Google Scholar] [CrossRef]
- Akinleye, A.; Rasool, Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019, 12, 92. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.; Li, D.; Zhu, X. Cancer immunotherapy: Pros, cons and beyond. Biomed. Pharmacother. 2020, 124, 109821. [Google Scholar] [CrossRef] [PubMed]
- Jiao, P.; Geng, Q.; Jin, P.; Su, G.; Teng, H.; Dong, J.; Yan, B. Small Molecules as PD-1/PD-L1 Pathway Modulators for Cancer Immunotherapy. Curr. Pharm. Des. 2019, 24, 4911–4920. [Google Scholar] [CrossRef] [PubMed]
- Shaabani, S.; Huizinga, H.P.S.; Butera, R.; Kouchi, A.; Guzik, K.; Magieramularz, K.; Holak, T.A.; Domling, A. A patent review on PD-1/PD-L1 antagonists: Small molecules, peptides, and macrocycles (2015–2018). Expert Opin. Ther. Pat. 2018, 28, 665–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Li, J. Dimerization of Transmembrane Proteins in Cancer Immunotherapy. Membranes 2023, 13, 393. https://doi.org/10.3390/membranes13040393
Li L, Li J. Dimerization of Transmembrane Proteins in Cancer Immunotherapy. Membranes. 2023; 13(4):393. https://doi.org/10.3390/membranes13040393
Chicago/Turabian StyleLi, Lei, and Jingying Li. 2023. "Dimerization of Transmembrane Proteins in Cancer Immunotherapy" Membranes 13, no. 4: 393. https://doi.org/10.3390/membranes13040393
APA StyleLi, L., & Li, J. (2023). Dimerization of Transmembrane Proteins in Cancer Immunotherapy. Membranes, 13(4), 393. https://doi.org/10.3390/membranes13040393