
Breakdown of Phospholipid Asymmetry Triggers ADAM17-Mediated Rescue Events in Cells Undergoing Apoptosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture
2.3. Expression Vectors
2.4. Biotinylation
2.5. Transfection and AP-Substrate Shedding Assay in HEK293T Cells
2.6. Annexin V Staining
2.7. Image Analysis and Image Statistics
2.8. Determination of Caspase Activity in HEK293T Cells
2.9. siRNA Transfection and Induction of Apoptosis
2.10. Epiregulin ELISA
2.11. Immunoblot Analysis
2.12. Real-Time PCR of Xkr8 Expression
2.13. Flow Cytometric Analysis (FACS) of PS Exposure
2.14. Cell Viability Assay
2.15. Statistics
3. Results
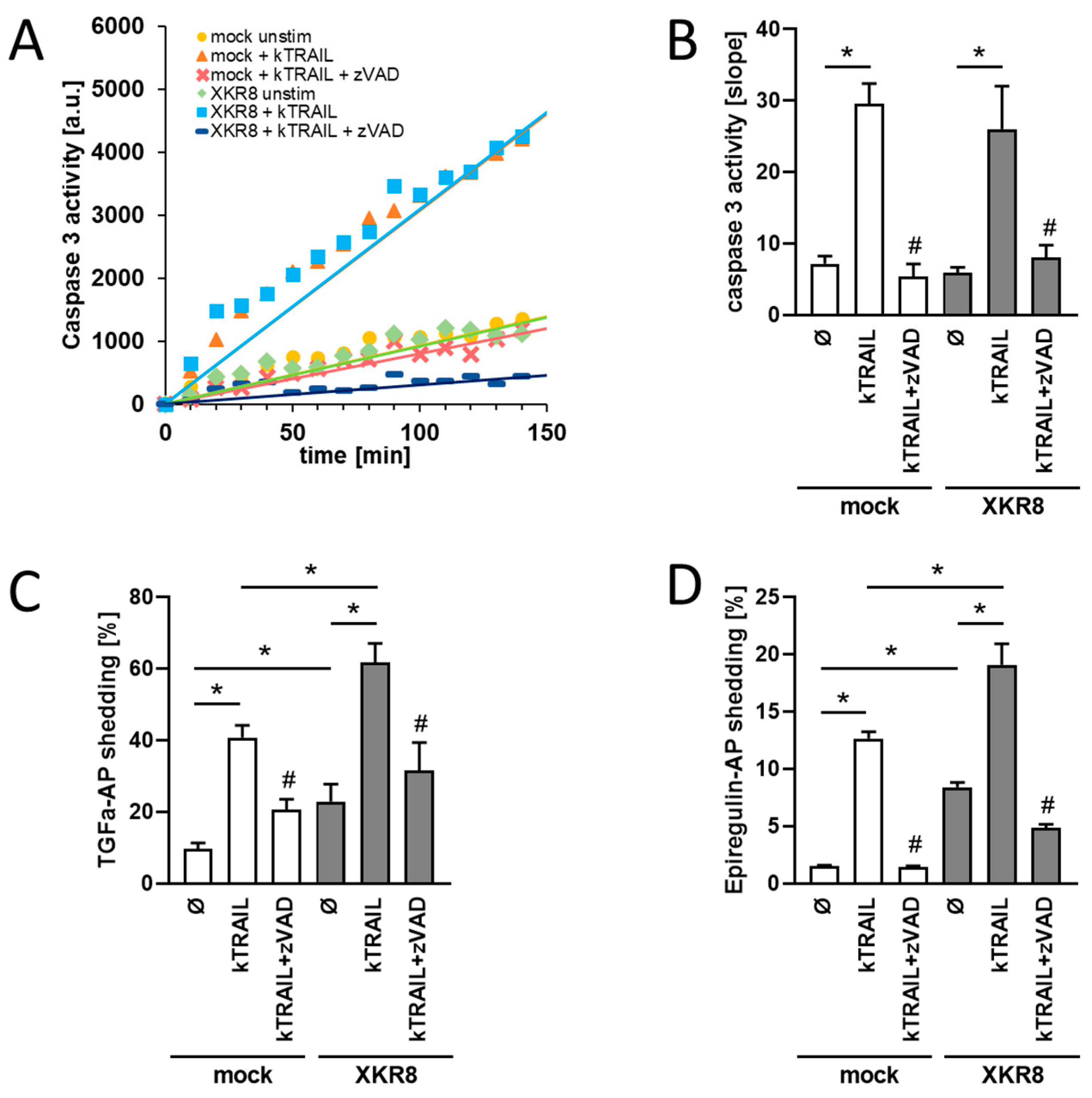
3.1. HEK293T Cells Overexpressing Xkr8 Display Increased Shedding of TGF-Alpha and EREG upon Induction of Apoptosis
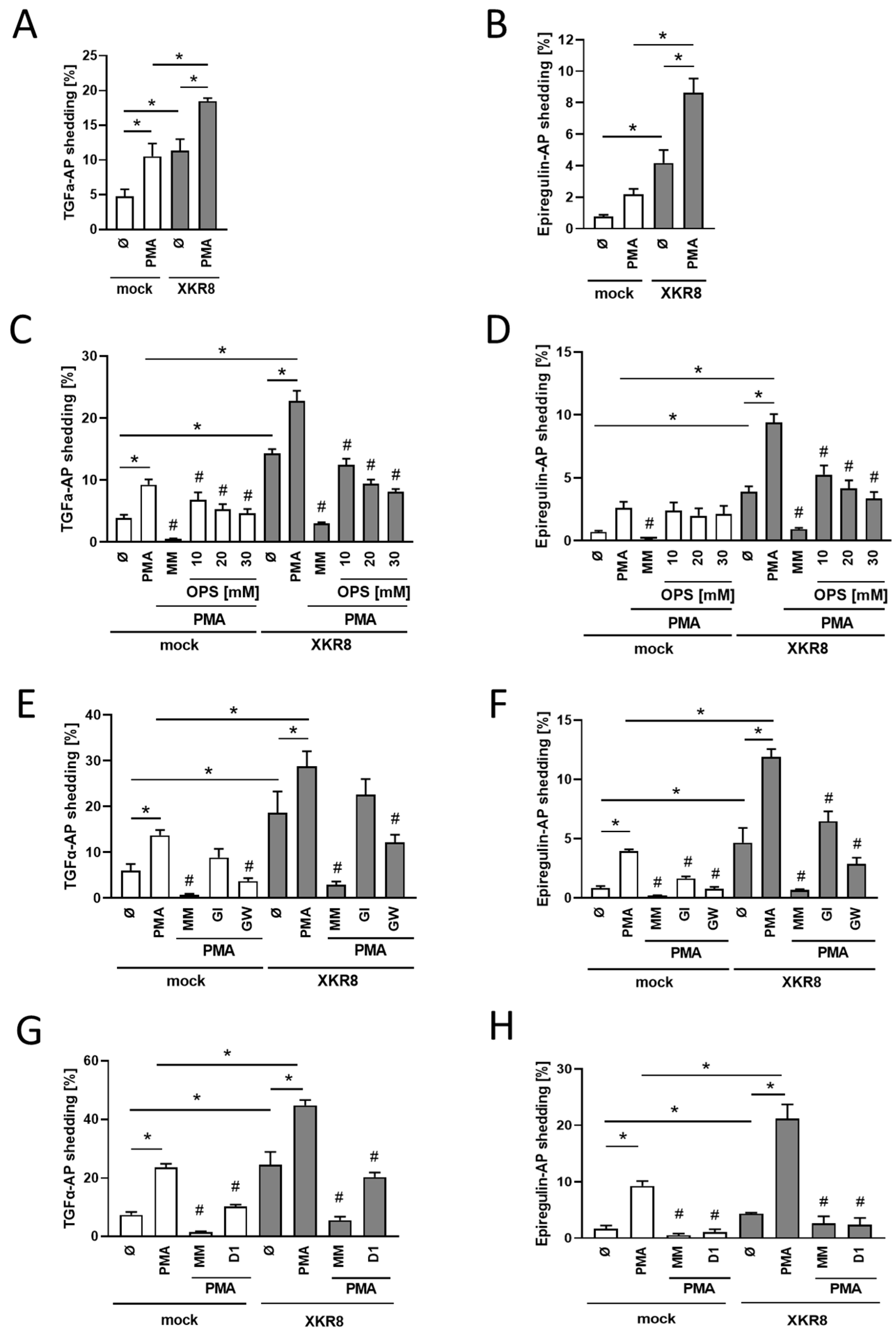
3.2. Xkr8 Overexpression Leads to Enhanced PMA-Induced Shedding of TGF-Alpha and EREG
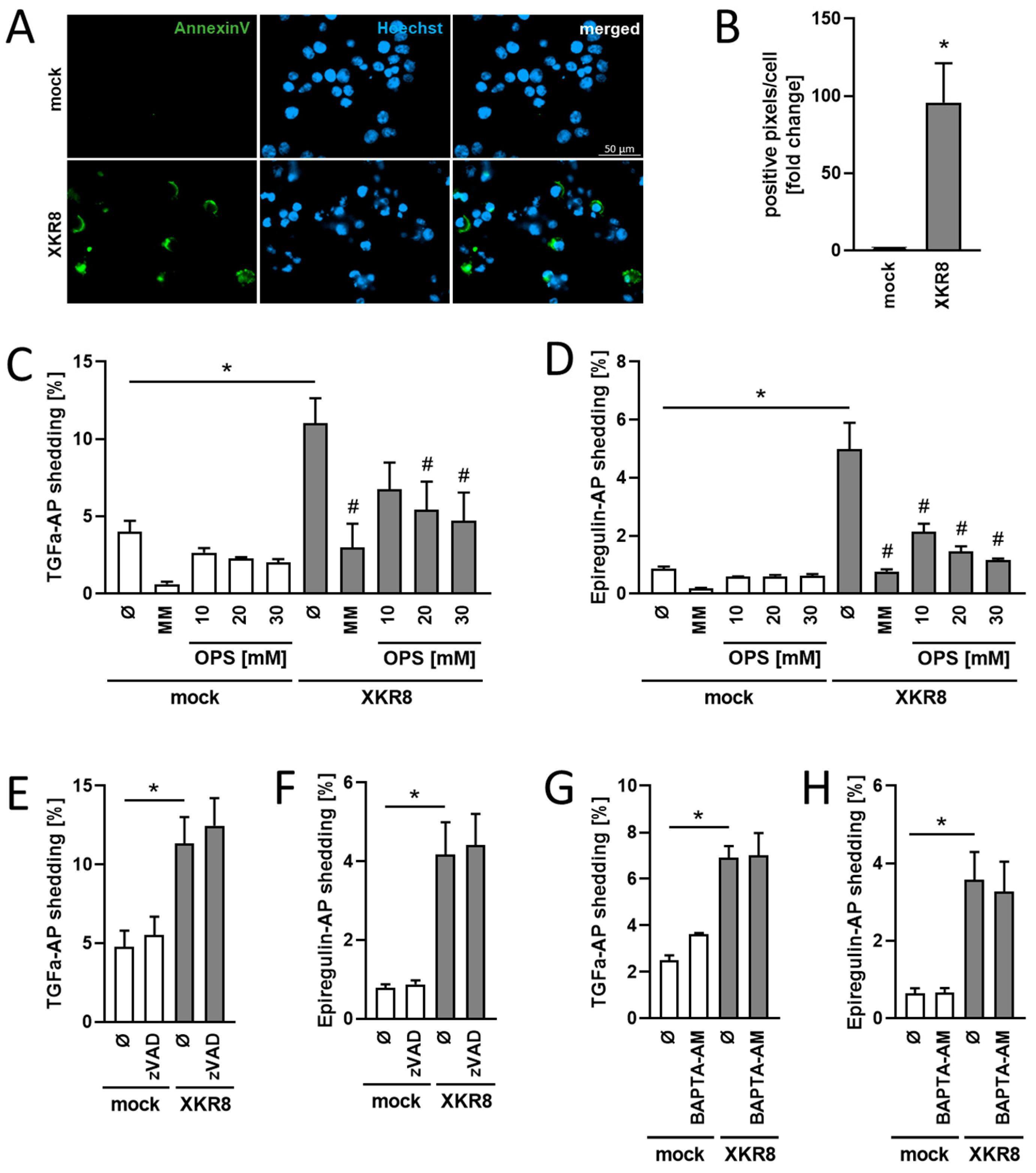
3.3. Increased Constitutive Substrate Cleavage in Xkr8-Overexpressing Cells
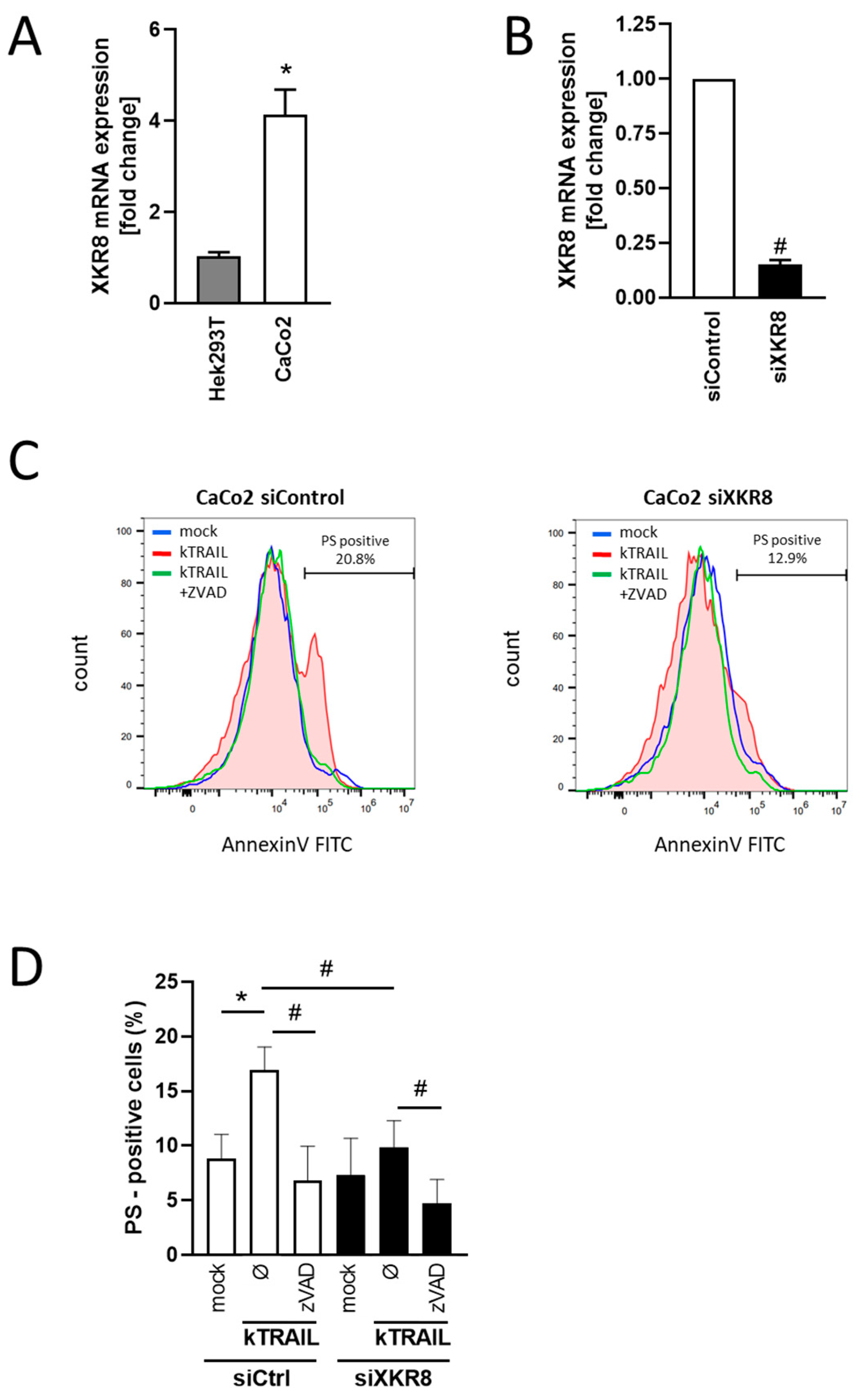
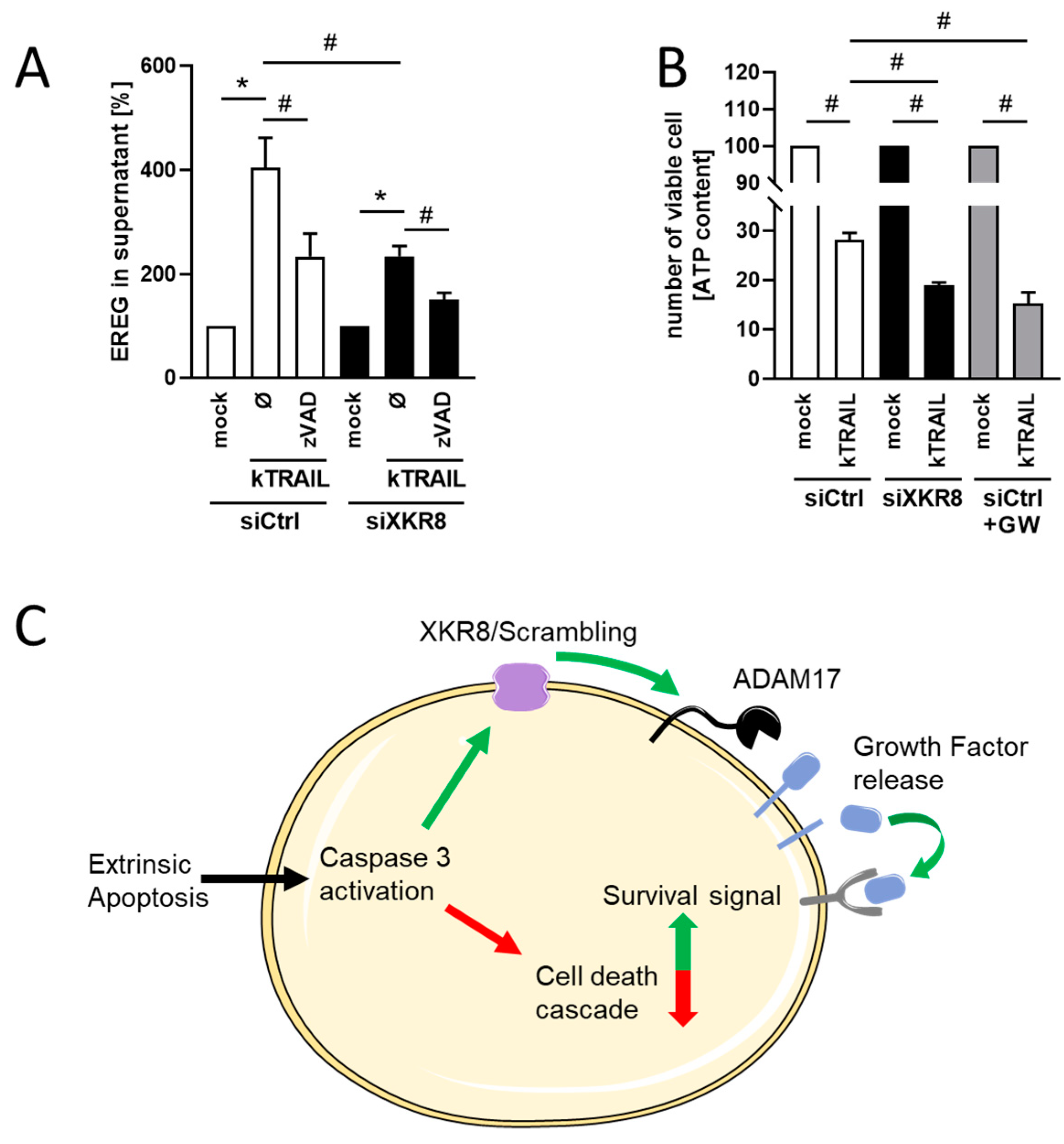
3.4. Downregulation of Xkr8 Diminishes PS-Externalization, Growth Factor Release and Cellular Resilience to Apoptosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Hankins, H.M.; Baldridge, R.D.; Xu, P.; Graham, T.R. Role of Flippases, Scramblases and Transfer Proteins in Phosphatidylserine Subcellular Distribution. Traffic 2015, 16, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Nagata, S.; Suzuki, J.; Segawa, K.; Fujii, T. Exposure of phosphatidylserine on the cell surface. Cell Death Differ. 2016, 23, 952–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenoir, G.; D’Ambrosio, J.M.; Dieudonné, T.; Čopič, A. Transport Pathways That Contribute to the Cellular Distribution of Phosphatidylserine. Front. Cell Dev. Biol. 2021, 9, 737907. [Google Scholar] [CrossRef]
- Bevers, E.M.; Williamson, P.L. Getting to the outer leaflet: Physiology of phosphatidylserine exposure at the plasma membrane. Physiol. Rev. 2016, 96, 605–645. [Google Scholar] [CrossRef] [PubMed]
- Doktorova, M.; Symons, J.L.; Levental, I. Structural and functional consequences of reversible lipid asymmetry in living membranes. Nat. Chem. Biol. 2020, 16, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.J.; Hossain, K.R.; Cao, K. Physiological roles of transverse lipid asymmetry of animal membranes. Biochim. Biophys. Acta-Biomembr. 2020, 1862, 183382. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.W.; Takatsu, H. Phosphatidylserine exposure in living cells. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 166–178. [Google Scholar] [CrossRef]
- Kalienkova, V.; Clerico Mosina, V.; Paulino, C. The Groovy TMEM16 Family: Molecular Mechanisms of Lipid Scrambling and Ion Conduction. J. Mol. Biol. 2021, 433, 166941. [Google Scholar] [CrossRef]
- Khelashvili, G.; Menon, A.K. Phospholipid Scrambling by G Protein-Coupled Receptors. Annu. Rev. Biophys. 2021, 51, 39–61. [Google Scholar] [CrossRef]
- Suzuki, J.; Denning, D.P.; Imanishi, E.; Horvitz, H.R.; Nagata, S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 2013, 341, 403–406. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, J.; Imanishi, E.; Nagata, S. Exposure of Phosphatidylserine by Xk-related Protein Family Members during Apoptosis. J. Biol. Chem. 2014, 289, 30257–30267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segawa, K.; Kurata, S.; Yanagihashi, Y.; Brummelkamp, T.R.; Matsuda, F.; Nagata, S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 2014, 344, 1164–1168. [Google Scholar] [CrossRef] [PubMed]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.L.; Jin, S.L.; Milla, M.E.; Bickett, D.M.; Burkhart, W.; Carter, H.L.; Chen, W.J.; Clay, W.C.; Didsbury, J.R.; Hassler, D.; et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature 1997, 385, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Weskamp, G.; Kelly, K.; Zhou, H.-M.; Higashiyama, S.; Peschon, J.; Hartmann, D.; Saftig, P.; Blobel, C.P. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol. 2004, 164, 769–779. [Google Scholar] [CrossRef]
- Blobel, C.P. ADAMs: Key components in EGFR signalling and development. Rev. Mol. Cell Biol. 2005, 6, 32–43. [Google Scholar] [CrossRef]
- Cheng, W.L.; Feng, P.H.; Lee, K.Y.; Chen, K.Y.; Sun, W.L.; Van Hiep, N.; Luo, C.S.; Wu, S.M. The Role of EREG/EGFR Pathway in Tumor Progression. Int. J. Mol. Sci. 2021, 22, 12828. [Google Scholar] [CrossRef]
- Riese, D.J.; Cullum, R.L. Epiregulin: Roles in Normal Physiology and Cancer. Semin. Cell Dev. Biol. 2014, 28, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Li, X.D.; Miao, S.Y.; Wang, G.L.; Yang, L.; Shu, Y.Q.; Yin, Y.M. Amphiregulin and epiregulin expression in colorectal carcinoma and the correlation with clinicopathological characteristics. Onkologie 2010, 33, 353–358. [Google Scholar] [CrossRef]
- Reiss, K.; Bhakdi, S. Pore-forming bacterial toxins and antimicrobial peptides as modulators of ADAM function. Med. Microbiol. Immunol. 2012, 201, 419–426. [Google Scholar] [CrossRef]
- Sommer, A.; Fries, A.; Cornelsen, I.; Speck, N.; Koch-Nolte, F.; Gimpl, G.; Andrä, J.; Bhakdi, S.; Reiss, K. Melittin modulates keratinocyte function through P2 receptor-dependent ADAM activation. J. Biol. Chem. 2012, 287, 23678–23689. [Google Scholar] [CrossRef] [Green Version]
- Horiuchi, K.; Le Gall, S.; Schulte, M.; Yamaguchi, T.; Reiss, K.; Murphy, G.; Toyama, Y.; Hartmann, D.; Saftig, P.; Blobel, C.P. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol. Biol. Cell 2007, 18, 176–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashida, K.; Bartlett, A.H.; Chen, Y.; Park, P.W. Molecular and cellular mechanisms of ectodomain shedding. Anat. Rec. 2010, 293, 925–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Gall, S.M.; Bobé, P.; Reiss, K.; Horiuchi, K.; Da Niu, X.-D.; Lundell, D.; Gibb, D.R.; Conrad, D.; Saftig, P.; Blobel, C.P.; et al. ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor alpha, L-selectin, and tumor necrosis factor alpha. Mol. Biol. Cell 2009, 20, 1785–1794. [Google Scholar] [CrossRef] [Green Version]
- Leitzke, S.; Seidel, J.; Ahrens, B.; Schreiber, R.; Kunzelmann, K.; Sperrhacke, M.; Bhakdi, S.; Reiss, K. Influence of Anoctamin-4 and -9 on ADAM10 and ADAM17 Sheddase Function. Membranes 2022, 12, 123. [Google Scholar] [CrossRef] [PubMed]
- Reiss, K.; Leitzke, S.; Seidel, J.; Sperrhacke, M.; Bhakdi, S. Scramblases as Regulators of Proteolytic ADAM Function. Membranes 2022, 12, 185. [Google Scholar] [CrossRef]
- Veit, M.; Koyro, K.I.; Ahrens, B.; Bleibaum, F.; Munz, M.; Rövekamp, H.; Andrä, J.; Schreiber, R.; Kunzelmann, K.; Sommer, A.; et al. Anoctamin-6 regulates ADAM sheddase function. Biochim. Biophys. Acta-Mol. Cell Res. 2018, 1865, 1598–1610. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.; Kordowski, F.; Büch, J.; Maretzky, T.; Evers, A.; Andrä, J.J.J.J.; Düsterhöft, S.; Michalek, M.; Lorenzen, I.; Somasundaram, P.; et al. Phosphatidylserine exposure is required for ADAM17 sheddase function. Nat. Commun. 2016, 7, 11523. [Google Scholar] [CrossRef] [Green Version]
- Sommer, A.; Bhakdi, S.; Reiss, K. How membrane asymmetry regulates ADAM17 sheddase function. Cell Cycle 2016, 15, 2995–2996. [Google Scholar] [CrossRef] [Green Version]
- Riethmueller, S.; Ehlers, J.C.; Lokau, J.; Düsterhöft, S.; Knittler, K.; Dombrowsky, G.; Grötzinger, J.; Rabe, B.; Rose-John, S.; Garbers, C. Cleavage Site Localization Differentially Controls Interleukin-6 Receptor Proteolysis by ADAM10 and ADAM17. Sci. Rep. 2016, 6, 25550. [Google Scholar] [CrossRef] [Green Version]
- Kodigepalli, K.M.; Bowers, K.; Sharp, A.; Nanjundan, M. Roles and regulation of phospholipid scramblases. FEBS Lett. 2015, 589, 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakuragi, T.; Kosako, H.; Nagata, S. Phosphorylation-mediated activation of mouse Xkr8 scramblase for phosphatidylserine exposure. Proc. Natl. Acad. Sci. USA 2019, 116, 2907–2912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, A.; Hundhausen, C.; Lambert, M.H.; Broadway, N.; Andrews, R.C.; Bickett, D.M.; Leesnitzer, M.A.; David Becherer, J. Metalloproteinase Inhibitors for the Disintegrin-Like Metalloproteinases ADAM10 and ADAM17 that Differentially Block Constitutive and Phorbol Ester-Inducible Shedding of Cell Surface Molecules. Comb. Chem. High Throughput Screen. 2005, 8, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Hundhausen, C.; Misztela, D.; Berkhout, T.A.; Broadway, N.; Saftig, P.; Reiss, K.; Hartmann, D.; Fahrenholz, F.; Postina, R.; Matthews, V.; et al. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood 2003, 102, 1186–1195. [Google Scholar] [CrossRef] [Green Version]
- Tape, C.J.; Willems, S.H.; Dombernowsky, S.L.; Stanley, P.L.; Fogarasi, M.; Ouwehand, W.; McCafferty, J.; Murphy, G. Cross-domain inhibition of TACE ectodomain. Proc. Natl. Acad. Sci. USA 2011, 108, 5578–5583. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, Y.; Li, Q.; Luo, Z.; Zhang, Z.; Huang, H.; Sun, J.; Zhang, L.X.T.; Sun, R.; Bain, D.J.; et al. Targeting Xkr8 via nanoparticle-mediated in situ co-delivery of siRNA and chemotherapy drugs for cancer immunochemotherapy. Nat. Nanotechnol. 2023, 18, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Van Geelen, C.; De Vries, E.; Le, T.; Van Weeghel, R.P.; De Jong, S. Differential modulation of the TRAIL receptors and the CD95 receptor in colon carcinoma cell lines. Br. J. Cancer 2003, 89, 363–373. [Google Scholar] [CrossRef]
- Sunaga, N.; Kaira, K. Epiregulin as a therapeutic target in non-small-cell lung cancer. Lung Cancer Targets Ther. 2015, 6, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wu, S.; Cen, Z.; Zhang, Y.; Chen, Y.; Huang, Y.; Cillo, A.R.; Prokopec, J.S.; Quarato, G.; Vignali, D.A.A.A.; et al. Mobilizing phospholipids on tumor plasma membrane implicates phosphatidylserine externalization blockade for cancer immunotherapy. Cell Rep. 2022, 41, 111582. [Google Scholar] [CrossRef] [PubMed]
- Bleibaum, F.; Sommer, A.; Veit, M.; Rabe, B.; Andrä, J.; Kunzelmann, K.; Nehls, C.; Correa, W.; Gutsmann, T.; Grötzinger, J.; et al. ADAM10 sheddase activation is controlled by cell membrane asymmetry. J. Mol. Cell Biol. 2019, 11, 979–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, J.; Imanishi, E.; Nagata, S. Xkr8 phospholipid scrambling complex in apoptotic phosphatidylserine exposure. Proc. Natl. Acad. Sci. USA 2016, 113, 9509–9514. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.W.; Nam, G.H.; Kim, I.S.; Park, S.Y. Xk-related protein 8 regulates myoblast differentiation and survival. FEBS J. 2017, 284, 3575–3588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takatsu, H.; Takayama, M.; Naito, T.; Takada, N.; Tsumagari, K.; Ishihama, Y.; Nakayama, K.; Shin, H.W. Phospholipid flippase ATP11C is endocytosed and downregulated following Ca2+ mediated protein kinase C activation. Nat. Commun. 2017, 8, 1423. [Google Scholar] [CrossRef] [Green Version]
- Grieve, A.G.; Xu, H.; Künzel, U.; Bambrough, P.; Sieber, B.; Freeman, M. Phosphorylation of iRhom2 at the plasma membrane controls mammalian TACE-dependent inflammatory and growth factor signalling. Elife 2017, 6, e23968. [Google Scholar] [CrossRef]
- Cavadas, M.; Oikonomidi, I.; Gaspar, C.J.; Burbridge, E.; Badenes, M.; Félix, I.; Bolado, A.; Hu, T.; Bileck, A.; Gerner, C.; et al. Phosphorylation of iRhom2 Controls Stimulated Proteolytic Shedding by the Metalloprotease ADAM17/TACE. Cell Rep. 2017, 21, 745–757. [Google Scholar] [CrossRef] [Green Version]
- Maretzky, T.; McIlwain, D.R.; Issuree, P.D.A.; Li, X.; Malapeira, J.; Amin, S.; Lang, P.A.; Mak, T.W.; Blobel, C.P. iRhom2 controls the substrate selectivity of stimulated ADAM17-dependent ectodomain shedding. Proc. Natl. Acad. Sci. USA 2013, 110, 11433–11438. [Google Scholar] [CrossRef]
- Murthy, A.; Defamie, V.; Smookler, D.S.; Di Grappa, M.A.; Horiuchi, K.; Federici, M.; Sibilia, M.; Blobel, C.P.; Khokha, R. Ectodomain shedding of EGFR ligands and TNFR1 dictates hepatocyte apoptosis during fulminant hepatitis in mice. J. Clin. Investig. 2010, 120, 2731–2744. [Google Scholar] [CrossRef] [Green Version]
- Sommer, A.; Düppe, M.; Baumecker, L.; Kordowski, F.; Büch, J.; Chico, J.F.; Fritsch, J.; Schütze, S.; Adam, D.; Sperrhacke, M.; et al. Extracellular sphingomyelinase activity impairs TNF-α-induced endothelial cell death via ADAM17 activation and TNF receptor 1 shedding. Oncotarget 2017, 8, 72584–72596. [Google Scholar] [CrossRef] [Green Version]
- Terrado, J.; Monnier, D.; Perrelet, D.; Vesin, D.; Jemelin, S.; Buurman, W.A.; Mattenberger, L.; King, B.; Kato, A.C.; Garcia, I. Soluble TNF receptors partially protect injured motoneurons in the postnatal CNS. Eur. J. Neurosci. 2000, 12, 3443–3447. [Google Scholar] [CrossRef]
- Xia, M.; Xue, S.B.; Xu, C.S. Shedding of TNFR1 in regenerative liver can be induced with TNF α and PMA. World J. Gastroenterol. 2002, 8, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Birge, R.B.; Boeltz, S.; Kumar, S.; Carlson, J.; Wanderley, J.; Calianese, D.; Barcinski, M.; Brekken, R.A.; Huang, X.; Hutchins, J.T.; et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016, 23, 962–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, W.; Fa, H.; Xiao, D.; Wang, J. Targeting phosphatidylserine for Cancer therapy: Prospects and challenges. Theranostics 2020, 10, 9214–9229. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sperrhacke, M.; Leitzke, S.; Ahrens, B.; Reiss, K. Breakdown of Phospholipid Asymmetry Triggers ADAM17-Mediated Rescue Events in Cells Undergoing Apoptosis. Membranes 2023, 13, 720. https://doi.org/10.3390/membranes13080720
Sperrhacke M, Leitzke S, Ahrens B, Reiss K. Breakdown of Phospholipid Asymmetry Triggers ADAM17-Mediated Rescue Events in Cells Undergoing Apoptosis. Membranes. 2023; 13(8):720. https://doi.org/10.3390/membranes13080720
Chicago/Turabian StyleSperrhacke, Maria, Sinje Leitzke, Björn Ahrens, and Karina Reiss. 2023. "Breakdown of Phospholipid Asymmetry Triggers ADAM17-Mediated Rescue Events in Cells Undergoing Apoptosis" Membranes 13, no. 8: 720. https://doi.org/10.3390/membranes13080720
APA StyleSperrhacke, M., Leitzke, S., Ahrens, B., & Reiss, K. (2023). Breakdown of Phospholipid Asymmetry Triggers ADAM17-Mediated Rescue Events in Cells Undergoing Apoptosis. Membranes, 13(8), 720. https://doi.org/10.3390/membranes13080720