
Antifibrotic Therapies and Progressive Fibrosing Interstitial Lung Disease (PF-ILD): Building on INBUILD


Abstract
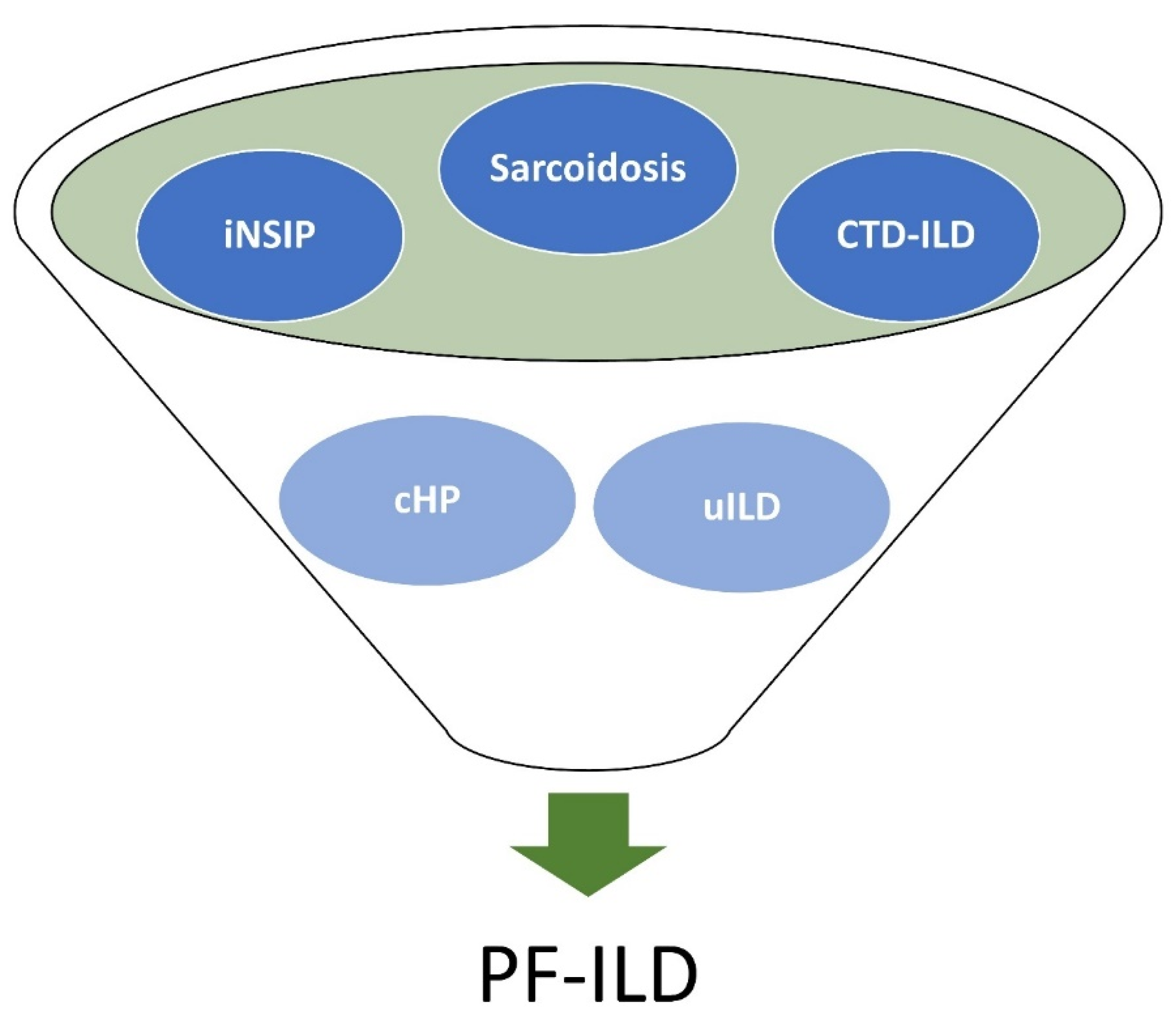
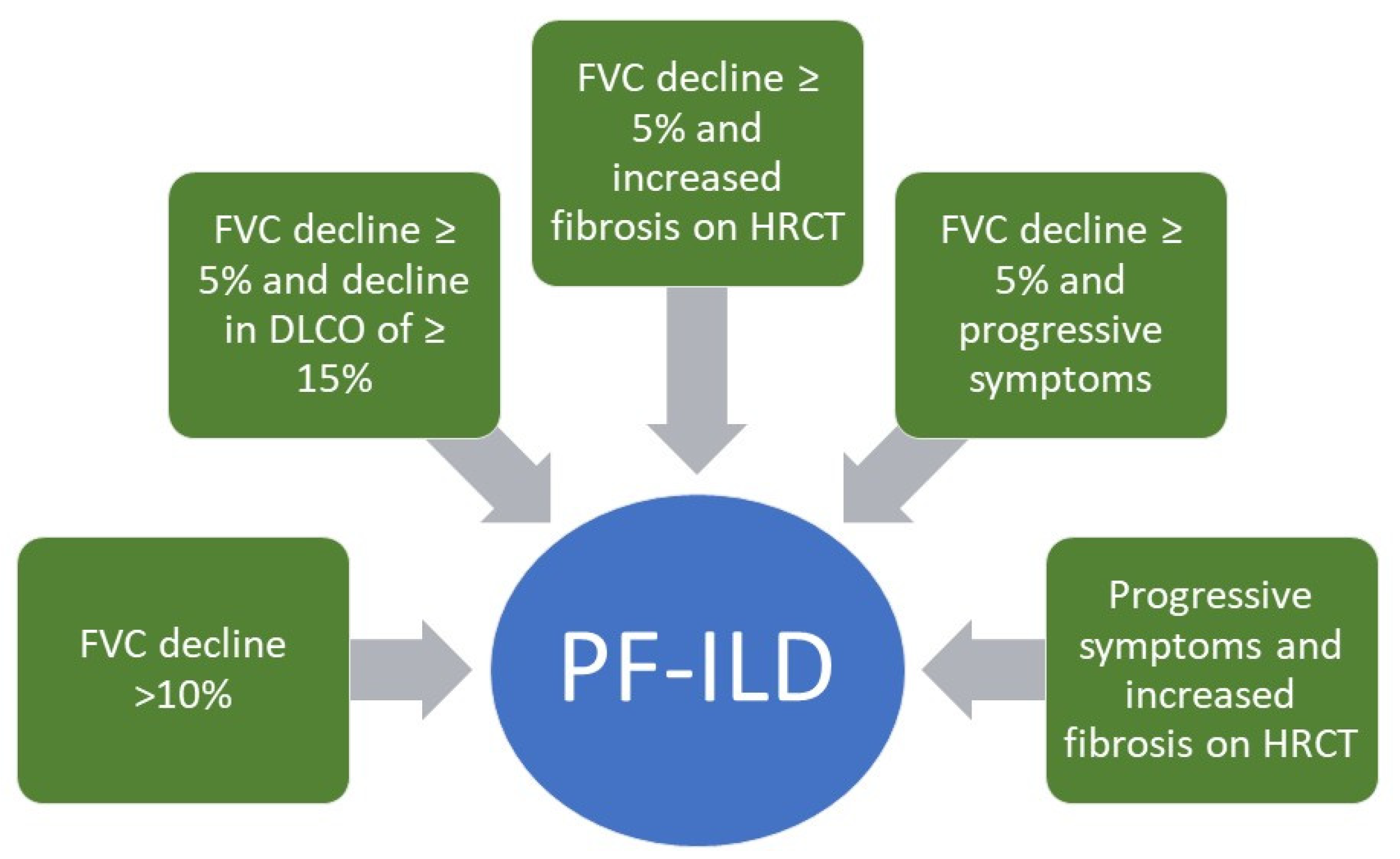
:1. Introduction
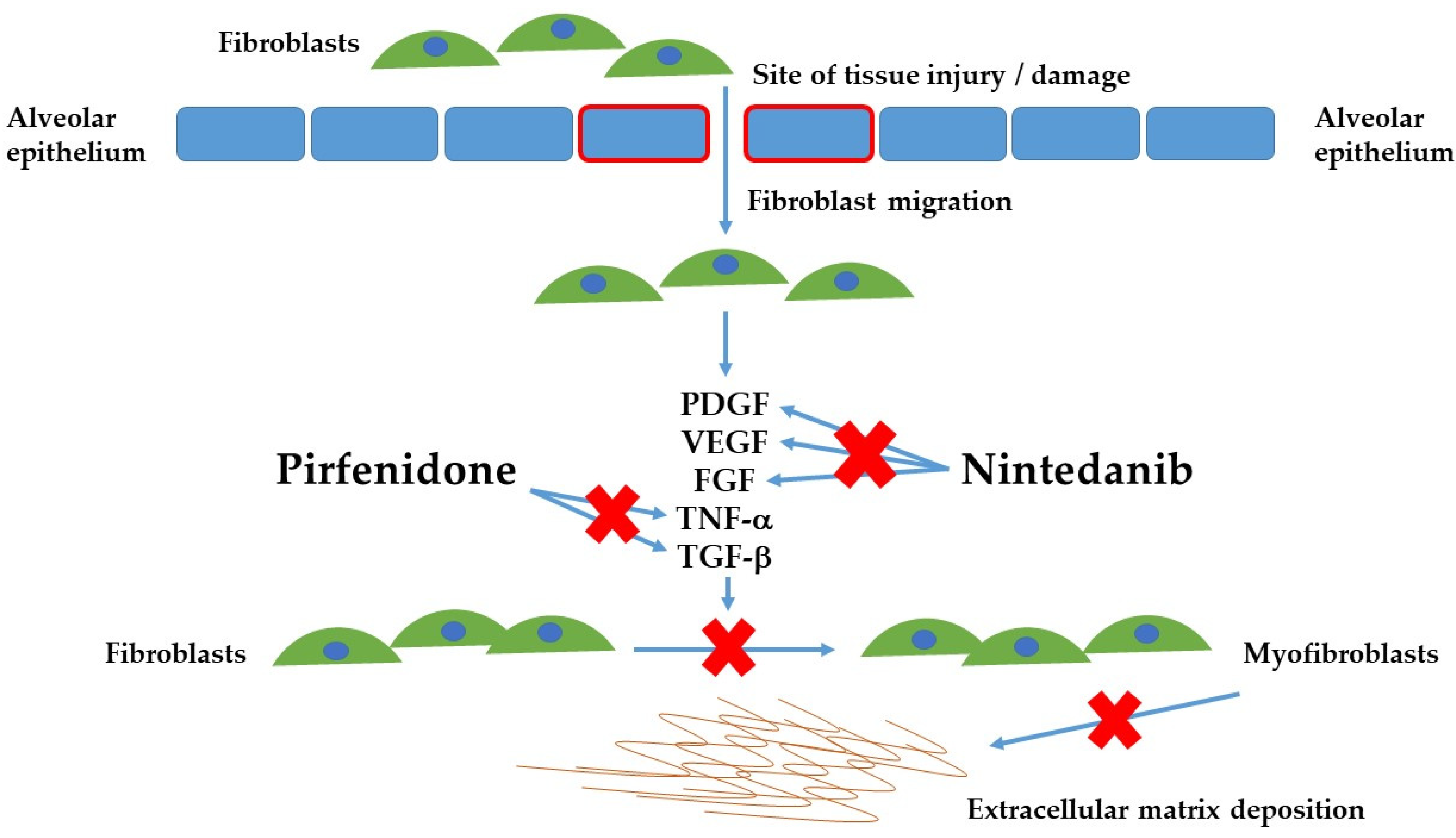
2. Pathogenesis of Fibrosis and Genetic Variants
3. Nintedanib
4. Pirfenidone
5. A Treatment Paradigm Change
Author Contributions
Funding
Conflicts of Interest
References
- Travis, W.D.; Costabel, U.; Hansell, D.M.; Talmadge, E.K., Jr.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. An Official American Thoracic Society/European Respiratory Society Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef]
- Duchemann, B.; Annesi-Maesano, I.; De Naurois, C.J.; Sanyal, S.; Brillet, P.-Y.; Brauner, M.; Kambouchner, M.; Huynh, S.; Naccache, J.M.; Borie, R.; et al. Prevalence and incidence of interstitial lung diseases in a multi-ethnic county of Greater Paris. Eur. Respir. J. 2017, 50, 1602419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-based Guidelines for Diagnosis and Management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, T.E.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [Green Version]
- Raghu, G.; Rochwerg, B.; Zhang, Y.; Garcia, C.A.C.; Azuma, A.; Behr, J.; Brozek, J.L.; Collard, H.R.; Cunningham, W.; Homma, S.; et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2015, 192, e3–e19. [Google Scholar] [CrossRef]
- Cottin, V.; Hirani, N.A.; Hotchkin, D.L.; Nambiar, A.M.; Ogura, T.; Otaola, M.; Skowasch, D.; Park, J.S.; Poonyagariyagorn, H.K.; Wuyts, W.; et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur. Respir. Rev. 2018, 27, 180076. [Google Scholar] [CrossRef] [Green Version]
- Collins, B.F.; Raghu, G. Antifibrotic therapy for fibrotic lung disease beyond idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2019, 28, 190022. [Google Scholar] [CrossRef]
- Yunt, Z.X.; Chung, J.H.; Hobbs, S.; Fernandez-Perez, E.R.; Olson, A.L.; Huie, T.J.; Keith, R.C.; Janssen, W.J.; Goldstein, B.L.; Lynch, D.A.; et al. High resolution computed tomography pattern of usual interstitial pneumonia in rheumatoid arthritis-associated interstitial lung disease: Relationship to survival. Respir. Med. 2017, 126, 100–104. [Google Scholar] [CrossRef] [Green Version]
- Churg, A.; Sin, D.D.; Everett, D.; Brown, K.; Cool, C. Pathologic Patterns and Survival in Chronic Hypersensitivity Pneumonitis. Am. J. Surg. Pathol. 2009, 33, 1765–1770. [Google Scholar] [CrossRef]
- Churg, A.; Muller, N.L.; Flint, J.; Wright, J.L. Chronic Hypersensitivity Pneumonitis. Am. J. Surg. Pathol. 2006, 30, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Cottin, V. Treatment of progressive fibrosing interstitial lung diseases: A milestone in the management of interstitial lung diseases. Eur. Respir. Rev. 2019, 28, 190109. [Google Scholar] [CrossRef]
- Flaherty, K.R.; Brown, K.K.; Wells, A.U.; Clerisme-Beaty, E.; Collard, H.R.; Cottin, V.; Devaraj, A.; Inoue, Y.; Le Maulf, F.; Richeldi, L.; et al. Design of the PF-ILD trial: A double-blind, randomised, placebo-controlled phase III trial of nintedanib in patients with progressive fibrosing interstitial lung disease. BMJ Open Respir. Res. 2017, 4, e000212. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strieter, R.M.; Mehrad, B. New Mechanisms of Pulmonary Fibrosis. Chest 2009, 136, 1364–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, T.M.; Wells, A.U.; Laurent, G.J. Idiopathic pulmonary fibrosis: Multiple causes and multiple mechanisms? Eur. Respir. J. 2007, 30, 835–839. [Google Scholar] [CrossRef] [Green Version]
- Wollin, L.; Distler, J.H.; Redente, E.F.; Riches, D.W.H.; Stowasser, S.; Schlenker-Herceg, R.; Maher, T.; Kolb, M. Potential of nintedanib in treatment of progressive fibrosing interstitial lung diseases. Eur. Respir. J. 2019, 54, 1900161. [Google Scholar] [CrossRef]
- Bagnato, G.; Harari, S. Cellular interactions in the pathogenesis of interstitial lung diseases. Eur. Respir. Rev. 2015, 24, 102–114. [Google Scholar] [CrossRef]
- Andersson-Sjöland, A.; de Alba, C.G.; Nihlberg, K.; Becerril, C.; Ramírez, R.; Pardo, A.; Westergren-Thorsson, G.; Selman, M. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2008, 40, 2129–2140. [Google Scholar] [CrossRef]
- Fernandez, E.I.; Eickelberg, O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet 2012, 380, 680–688. [Google Scholar] [CrossRef]
- Huang, X.; Yang, N.; Fiore, V.F.; Barker, T.H.; Sun, Y.; Morris, S.W.; Ding, Q.; Thannickal, V.J.; Zhou, Y. Matrix Stiffness–Induced Myofibroblast Differentiation Is Mediated by Intrinsic Mechanotransduction. Am. J. Respir. Cell Mol. Biol. 2012, 47, 340–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, J.T.; A Spiteri, M. Growth factors in idiopathic pulmonary fibrosis: Relative roles. Respir. Res. 2002, 3, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morais, A.; Beltrão, M.; Sokhatska, O.; Costa, D.; Melo, N.; Mota, P.; Marques, A.; Delgado, L. Serum metalloproteinases 1 and 7 in the diagnosis of idiopathic pulmonary fibrosis and other interstitial pneumonias. Respir. Med. 2015, 109, 1063–1068. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann-Vold, A.-M.; Weigt, S.S.; Saggar, R.; Palchevskiy, V.; Volkmann, E.R.; Liang, L.L.; Ross, D.; Ardehali, A.; Lynch, J.P.; Belperio, J.A. Endotype-phenotyping may predict a treatment response in progressive fibrosing interstitial lung disease. EBioMedicine 2019, 50, 379–386. [Google Scholar] [CrossRef] [Green Version]
- Gibson, C.D.; Kugler, M.C.; Deshwal, H.; Munger, J.S.; Condos, R. Advances in Targeted Therapy for Progressive Fibrosing Interstitial Lung Disease. Lung 2020, 198, 597–608. [Google Scholar] [CrossRef]
- Spagnolo, P.; Distler, O.; Ryerson, C.J.; Tzouvelekis, A.; Lee, J.S.; Bonella, F.; Bouros, D.; Hoffmann-Vold, A.-M.; Crestani, B.; Matteson, E.L. Mechanisms of progressive fibrosis in connective tissue disease (CTD)-associated interstitial lung diseases (ILDs). Ann. Rheum. Dis. 2021, 80, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Juge, P.A.; Lee, J.S.; Ebstein, E.; Furukawa, H.; Dobrinskikh, E.; Gazal, S.; Kannengiesser, C.; Ottaviani, S.; Oka, S.; Tohma, S.; et al. MUC5B Promoter Variant and Rheumatoid Arthritis with Interstitial Lung Disease. N. Engl. J. Med. 2018, 379, 2209–2219. [Google Scholar] [CrossRef] [PubMed]
- Ley, B.; Newton, C.; Arnould, I.; Elicker, B.M.; Henry, T.S.; Vittinghoff, E.; A Golden, J.; Jones, K.D.; Batra, K.; Torrealba, J.; et al. The MUC5B promoter polymorphism and telomere length in patients with chronic hypersensitivity pneumonitis: An observational cohort-control study. Lancet Respir. Med. 2017, 5, 639–647. [Google Scholar] [CrossRef]
- Stuart, B.D.; Lee, J.S.; Kozlitina, J.; Noth, I.; Devine, M.S.; Glazer, C.S.; Torres, F.; Kaza, V.; E Girod, C.; Jones, K.D.; et al. Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: An observational cohort study with independent validation. Lancet Respir. Med. 2014, 2, 557–565. [Google Scholar] [CrossRef] [Green Version]
- Hilberg, F.; Roth, G.J.; Krssak, M.; Kautschitsch, S.; Sommergruber, W.; Tontsch-Grunt, U.; Garin-Chesa, P.; Bader, G.; Zoephel, A.; Quant, J.; et al. BIBF 1120: Triple Angiokinase Inhibitor with Sustained Receptor Blockade and Good Antitumor Efficacy. Cancer Res. 2008, 68, 4774–4782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richeldi, L.; Costabel, U.; Selman, M.; Kim, D.S.; Hansell, D.M.; Nicholson, A.G.; Brown, K.K.; Flaherty, K.R.; Noble, P.W.; Raghu, G.; et al. Efficacy of a Tyrosine Kinase Inhibitor in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2011, 365, 1079–1087. [Google Scholar] [CrossRef] [Green Version]
- Richeldi, L.; Kreuter, M.; Selman, M.; Crestani, B.; Kirsten, A.-M.; A Wuyts, W.; Xu, Z.; Bernois, K.; Stowasser, S.; Quaresma, M.; et al. Long-term treatment of patients with idiopathic pulmonary fibrosis with nintedanib: Results from the TOMORROW trial and its open-label extension. Thorax 2017, 73, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Distler, O.; Highland, K.B.; Gahlemann, M.; Azuma, A.; Fischer, A.; Mayes, M.D.; Raghu, G.; Sauter, W.; Girard, M.; Alves, M.; et al. Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease. N. Engl. J. Med. 2019, 380, 2518–2528. [Google Scholar] [CrossRef]
- Wells, A.U.; Flaherty, K.R.; Brown, K.K.; Inoue, Y.; Devaraj, A.; Richeldi, L.; Moua, T.; Crestani, B.; A Wuyts, W.; Stowasser, S.; et al. Nintedanib in patients with progressive fibrosing interstitial lung diseases—Subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: A randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir. Med. 2020, 8, 453–460. [Google Scholar] [CrossRef]
- Feitelson, M.A.; Arzumanyan, A.; Kulathinal, R.J.; Blain, S.W.; Holcombe, R.F.; Mahajna, J.; Marino, M.; Martinez-Chantar, M.L.; Nawroth, R.; Sanchez-Garcia, I.; et al. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Semin. Cancer Biol. 2015, 35, S25–S54. [Google Scholar] [CrossRef]
- Iyer, S.N.; Gurujeyalakshmi, G.; Giri, S.N. Effects of pirfenidone on transforming growth factor-beta gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J. Pharmacol. Exp. Ther. 1999, 291, 367–373. [Google Scholar]
- Macías-Barragán, J.; Sandoval-Rodríguez, A.; Navarro-Partida, J.; Armendáriz-Borunda, J. The multifaceted role of pirfenidone and its novel targets. Fibrogenes. Tissue Repair 2010, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; E King, T.; Lancaster, L.; A Sahn, S.; Szwarcberg, J.; et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- Behr, J.; Neuser, P.; Prasse, A.; Kreuter, M.; Rabe, K.; Schade-Brittinger, C.; Wagner, J.; Günther, A. Exploring efficacy and safety of oral Pirfenidone for progressive, non-IPF lung fibrosis (RELIEF): A randomized, double-blind, placebo-controlled, parallel group, multi-center, phase II trial. BMC Pulm. Med. 2017, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behr, J.; Prasse, A.; Kreuter, M.; Johow, J.; Rabe, K.F.; Bonella, F.; Bonnet, R.; Grohe, C.; Held, M.; Wilkens, H.; et al. Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): A double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2021, 9, 476–486. [Google Scholar] [CrossRef]
- Vancheri, C.; Kreuter, M.; Richeldi, L.; Ryerson, C.J.; Valeyre, D.; Grutters, J.C.; Wiebe, S.; Stansen, W.; Quaresma, M.; Stowasser, S.; et al. Nintedanib with Add-on Pirfenidone in Idiopathic Pulmonary Fibrosis. Results of the INJOURNEY Trial. Am. J. Respir. Crit. Care Med. 2018, 197, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M.; Corte, T.J.; Fischer, A.; Kreuter, M.; Lederer, D.J.; Molina-Molina, M.; Axmann, J.; Kirchgaessler, K.U.; Samara, K.; Gilberg, F.; et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir. Med. 2020, 8, 147–157. [Google Scholar] [CrossRef]
- Nathan, S.D.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glaspole, I.; Glassberg, M.K.; Kardatzke, D.R.; Daigl, M.; Kirchgaessler, K.U.; Lancaster, L.H.; et al. Effect of pirfenidone on mortality: Pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir. Med. 2017, 5, 33–41. [Google Scholar] [CrossRef]
- Nathan, S.D.; Costabel, U.; Glaspole, I.; Glassberg, M.K.; Lancaster, L.H.; Lederer, D.J.; Pereira, C.A.; Trzaskoma, B.; Morgenthien, E.A.; Limb, S.L.; et al. Efficacy of Pirfenidone in the Context of Multiple Disease Progression Events in Patients With Idiopathic Pulmonary Fibrosis. Chest 2019, 155, 712–719. [Google Scholar] [CrossRef] [Green Version]
- Khanna, D.; Albera, C.; Fischer, A.; Khalidi, N.; Raghu, G.; Chung, L.; Chen, D.; Schiopu, E.; Tagliaferri, M.; Seibold, J.R.; et al. An Open-label, Phase II Study of the Safety and Tolerability of Pirfenidone in Patients with Scleroderma-associated Interstitial Lung Disease: The LOTUSS Trial. J. Rheumatol. 2016, 43, 1672–1679. [Google Scholar] [CrossRef] [Green Version]
- George, P.M.; Spagnolo, P.; Kreuter, M.; Altinisik, G.; Bonifazi, M.; Martinez, F.J.; Molyneaux, P.L.; A Renzoni, E.; Richeldi, L.; Tomassetti, S.; et al. Progressive fibrosing interstitial lung disease: Clinical uncertainties, consensus recommendations, and research priorities. Lancet Respir. Med. 2020, 8, 925–934. [Google Scholar] [CrossRef]
- Nunes, H.; Schubel, K.; Piver, D.; Magois, E.; Feuillet, S.; Uzunhan, Y.; Carton, Z.; Tazi, A.; Levy, P.; Brillet, P.-Y.; et al. Nonspecific interstitial pneumonia: Survival is influenced by the underlying cause. Eur. Respir. J. 2014, 45, 746–755. [Google Scholar] [CrossRef] [Green Version]
- Wijsenbeek, M.; Kreuter, M.; Olson, A.; Fischer, A.; Bendstrup, E.; Wells, C.D.; Denton, C.P.; Mounir, B.; Zouad-Lejour, L.; Quaresma, M.; et al. Progressive fibrosing interstitial lung diseases: Current practice in diagnosis and management. Curr. Med. Res. Opin. 2019, 35, 2015–2024. [Google Scholar] [CrossRef] [Green Version]
- Fischer, A.; Brown, K.K.; Du Bois, R.M.; Frankel, S.K.; Cosgrove, G.P.; Fernandez-Perez, E.R.; Huie, T.J.; Krishnamoorthy, M.; Meehan, R.T.; Olson, A.L.; et al. Mycophenolate Mofetil Improves Lung Function in Connective Tissue Disease-associated Interstitial Lung Disease. J. Rheumatol. 2013, 40, 640–646. [Google Scholar] [CrossRef]
- The Idiopathic Pulmonary Fibrosis Clinical Research Network Prednisone, Azathioprine, and N-Acetylcysteine for Pulmonary Fibrosis. N. Engl. J. Med. 2012, 366, 1968–1977. [CrossRef]
- Goos, T.; De Sadeleer, L.; Yserbyt, J.; Verleden, G.; Vermant, M.; Verleden, S.; Wuyts, W. Progression in the Management of Non-Idiopathic Pulmonary Fibrosis Interstitial Lung Diseases, Where Are We Now and Where We Would Like to Be. J. Clin. Med. 2021, 10, 1330. [Google Scholar] [CrossRef] [PubMed]
- Proesmans, V.L.J.; Drent, M.; Elfferich, M.D.P.; Wijnen, P.A.H.M.; Jessurun, N.T.; Bast, A. Self-reported Gastrointestinal Side Effects of Antifibrotic Drugs in Dutch Idiopathic Pulmonary Fibrosis patients. Lung 2019, 197, 551–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cottin, V.; Koschel, D.; Günther, A.; Albera, C.; Azuma, A.; Sköld, C.M.; Tomassetti, S.; Hormel, P.; Stauffer, J.L.; Strombom, I.; et al. Long-term safety of pirfenidone: Results of the prospective, observational PASSPORT study. ERJ Open Res. 2018, 4, 00084–02018. [Google Scholar] [CrossRef]
- Nathan, S.D.; Lancaster, L.H.; Albera, C.; Glassberg, M.K.; Swigris, J.J.; Gilberg, F.; Kirchgaessler, K.-U.; Limb, S.L.; Petzinger, U.; Noble, P.W. Dose modification and dose intensity during treatment with pirfenidone: Analysis of pooled data from three multinational phase III trials. BMJ Open Respir. Res. 2018, 5, e000323. [Google Scholar] [CrossRef] [Green Version]
- Corral, M.; Deyoung, K.; Kong, A.M. Treatment patterns, healthcare resource utilization, and costs among patients with idiopathic pulmonary fibrosis treated with antifibrotic medications in US-based commercial and Medicare Supplemental claims databases: A retrospective cohort study. BMC Pulm. Med. 2020, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Trial Name | Intervention | Phase and Study Design | Patient Enrollment | Primary Outcome (s) | Secondary Outcome (s) |
|---|---|---|---|---|---|
| Nintedanib | |||||
| TOMORROW [8,32] (NCT00514683, NCT01170065) | Nintedanib (50 mg daily, 50 mg twice daily, 100 mg twice daily, 150 mg twice daily) versus placebo | Phase 2, randomized, double-blind, placebo-controlled | 432 patients with IPF | Annual rate of FVC decline −60 mL in nintedanib 150 mg twice daily group versus 190 mL in placebo group | Lower incidence of AE-IPF, small decrease in SGRQ with nintedanib 150 mg twice daily |
| INPULSIS I [4,8] (NCT01335464) | Nintedanib (Assigned in 3:2 ratio to receive either 150 mg twice daily) versus placebo | Phase 3, randomized, double-blind, placebo-controlled | 515 patients with IPF | Annual rate of decline FVC −114.7 mL nintedanib versus −239.9 mL placebo (p < 0.01) | No significant difference in time to first AE-IPF or proportion with exacerbation |
| INPULSIS II [4,8] (NCT01335477) | Nintedanib (Assigned in 3:2 ratio to receive either 150 mg twice daily) versus placebo | Phase 3, randomized, double-blind, placebo-controlled | 551 patients with IPF | Annual rate of decline FVC −113.6 mL nintedanib versus −207.3 mL placebo (p < 0.01) | Increase in time to first AE-IPF in nintedanib group and lower proportion with exacerbation in nintedanib group |
| SENSCIS [33] (NCT02597933) | Nintedanib (150 mg twice daily) versus placebo | Phase 3, randomized, double-blind, placebo-controlled | 580 patients with SSc-ILD (48.4% who were concurrently taking mycophenolate mofetil) | Annual rate of decline FVC −52.4 mL nintedanib versus −93.3 ml placebo (p = 0.04). | Change from baseline in the total score on the SGRQ did not differ significantly |
| INBUILD [14] (NCT02999178) | Nintedanib (150 mg twice daily) versus placebo | Phase 3, randomized, double-blind, placebo-controlled | 663 patients with fibrosing lung disease on HRCT | Adjusted rate of decline in the FVC was −80.8 ml per year with nintedanib and −187.8 mL per year with placebo | No significant changes in quality of life metrics as determined by the King’s Brief ILD Questionnaire |
| Trial Name | Intervention | Phase and Study Design | Patient Enrollment | Primary Outcome(s) | Secondary Outcome(s) |
|---|---|---|---|---|---|
| Pirfenidone | |||||
| CAPACITY I [8,38] (NCT00287716) | Pirfenidone versus placebo (randomized 2:1:2 pirfenidone 2403 mg daily, 1197 mg daily, or placebo) | Phase 3, randomized, double-blind, placebo-controlled | 435 patients with IPF | Mean decline FVC −8% pirfenidone versus −12.4% placebo (p < 0.01) | Decreased proportion of patients with >10% decline in FVC |
| CAPACITY II [8,38] (NCT00287729) | Pirfenidone versus placebo (randomized 1:1 pirfenidone 2403 mg daily or placebo) | Phase 3, randomized, double-blind, placebo-controlled | 344 patients with IPF | Mean decline FVC −9% pirfenidone versus −9.6% placebo (p = 0.5) | Reduced decline in 6MWD |
| ASCEND [5,8] (NCT01366209) | Pirfenidone (801 mg three times daily) versus placebo | Phase 3, randomized, double-blind, placebo-controlled | 555 patients with IPF | Proportion of patients with ≥10% decline in FVC or death reduced by 47.9% pirfenidone versus placebo | Decreased decline in 6MWD, improved progression free survival |
| RELIEF [39,40] (EudraCT 2014-000861-32) | Pirfenidone (801 mg three times daily) versus placebo | Phase 2, randomized, double-blind, placebo-controlled | 127 patients with progressive fibrosing ILD | Change in percentage of predicted FVC from baseline to week 48 by home spirometry, planned statistical model unable to be applied | Pirfenidone group less likely to have declines in FVC. Pirfenidone intervention also noted improvement in DLCO and 6MWD |
| INJOURNEY [8,41] (NCT02579603) | Nintedanib (150 mg twice daily) alone versus nintedanib and pirfenidone (801 mg three times daily) | Phase 4, randomized, open-label, parallel-group study | 105 patients with IPF | Overall manageable safety and side effect profile (main side effect was gastrointestinal adverse effects) | Mean change in FVC were −13.3 mL and −40.9 mL in nintedanib with add-on pirfenidone vs. nintedanib alone |
| Maher et al. [42] (NCT03099187) | Pirfenidone (801 mg three times daily) versus placebo | Phase 2, randomized, double-blind, placebo-controlled | 253 patients with progressive fibrosing ILD | Median change in FVC measured by home spirometry was −87.7 mL pirfenidone versus −157.1 mL placebo | Pirfenidone group less likely to have a decline in FVC > 5% (OR 0.42, p = 0.001) or >10% (OR 0.44, p = 0.011). Pirfenidone group also noted improvements in DLCO and 6MWD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shumar, J.N.; Chandel, A.; King, C.S. Antifibrotic Therapies and Progressive Fibrosing Interstitial Lung Disease (PF-ILD): Building on INBUILD. J. Clin. Med. 2021, 10, 2285. https://doi.org/10.3390/jcm10112285
Shumar JN, Chandel A, King CS. Antifibrotic Therapies and Progressive Fibrosing Interstitial Lung Disease (PF-ILD): Building on INBUILD. Journal of Clinical Medicine. 2021; 10(11):2285. https://doi.org/10.3390/jcm10112285
Chicago/Turabian StyleShumar, John N., Abhimanyu Chandel, and Christopher S. King. 2021. "Antifibrotic Therapies and Progressive Fibrosing Interstitial Lung Disease (PF-ILD): Building on INBUILD" Journal of Clinical Medicine 10, no. 11: 2285. https://doi.org/10.3390/jcm10112285
APA StyleShumar, J. N., Chandel, A., & King, C. S. (2021). Antifibrotic Therapies and Progressive Fibrosing Interstitial Lung Disease (PF-ILD): Building on INBUILD. Journal of Clinical Medicine, 10(11), 2285. https://doi.org/10.3390/jcm10112285