
Post-COVID-19 Pulmonary Fibrosis: Novel Sequelae of the Current Pandemic


 ,
,
Abstract
:1. Introduction
2. Definition and Classification of Interstitial Lung Disease (ILD)
3. Post-COVID Pulmonary Fibrosis
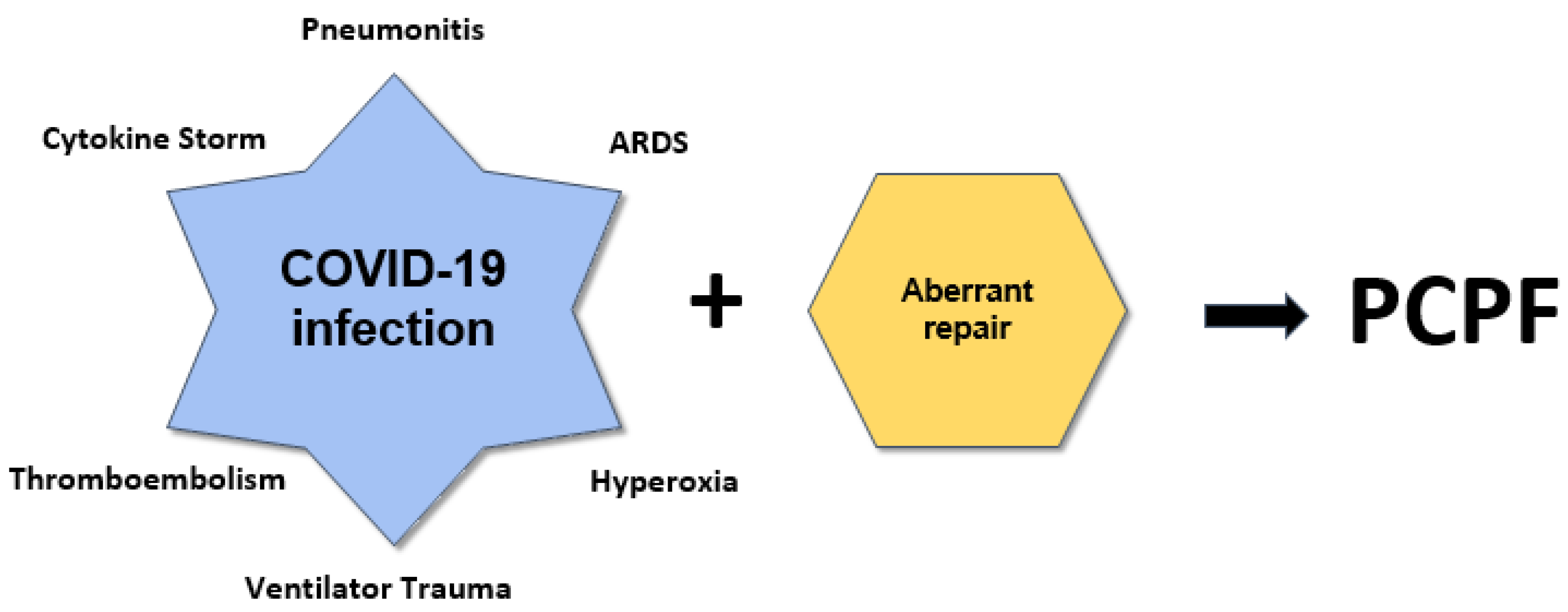
4. Etiology and Pathophysiology
4.1. Viral Pneumonia and Pneumonitis
4.2. Post-ARDS Pulmonary Fibrosis
4.3. Direct Trauma from Mechanical Ventilation
4.4. Thromboembolism
4.5. Pro-Inflammatory State
4.6. Role of Oxygen
4.7. Other Possibilities
5. Patterns of Progression of PCPF
6. Risk Factors for PCPF
7. Strategies for Mitigation of Risk
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Polak, S.B.; Van Gool, I.C.; Cohen, D.; von der Thüsen, J.H.; van Paassen, J. A systematic review of pathological findings in COVID-19: A pathophysiological timeline and possible mechanisms of disease progression. Mod. Pathol. 2020, 33, 2128–2138. [Google Scholar] [CrossRef]
- George, P.M.; Wells, A.U.; Jenkins, R.G. Pulmonary fibrosis and COVID-19: The potential role for antifibrotic therapy. Lancet Respir. Med. 2020, 8, 807–815. [Google Scholar] [CrossRef]
- Kalchiem-Dekel, O.; Galvin, J.R.; Burke, A.P.; Atamas, S.P.; Todd, N.W. Interstitial lung disease and pulmonary fibrosis: A practical approach for general medicine physicians with focus on the medical history. J. Clin. Med. 2018, 7, 476. [Google Scholar] [CrossRef] [Green Version]
- Lechowicz, K.; Drozdzal, S.; Machaj, F.; Rosik, J.; Szostak, B.; Zegan-Baranska, M.; Biernawska, J.; Dabrowski, W.; Rotter, I.; Kotfis, K. COVID-19: The potential treatment of pulmonary fibrosis associated with SARS-CoV-2 infection. J. Clin. Med. 2020, 9, 1917. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Maeno, T.; Nomura, M.; Aoyagi-Ikeda, K.; Matsui, H.; Hara, K.; Tanaka, T.; Iso, T.; Suga, T.; Kurabayashi, M. Hypoxia-inducible factor-1α mediates TGF-β-induced PAI-1 production in alveolar macrophages in pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L740–L752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, N.W.; Luzina, I.G.; Atamas, S.P. Molecular and cellular mechanisms of pulmonary fibrosis. Fibrogenesis Tissue Repair 2012, 5, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mac Sweeney, R.; McAuley, D.F. Acute respiratory distress syndrome. Lancet 2016, 388, 2416–2430. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.-W.; Rizzo, S.; Ma, L.-H.; Qiu, X.-Y.; Warth, A.; Seki, N.; Hasegawa, M.; Zou, J.-W.; Li, Q.; Femia, M. Pulmonary ground-glass opacity: Computed tomography features, histopathology and molecular pathology. Transl. Lung Cancer Res. 2017, 6, 68. [Google Scholar] [CrossRef] [Green Version]
- Azadeh, N.; Limper, A.H.; Carmona, E.M.; Ryu, J.H. The role of infection in interstitial lung diseases: A review. Chest 2017, 152, 842–852. [Google Scholar] [CrossRef]
- Venkataraman, T.; Frieman, M.B. The role of epidermal growth factor receptor (EGFR) signaling in SARS coronavirus-induced pulmonary fibrosis. Antivir. Res. 2017, 143, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.K.; Moore, B.B. Viral infection and aging as cofactors for the development of pulmonary fibrosis. Expert Rev. Respir. Med. 2010, 4, 759–771. [Google Scholar] [CrossRef] [Green Version]
- Sheng, G.; Chen, P.; Wei, Y.; Yue, H.; Chu, J.; Zhao, J.; Wang, Y.; Zhang, W.; Zhang, H.L. Viral infection increases the risk of idiopathic pulmonary fibrosis: A meta-analysis. Chest 2020, 157, 1175–1187. [Google Scholar] [CrossRef] [Green Version]
- Crisan-Dabija, R.; Pavel, C.A.; Popa, I.V.; Tarus, A.; Burlacu, A. “A chain only as strong as its weakest link”: An up-to-date literature review on the bidirectional interaction of pulmonary fibrosis and COVID-19. J. Proteome Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, S.; Natt, B.; Bime, C.; Knox, K.S.; Glassberg, M.K. Antifibrotics in COVID-19 lung disease: Let us stay focused. Front. Med. 2020, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Wallace, W.A.; Fitch, P.M.; Simpson, A.J.; Howie, S.E. Inflammation-associated remodelling and fibrosis in the lung—A process and an end point. Int. J. Exp. Pathol. 2007, 88, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.H.; Juan, Y.H.; Hu, H.C.; Kao, K.C.; Lee, C.S. Reversal of lung fibrosis: An unexpected finding in survivor of acute respiratory distress syndrome. QJM 2018, 111, 47–48. [Google Scholar] [CrossRef] [Green Version]
- Noble, P.W.; Barkauskas, C.E.; Jiang, D. Pulmonary fibrosis: Patterns and perpetrators. J. Clin. Investig. 2012, 122, 2756–2762. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.W.; Fidler, L.; Marcoux, V.; Johannson, K.A.; Assayag, D.; Fisher, J.H.; Hambly, N.; Kolb, M.; Morisset, J.; Shapera, S.; et al. Practical considerations for the diagnosis and treatment of fibrotic interstitial lung disease during the coronavirus disease 2019 pandemic. Chest 2020, 158, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, K.M.; Raghu, G.; Tzilas, V.; Bouros, D. Management of patients with interstitial lung disease in the midst of the COVID-19 pandemic. Respiration 2020, 99, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dong, C.; Hu, Y.; Li, C.; Ren, Q.; Zhang, X.; Shi, H.; Zhou, M. Temporal changes of CT findings in 90 patients with COVID-19 pneumonia: A longitudinal study. Radiology 2020, 296, E55–E64. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Khan, A.; Zhou, W.; Dai, Y.; Md, E.; Chen, R.; Cheng, G. Follow-up study of clinical and chest CT scans in confirmed COVID-19 patients. Radiol. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Barisione, E.; Grillo, F.; Ball, L.; Bianchi, R.; Grosso, M.; Morbini, P.; Pelosi, P.; Patroniti, N.A.; De Lucia, A.; Orengo, G.; et al. Fibrotic progression and radiologic correlation in matched lung samples from COVID-19 post-mortems. Virchows Arch. 2020. [Google Scholar] [CrossRef]
- Huang, W.; Wu, Q.; Chen, Z.; Xiong, Z.; Wang, K.; Tian, J.; Zhang, S. The potential indicators for pulmonary fibrosis in survivors of severe COVID-19. J. Infect. 2020. [Google Scholar] [CrossRef]
- Revel, M.P.; Parkar, A.P.; Prosch, H.; Silva, M.; Sverzellati, N.; Gleeson, F.; Brady, A.; The European Society of Radiology (ESR); The European Society of Thoracic Imaging (ESTI). COVID-19 patients and the radiology department—Advice from the European Society of Radiology (ESR) and the European Society of Thoracic Imaging (ESTI). Eur. Radiol. 2020, 30, 4903–4909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wu, J.; Wang, S.; Li, X.; Zhou, J.; Huang, B.; Luo, D.; Cao, Q.; Chen, Y.; Chen, S.; et al. Progression to fibrosing diffuse alveolar damage in a series of 30 minimally invasive autopsies with COVID-19 pneumonia in Wuhan, China. Histopathology 2020. [Google Scholar] [CrossRef]
- Ojo, A.S.; Balogun, S.A.; Williams, O.T.; Ojo, O.S. Pulmonary fibrosis in COVID-19 survivors: Predictive factors and risk reduction strategies. Pulm. Med. 2020, 2020, 6175964. [Google Scholar] [CrossRef] [PubMed]
- George, P.M.; Barratt, S.L.; Condliffe, R.; Desai, S.R.; Devaraj, A.; Forrest, I.; Gibbons, M.A.; Hart, N.; Jenkins, R.G.; McAuley, D.F.; et al. Respiratory follow-up of patients with COVID-19 pneumonia. Thorax 2020. [Google Scholar] [CrossRef] [PubMed]
- Scelfo, C.; Fontana, M.; Casalini, E.; Menzella, F.; Piro, R.; Zerbini, A.; Spaggiari, L.; Ghidorsi, L.; Ghidoni, G.; Facciolongo, N.C. A dangerous consequence of the recent pandemic: Early lung fibrosis following COVID-19 pneumonia—Case reports. Ther. Clin. Risk Manag. 2020, 16, 1039–1046. [Google Scholar] [CrossRef]
- Combet, M.; Pavot, A.; Savale, L.; Humbert, M.; Monnet, X. Rapid onset honeycombing fibrosis in spontaneously breathing patient with COVID-19. Eur. Respir. J. 2020, 56. [Google Scholar] [CrossRef]
- Tale, S.; Ghosh, S.; Meitei, S.P.; Kolli, M.; Garbhapu, A.K.; Pudi, S. Post COVID-19 pneumonia pulmonary fibrosis. QJM 2020. [Google Scholar] [CrossRef]
- Vasarmidi, E.; Tsitoura, E.; Spandidos, D.A.; Tzanakis, N.; Antoniou, K.M. Pulmonary fibrosis in the aftermath of the COVID-19 era (Review). Exp. Ther. Med. 2020, 20, 2557–2560. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Benitez, N.E.; Laffey, J.G.; Parotto, M.; Spieth, P.M.; Villar, J.; Zhang, H.; Slutsky, A.S. Mechanical ventilation-associated lung fibrosis in acute respiratory distress syndrome: A significant contributor to poor outcome. Anesthesiology 2014, 121, 189–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrera-Benitez, N.E.; Parotto, M.; Post, M.; Han, B.; Spieth, P.M.; Cheng, W.E.; Valladares, F.; Villar, J.; Liu, M.; Sato, M.; et al. Mechanical stress induces lung fibrosis by epithelial-mesenchymal transition. Crit. Care Med. 2012, 40, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Spagnolo, P.; Balestro, E.; Aliberti, S.; Cocconcelli, E.; Biondini, D.; Casa, G.D.; Sverzellati, N.; Maher, T.M. Pulmonary fibrosis secondary to COVID-19: A call to arms? Lancet Respir. Med. 2020, 8, 750–752. [Google Scholar] [CrossRef]
- Otoupalova, E.; Smith, S.; Cheng, G.; Thannickal, V.J. Oxidative stress in pulmonary fibrosis. Compr. Physiol. 2020, 10, 509–547. [Google Scholar] [CrossRef] [Green Version]
- Marini, J.J.; Gattinoni, L. Management of COVID-19 respiratory distress. JAMA 2020, 323, 2329–2330. [Google Scholar] [CrossRef]
- Brochard, L.; Slutsky, A.; Pesenti, A. Mechanical ventilation to minimize progression of lung injury in acute respiratory failure. Am. J. Respir. Crit. Care Med. 2017, 195, 438–442. [Google Scholar] [CrossRef]
- Frizzelli, A.; Tuttolomondo, D.; Aiello, M.; Majori, M.; Bertorelli, G.; Chetta, A. What happens to people’s lungs when they get coronavirus disease 2019? Acta Biomed. 2020, 91, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.D.; Wunderink, R.G. Viral pneumonia and acute respiratory distress syndrome. Clin. Chest Med. 2017, 38, 113–125. [Google Scholar] [CrossRef]
- Booth, C.M.; Stewart, T.E. Severe acute respiratory syndrome and critical care medicine: The Toronto experience. Crit. Care Med. 2005, 33, S53–S60. [Google Scholar] [CrossRef]
- Chen, C.Y.; Lee, C.H.; Liu, C.Y.; Wang, J.H.; Wang, L.M.; Perng, R.P. Clinical features and outcomes of severe acute respiratory syndrome and predictive factors for acute respiratory distress syndrome. J. Chin. Med. Assoc. 2005, 68, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Fowler, R.A.; Lapinsky, S.E.; Hallett, D.; Detsky, A.S.; Sibbald, W.J.; Slutsky, A.S.; Stewart, T.E.; Toronto SARS Critical Care Group. Critically ill patients with severe acute respiratory syndrome. JAMA 2003, 290, 367–373. [Google Scholar] [CrossRef]
- Hui, D.S.; Memish, Z.A.; Zumla, A. Severe acute respiratory syndrome vs. the Middle East respiratory syndrome. Curr. Opin. Pulm. Med. 2014, 20, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Lien, T.C.; Sung, C.S.; Lee, C.H.; Kao, H.K.; Huang, Y.C.; Liu, C.Y.; Perng, R.P.; Wang, J.H. Characteristic features and outcomes of severe acute respiratory syndrome found in severe acute respiratory syndrome intensive care unit patients. J. Crit. Care 2008, 23, 557–564. [Google Scholar] [CrossRef]
- Wang, J.T.; Sheng, W.H.; Fang, C.T.; Chen, Y.C.; Wang, J.L.; Yu, C.J.; Chang, S.C.; Yang, P.C. Clinical manifestations, laboratory findings, and treatment outcomes of SARS patients. Emerg. Infect. Dis. 2004, 10, 818–824. [Google Scholar] [CrossRef]
- Cantan, B.; Luyt, C.E.; Martin-Loeches, I. Influenza infections and emergent viral infections in intensive care unit. Semin. Respir. Crit. Care Med. 2019, 40, 488–497. [Google Scholar] [CrossRef] [Green Version]
- Delaney, J.W.; Fowler, R.A. 2009 influenza A (H1N1): A clinical review. Hosp. Pract. (1995) 2010, 38, 74–81. [Google Scholar]
- Duggal, A.; Pinto, R.; Rubenfeld, G.; Fowler, R.A. Global variability in reported mortality for critical illness during the 2009–10 influenza A(H1N1) pandemic: A systematic review and meta-regression to guide reporting of outcomes during disease outbreaks. PLoS ONE 2016, 11, e0155044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwyer, D.E.; Lynfield, R.; Losso, M.H.; Davey, R.T.; Cozzi-Lepri, A.; Wentworth, D.; Uyeki, T.M.; Gordin, F.; Angus, B.; Qvist, T.; et al. Comparison of the outcomes of individuals with medically attended influenza A and B virus infections enrolled in 2 international cohort studies over a 6-year period: 2009–2015. Open Forum Infect. Dis. 2017, 4, ofx212. [Google Scholar] [CrossRef]
- Kumar, A.; Zarychanski, R.; Pinto, R.; Cook, D.J.; Marshall, J.; Lacroix, J.; Stelfox, T.; Bagshaw, S.; Choong, K.; Lamontagne, F.; et al. Critically ill patients with 2009 influenza A(H1N1) infection in Canada. JAMA 2009, 302, 1872–1879. [Google Scholar] [CrossRef] [Green Version]
- Sarda, C.; Palma, P.; Rello, J. Severe influenza: Overview in critically ill patients. Curr. Opin. Crit. Care 2019, 25, 449–457. [Google Scholar] [CrossRef]
- Zha, L.; Shen, Y.; Pan, L.; Han, M.; Yang, G.; Teng, X.; Tefsen, B. Follow-up study on pulmonary function and radiological changes in critically ill patients with COVID-19. J. Infect. 2020. [Google Scholar] [CrossRef] [PubMed]
- Senga, M.; Arabi, Y.M.; Fowler, R.A. Clinical spectrum of the Middle East respiratory syndrome coronavirus (MERS-CoV). J. Infect. Public Health 2017, 10, 191–194. [Google Scholar] [CrossRef] [Green Version]
- Arnold, D.T.; Hamilton, F.W.; Milne, A.; Morley, A.J.; Viner, J.; Attwood, M.; Noel, A.; Gunning, S.; Hatrick, J.; Hamilton, S.; et al. Patient outcomes after hospitalisation with COVID-19 and implications for follow-up: Results from a prospective UK cohort. Thorax 2020. [Google Scholar] [CrossRef]
- Burnham, E.L.; Hyzy, R.C.; Paine, R., 3rd; Coley, C., 2nd; Kelly, A.M.; Quint, L.E.; Lynch, D.; Janssen, W.J.; Moss, M.; Standiford, T.J. Chest CT features are associated with poorer quality of life in acute lung injury survivors. Crit. Care Med. 2013, 41, 445–456. [Google Scholar] [CrossRef] [Green Version]
- Mo, X.; Jian, W.; Su, Z.; Chen, M.; Peng, H.; Peng, P.; Lei, C.; Chen, R.; Zhong, N.; Li, S. Abnormal pulmonary function in COVID-19 patients at time of hospital discharge. Eur. Respir. J. 2020, 55. [Google Scholar] [CrossRef]
- Venkataraman, T.; Coleman, C.M.; Frieman, M.B. Overactive epidermal growth factor receptor signaling leads to increased fibrosis after severe acute respiratory syndrome coronavirus infection. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, S.; Hirsch, J.S.; Narasimhan, M.; Crawford, J.M.; McGinn, T.; Davidson, K.W.; Barnaby, D.P.; Becker, L.B.; Chelico, J.D.; Cohen, S.L.; et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA 2020, 323, 2052–2059. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [Green Version]
- Chand, S.; Kapoor, S.; Orsi, D.; Fazzari, M.J.; Tanner, T.G.; Umeh, G.C.; Islam, M.; Dicpinigaitis, P.V. COVID-19-associated critical illness-report of the first 300 patients admitted to intensive care units at a New York City Medical Center. J. Intensive Care Med. 2020, 35, 963–970. [Google Scholar] [CrossRef]
- Burnham, E.L.; Janssen, W.J.; Riches, D.W.; Moss, M.; Downey, G.P. The fibroproliferative response in acute respiratory distress syndrome: Mechanisms and clinical significance. Eur. Respir. J. 2014, 43, 276–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Thoracic Society. International consensus conferences in intensive care medicine: Ventilator-associated Lung Injury in ARDS. This official conference report was cosponsored by the American Thoracic Society, The European Society of Intensive Care Medicine, and The Societe de Reanimation de Langue Francaise, and was approved by the ATS Board of Directors, July 1999. Am. J. Respir. Crit. Care Med. 1999, 160, 2118–2124. [Google Scholar] [CrossRef] [Green Version]
- Kuchnicka, K.; Maciejewski, D. Ventilator-associated lung injury. Anaesthesiol. Intensive Ther. 2013, 45, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Abou-Ismail, M.Y.; Diamond, A.; Kapoor, S.; Arafah, Y.; Nayak, L. The hypercoagulable state in COVID-19: Incidence, pathophysiology, and management. Thromb. Res. 2020, 194, 101–115. [Google Scholar] [CrossRef]
- Sode, B.F.; Dahl, M.; Nielsen, S.F.; Nordestgaard, B.G. Venous thromboembolism and risk of idiopathic interstitial pneumonia: A nationwide study. Am. J. Respir. Crit. Care Med. 2010, 181, 1085–1092. [Google Scholar] [CrossRef]
- Grosse, C.; Grosse, A.; Salzer, H.J.F.; Dünser, M.W.; Motz, R.; Langer, R. Analysis of cardiopulmonary findings in COVID-19 fatalities: High incidence of pulmonary artery thrombi and acute suppurative bronchopneumonia. Cardiovasc. Pathol. 2020, 49, 107263. [Google Scholar] [CrossRef]
- Radermecker, C.; Detrembleur, N.; Guiot, J.; Cavalier, E.; Henket, M.; d’Emal, C.; Vanwinge, C.; Cataldo, D.; Oury, C.; Delvenne, P.; et al. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID-19. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Chrysanthopoulou, A.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Mikroulis, D.; Konstantinidis, T.; Sivridis, E.; Koffa, M.; Giatromanolaki, A.; Boumpas, D.T. Neutrophil extracellular traps promote differentiation and function of fibroblasts. J. Pathol. 2014, 233, 294–307. [Google Scholar] [CrossRef]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodríguez, L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef]
- Sun, X.; Wang, T.; Cai, D.; Hu, Z.; Liao, H.; Zhi, L.; Wei, H.; Zhang, Z.; Qiu, Y.; Wang, J. Cytokine storm intervention in the early stages of COVID-19 pneumonia. Cytokine Growth Factor Rev. 2020, 53, 38–42. [Google Scholar] [CrossRef] [PubMed]
- McElvaney, O.J.; McEvoy, N.L.; McElvaney, O.F.; Carroll, T.P.; Murphy, M.P.; Dunlea, D.M.; Ní Choileáin, O.; Clarke, J.; O’Connor, E.; Hogan, G. Characterization of the inflammatory response to severe COVID-19 illness. Am. J. Respir. Crit. Care Med. 2020, 202, 812–821. [Google Scholar] [CrossRef]
- Sheppard, D. Transforming growth factor beta: A central modulator of pulmonary and airway inflammation and fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 413–417. [Google Scholar] [CrossRef]
- Tamaki, S.; Mano, T.; Sakata, Y.; Ohtani, T.; Takeda, Y.; Kamimura, D.; Omori, Y.; Tsukamoto, Y.; Ikeya, Y.; Kawai, M. Interleukin-16 promotes cardiac fibrosis and myocardial stiffening in heart failure with preserved ejection fraction. PLoS ONE 2013, 8, e68893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalbandian, A.; Sehgal, K.; Gupta, A.; Madhavan, M.V.; McGroder, C.; Stevens, J.S.; Cook, J.R.; Nordvig, A.S.; Shalev, D.; Sehrawat, T.S. Post-acute COVID-19 syndrome. Nat. Med. 2021, 27, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Tzouvelekis, A.; Harokopos, V.; Paparountas, T.; Oikonomou, N.; Chatziioannou, A.; Vilaras, G.; Tsiambas, E.; Karameris, A.; Bouros, D.; Aidinis, V. Comparative expression profiling in pulmonary fibrosis suggests a role of hypoxia-inducible factor-1alpha in disease pathogenesis. Am. J. Respir. Crit. Care Med. 2007, 176, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Higgins, D.F.; Kimura, K.; Bernhardt, W.M.; Shrimanker, N.; Akai, Y.; Hohenstein, B.; Saito, Y.; Johnson, R.S.; Kretzler, M.; Cohen, C.D. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Investig. 2007, 117, 3810–3820. [Google Scholar] [CrossRef]
- Manresa, M.C.; Godson, C.; Taylor, C.T. Hypoxia-sensitive pathways in inflammation-driven fibrosis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R1369–R1380. [Google Scholar] [CrossRef] [Green Version]
- Budinger, G.R.S.; Mutlu, G.M. Balancing the risks and benefits of oxygen therapy in critically III adults. Chest 2013, 143, 1151–1162. [Google Scholar] [CrossRef] [Green Version]
- Tobin, M.J. Basing respiratory management of coronavirus on physiological principles. Am. J. Respir. Crit. Care Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Goobie, G.C.; Gregory, A.D.; Kass, D.J.; Zhang, Y. TOLL-interacting protein in pulmonary diseases: Abiding by the goldilocks principle. Am. J. Respir. Cell Mol. Biol. 2020. [Google Scholar] [CrossRef]
- Molyneaux, P.L.; Cox, M.J.; Wells, A.U.; Kim, H.C.; Ji, W.; Cookson, W.O.; Moffatt, M.F.; Kim, D.S.; Maher, T.M. Changes in the respiratory microbiome during acute exacerbations of idiopathic pulmonary fibrosis. Respir. Res. 2017, 18, 29. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.Y.; Qiao, K.; Liu, F.; Wu, B.; Xu, X.; Jiao, G.Q.; Lu, R.G.; Li, H.X.; Zhao, J.; Huang, J.; et al. Lung transplantation as therapeutic option in acute respiratory distress syndrome for coronavirus disease 2019-related pulmonary fibrosis. Chin. Med. J. 2020, 133, 1390–1396. [Google Scholar] [CrossRef]
- Antonio, G.E.; Wong, K.T.; Hui, D.S.; Wu, A.; Lee, N.; Yuen, E.H.; Leung, C.B.; Rainer, T.H.; Cameron, P.; Chung, S.S.; et al. Thin-section CT in patients with severe acute respiratory syndrome following hospital discharge: Preliminary experience. Radiology 2003, 228, 810–815. [Google Scholar] [CrossRef]
- Zhang, P.; Li, J.; Liu, H.; Han, N.; Ju, J.; Kou, Y.; Chen, L.; Jiang, M.; Pan, F.; Zheng, Y.; et al. Long-term bone and lung consequences associated with hospital-acquired severe acute respiratory syndrome: A 15-year follow-up from a prospective cohort study. Bone Res. 2020, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Liu, Y.; Xu, D.; Zhang, R.; Lan, L.; Xu, H. Prediction of the development of pulmonary fibrosis using serial thin-section CT and clinical features in patients discharged after treatment for COVID-19 pneumonia. Korean J. Radiol. 2020, 21, 746–755. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.J.; Xu, J.; Yin, J.M.; Li, L.; Hou, W.; Zhang, L.L.; Zhou, Z.; Yu, Y.Z.; Li, H.J.; Feng, Y.M.; et al. Lower circulating interferon-gamma is a risk factor for lung fibrosis in COVID-19 patients. Front. Immunol. 2020, 11, 585647. [Google Scholar] [CrossRef] [PubMed]
- Das, K.M.; Lee, E.Y.; Singh, R.; Enani, M.A.; Al Dossari, K.; Van Gorkom, K.; Larsson, S.G.; Langer, R.D. Follow-up chest radiographic findings in patients with MERS-CoV after recovery. Indian J. Radiol. Imaging 2017, 27, 342. [Google Scholar] [CrossRef] [PubMed]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the treatment of Covid-19—Final report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Rochwerg, B.; Agarwal, A.; Zeng, L.; Leo, Y.S.; Appiah, J.A.; Agoritsas, T.; Bartoszko, J.; Brignardello-Petersen, R.; Ergan, B.; Ge, L.; et al. Remdesivir for severe covid-19: A clinical practice guideline. BMJ 2020, 370, m2924. [Google Scholar] [CrossRef]
- Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; Elmahi, E.; et al. Dexamethasone in hospitalized patients with Covid-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef]
- Huang, C.; Huang, L.; Wang, Y.; Li, X.; Ren, L.; Gu, X.; Kang, L.; Guo, L.; Liu, M.; Zhou, X. 6-month consequences of COVID-19 in patients discharged from hospital: A cohort study. Lancet 2021, 397, 220–232. [Google Scholar] [CrossRef]
- Lusczek, E.R.; Ingraham, N.E.; Karam, B.S.; Proper, J.; Siegel, L.; Helgeson, E.S.; Lotfi-Emran, S.; Zolfaghari, E.J.; Jones, E.; Usher, M.G. Characterizing COVID-19 clinical phenotypes and associated comorbidities and complication profiles. PLoS ONE 2021, 16, e0248956. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Study | N= | Criteria for Defining PF | Age | CMs | Medications | Ventilation | Labs |
|---|---|---|---|---|---|---|---|
| Yu, et al., 2020 | 32 | Based on f/u CT; Pts put into 1 of 2 groups. Fibrosis group had evidence of fibrosis. | Older (Median 54 y.o, vs. 37) | HTN | Longer L/O steroid therapy, longer L/O antiviral therapy | – | Higher CRP, higher IL-6, lower lymphocytes |
| Hu, et al., 2020 | 76 | Based on CT at discharge; Pts put into 1 of 2 groups. Fibrosis group showed presence of PF. | Older (Median 58 y.o, vs. 39) | HTN | – | – | Higher CRP, lower IFN-y, lower lymphocytes |
| Huang, et al., 2020 | 81 | Based on f/u CT; Pts put into 1 of 2 groups: PF group showed fibrotic changes. | Older (Median 63 y.o, vs. 51) | HTN | More likely to have needed steroids | Higher rate of ventilation | Higher CRP, higher D-Dimer, lower lymphocytes |
| Li, Y. et al., 2020 | 30 | Based on histopathology; Samples put into fibrosing DAD, acute, or organizing groups. | Younger * (Median 64 y.o, vs. 77) | – | – | Longer ventilation ** | – |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ambardar, S.R.; Hightower, S.L.; Huprikar, N.A.; Chung, K.K.; Singhal, A.; Collen, J.F. Post-COVID-19 Pulmonary Fibrosis: Novel Sequelae of the Current Pandemic. J. Clin. Med. 2021, 10, 2452. https://doi.org/10.3390/jcm10112452
Ambardar SR, Hightower SL, Huprikar NA, Chung KK, Singhal A, Collen JF. Post-COVID-19 Pulmonary Fibrosis: Novel Sequelae of the Current Pandemic. Journal of Clinical Medicine. 2021; 10(11):2452. https://doi.org/10.3390/jcm10112452
Chicago/Turabian StyleAmbardar, Shiva Rattan, Stephanie L. Hightower, Nikhil A. Huprikar, Kevin K. Chung, Anju Singhal, and Jacob F. Collen. 2021. "Post-COVID-19 Pulmonary Fibrosis: Novel Sequelae of the Current Pandemic" Journal of Clinical Medicine 10, no. 11: 2452. https://doi.org/10.3390/jcm10112452
APA StyleAmbardar, S. R., Hightower, S. L., Huprikar, N. A., Chung, K. K., Singhal, A., & Collen, J. F. (2021). Post-COVID-19 Pulmonary Fibrosis: Novel Sequelae of the Current Pandemic. Journal of Clinical Medicine, 10(11), 2452. https://doi.org/10.3390/jcm10112452