
Engineering of CD19 Antibodies: A CD19-TRAIL Fusion Construct Specifically Induces Apoptosis in B-Cell Precursor Acute Lymphoblastic Leukemia (BCP-ALL) Cells In Vivo

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Antibodies
2.3. Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Western Blot Analysis
2.4. CD19 Binding Capacity
2.5. Expression of CD19, TRAIL-R1, and TRAIL-R2 on Cell Lines and PDX Samples
2.6. Cell Viability Assay
2.7. Analysis of Apoptosis Induction
2.8. Analysis of Drug Combination Effects
2.9. Animal Experiments
2.10. Statistical Analysis
3. Results
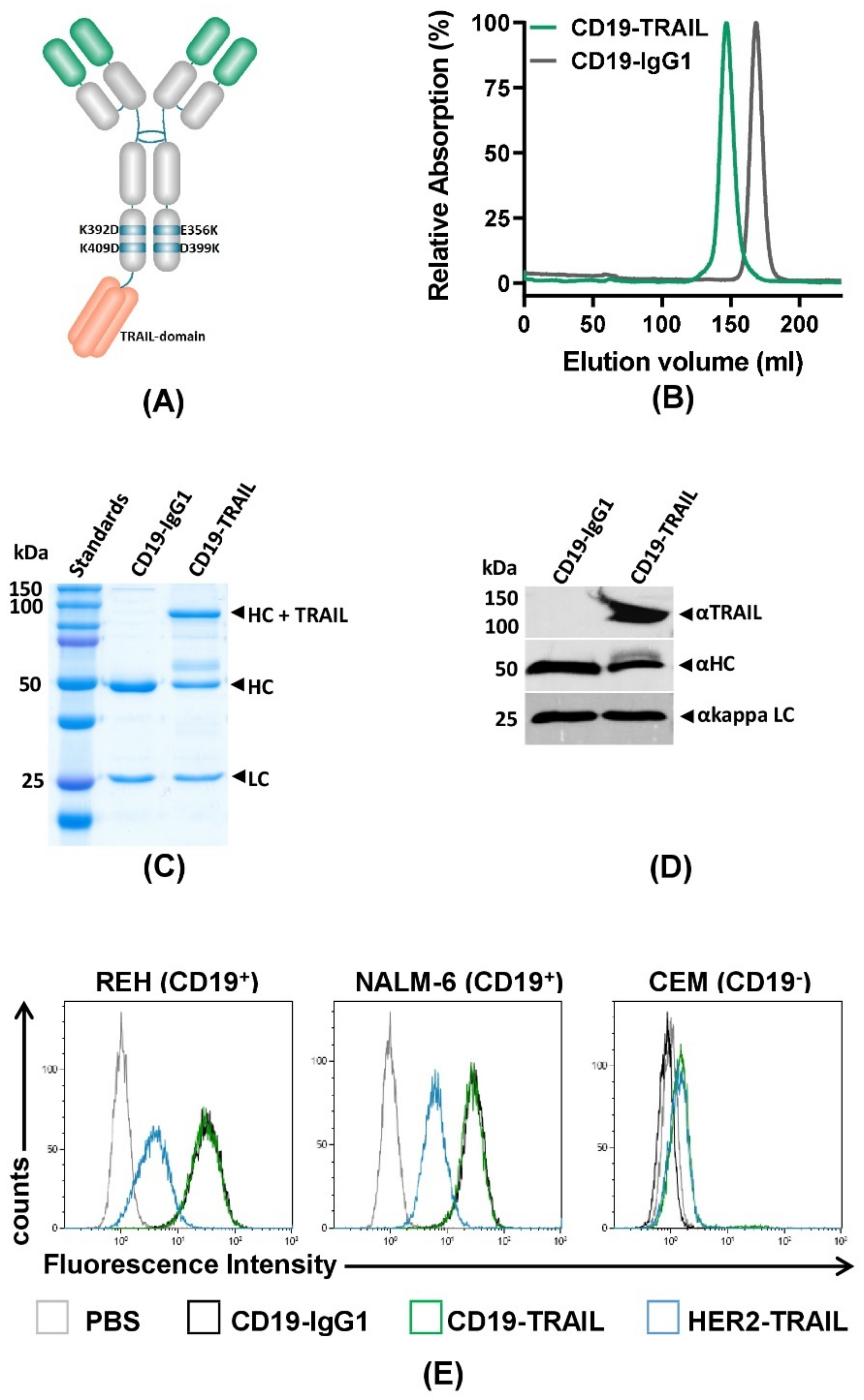
3.1. Generation of a CD19-TRAIL Fusion Construct with CD19 Specific Binding Capacity
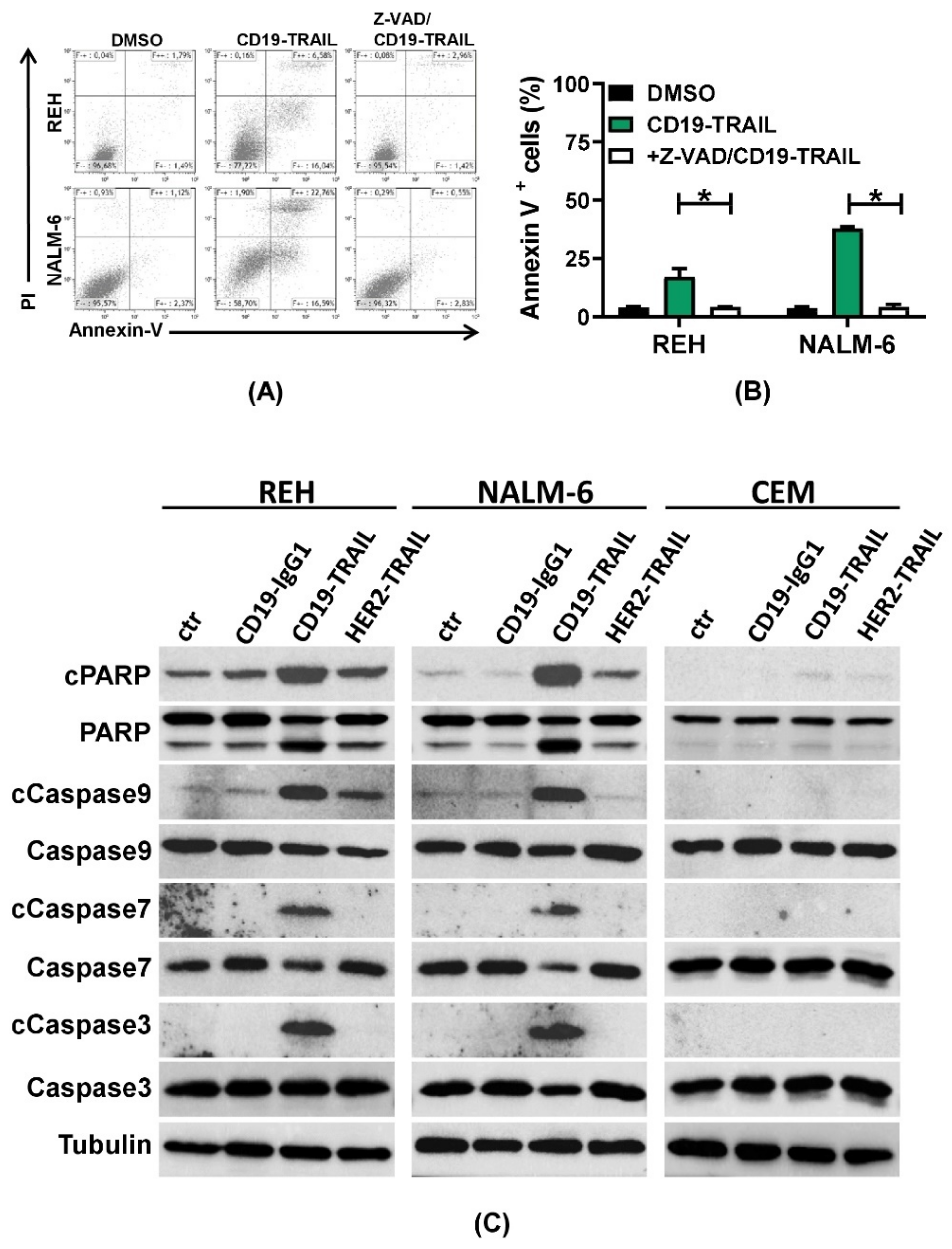
3.2. CD19-TRAIL Induces Direct Apoptotic Effects in CD19+ BCP-ALL Cells
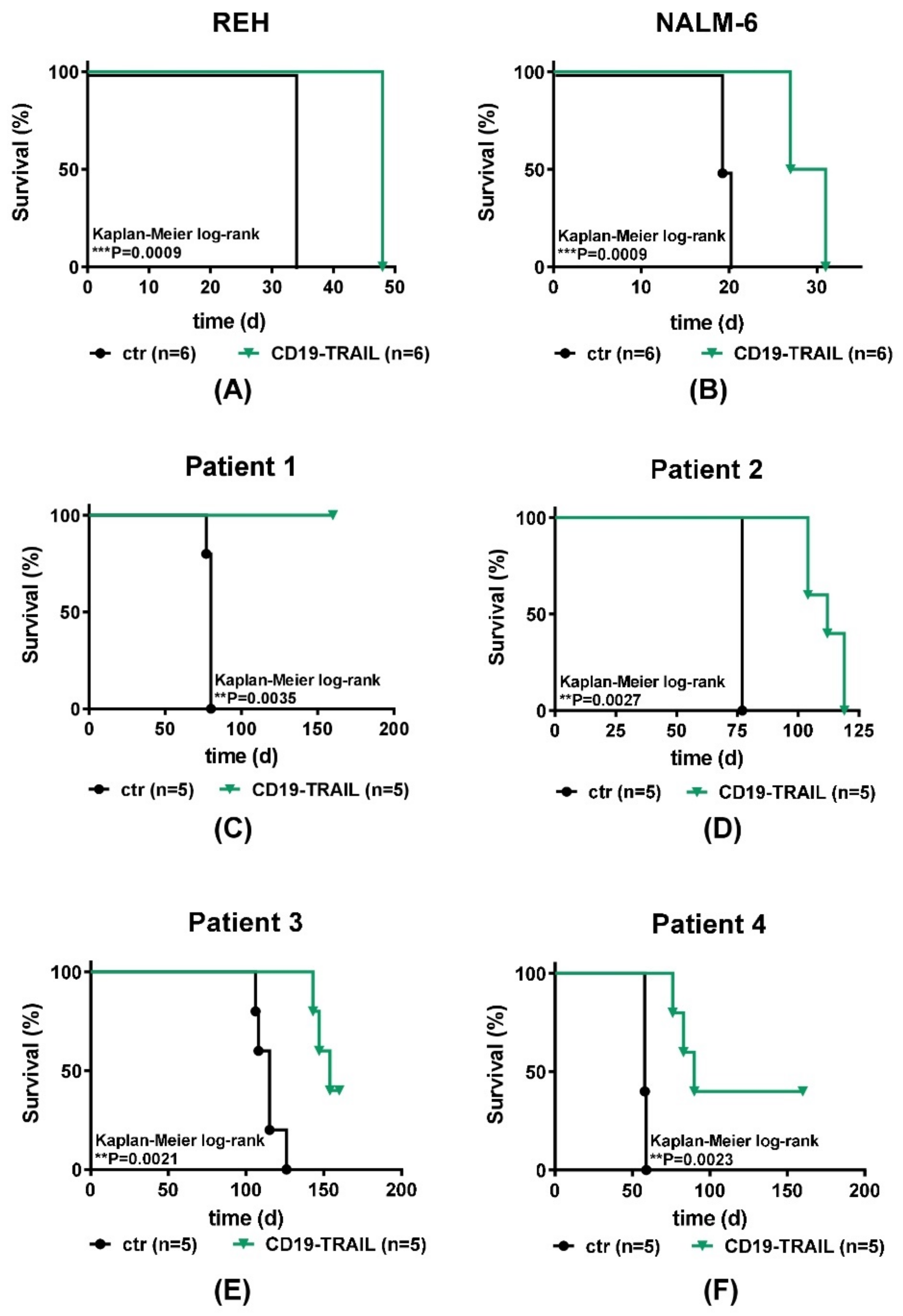
3.3. CD19-TRAIL Eradicates BCP-ALL Cells In Vivo in Xenograft Models
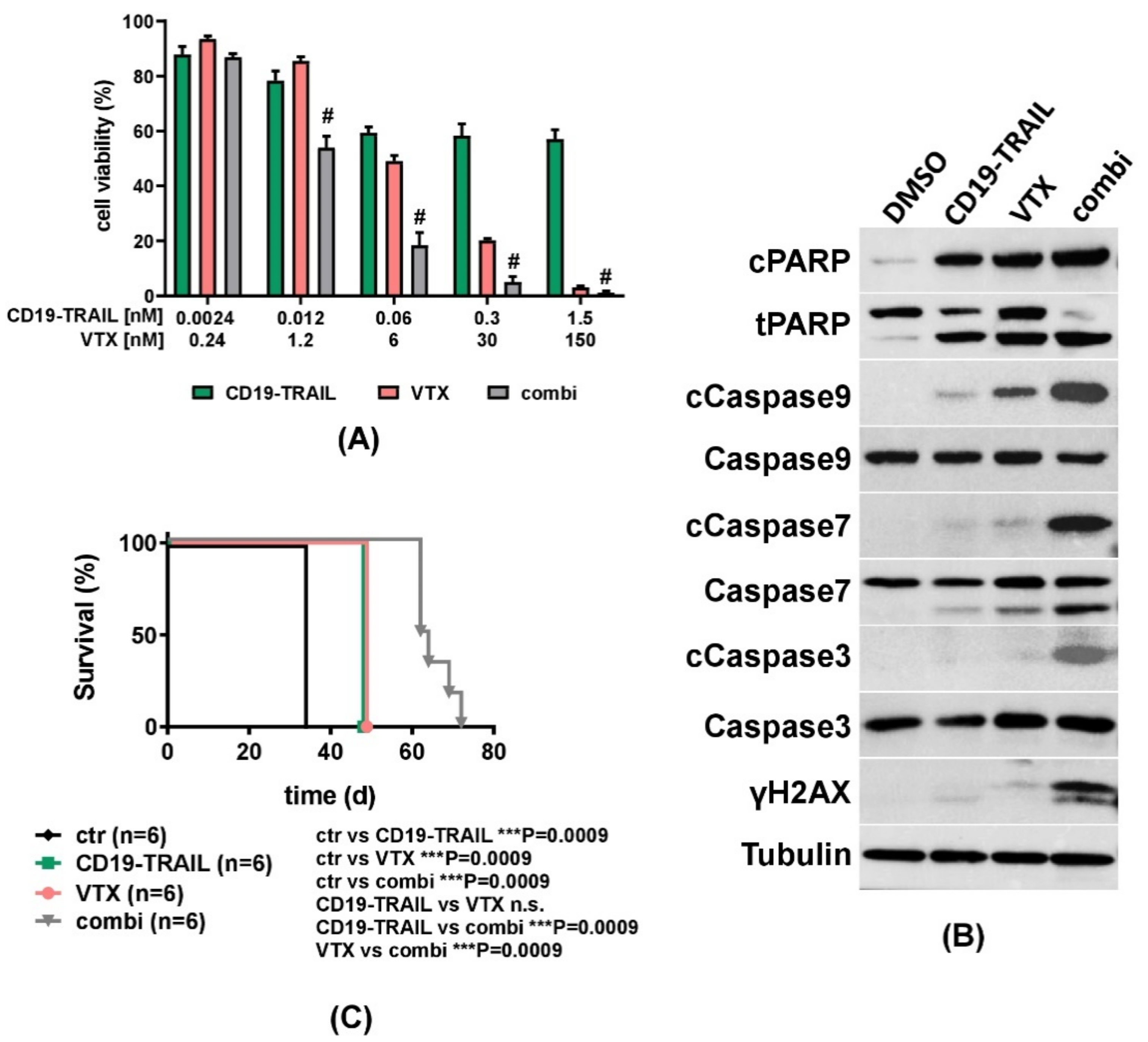
3.4. The Cytotoxic Effect of CD19-TRAIL Is Synergistically Enhanced by Venetoclax in BCP-ALL Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Locatelli, F.; Schrappe, M.; Bernardo, M.E.; Rutella, S. How I treat relapsed childhood acute lymphoblastic leukemia. Blood 2012, 120, 2807–2816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [Green Version]
- Bhojwani, D.; Pui, C.-H. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 2013, 14, e205–e217. [Google Scholar] [CrossRef]
- Wedekind, M.F.; Denton, N.L.; Chen, C.-Y.; Cripe, T.P. Pediatric Cancer Immunotherapy: Opportunities and Challenges. Pediatr. Drugs 2018, 20, 395–408. [Google Scholar] [CrossRef] [Green Version]
- Maury, S.; Chevret, S.; Thomas, X.; Heim, D.; Leguay, T.; Huguet, F.; Chevallier, P.; Hunault, M.; Boissel, N.; Escoffre-Barbe, M.; et al. Rituximab in B-Lineage Adult Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; O’Brien, S.; Ravandi, F.; Kantarjian, H. Monoclonal antibodies in acute lymphoblastic leukemia. Blood 2015, 125, 4010–4016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoelzer, D. Novel Antibody-Based Therapies for Acute Lymphoblastic Leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2011, 2011, 243–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadler, L.M.; Anderson, K.C.; Marti, G.; Bates, M.; Park, E.; Daley, J.F.; Schlossman, S.F. B4, a human B lymphocyte-associated antigen expressed on normal, mitogen-activated, and malignant B lymphocytes. J. Immunol. 1983, 131, 244–250. [Google Scholar]
- Scheuermann, R.H.; Racila, E. CD19 Antigen in Leukemia and Lymphoma Diagnosis and Immunotherapy. Leuk. Lymphoma 1995, 18, 385–397. [Google Scholar] [CrossRef]
- Mejstríková, E.; Hrusak, O.; Borowitz, M.J.; Whitlock, J.A.; Brethon, B.; Trippett, T.M.; Zugmaier, G.; Gore, L.; Von Stackelberg, A.; Locatelli, F. CD19-negative relapse of pediatric B-cell precursor acute lymphoblastic leukemia following blinatumomab treatment. Blood Cancer J. 2017, 7, 659. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.-M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Locatelli, F.; Zugmaier, G.; Mergen, N.; Bader, P.; Jeha, S.; Schlegel, P.-G.; Bourquin, J.-P.; Handgretinger, R.; Brethon, B.; Rossig, C.; et al. Blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia: Results of the RIALTO trial, an expanded access study. Blood Cancer J. 2020, 10, 1–5. [Google Scholar] [CrossRef]
- Anagnostou, T.; Riaz, I.B.; Hashmi, S.K.; Murad, M.H.; Kenderian, S.S. Anti-CD19 chimeric antigen receptor T-cell therapy in acute lymphocytic leukaemia: A systematic review and meta-analysis. Lancet Haematol. 2020, 7, e816–e826. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Lioure, B.; Kim, S.K.; Atallah, E.; Leguay, T.; Kelly, K.; Marolleau, J.-P.; Escoffre-Barbe, M.; Thomas, X.G.; Cortes, J.; et al. A Phase II Study of Coltuximab Ravtansine (SAR3419) Monotherapy in Patients with Relapsed or Refractory Acute Lymphoblastic Leukemia. Clin. Lymphoma Myeloma Leuk. 2016, 16, 139–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, H.M.; Bernett, M.J.; Pong, E.; Peipp, M.; Karki, S.; Chu, S.Y.; Richards, J.O.; Vostiar, I.; Joyce, P.F.; Repp, R.; et al. Potent In vitro and In vivo Activity of an Fc-Engineered Anti-CD19 Monoclonal Antibody against Lymphoma and Leukemia. Cancer Res. 2008, 68, 8049–8057. [Google Scholar] [CrossRef] [Green Version]
- Hekman, A.; Honselaar, A.; Vuist, W.M.J.; Sein, J.J.; Rodenhuis, S.; Huinink, W.W.T.B.; Somers, R.; Rümke, P.; Melief, C.J.M. Initial experience with treatment of human B cell lymphoma with anti-CD19 monoclonal antibody. Cancer Immunol. Immunother. 1991, 32, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Roßkopf, S.; Eichholz, K.M.; Winterberg, D.; Diemer, K.J.; Lutz, S.; Münnich, I.A.; Klausz, K.; Rösner, T.; Valerius, T.; Schewe, D.M.; et al. Enhancing CDC and ADCC of CD19 Antibodies by Combining Fc Protein-Engineering with Fc Glyco-Engineering. Antibodies 2020, 9, 63. [Google Scholar] [CrossRef]
- Hammer, O. CD19 as an attractive target for antibody-based therapy. mAbs 2012, 4, 571–577. [Google Scholar] [CrossRef] [Green Version]
- Schewe, D.M.; Alsadeq, A.; Sattler, C.; Lenk, L.; Vogiatzi, F.; Cario, G.; Vieth, S.; Valerius, T.; Rosskopf, S.; Meyersieck, F.; et al. An Fc-engineered CD19 antibody eradicates MRD in patient-derived MLL-rearranged acute lymphoblastic leukemia xenografts. Blood 2017, 130, 1543–1552. [Google Scholar] [CrossRef] [Green Version]
- Siegemund, M.; Schneider, F.; Hutt, M.; Seifert, O.; Muller, I.; Kulms, D.; Pfizenmaier, K.; Kontermann, R.E. IgG-single-chain TRAIL fusion proteins for tumour therapy. Sci. Rep. 2018, 8, 7808. [Google Scholar] [CrossRef]
- Pan, G.; Ni, J.; Wei, Y.-F.; Yu, G.-L.; Gentz, R.; Dixit, V.M. An Antagonist Decoy Receptor and a Death Domain-Containing Receptor for TRAIL. Science 1997, 277, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; O’Rourke, K.; Chinnaiyan, A.M.; Gentz, R.; Ebner, R.; Ni, J.; Dixit, V.M. The Receptor for the Cytotoxic Ligand TRAIL. Science 1997, 276, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H.; Degli-Esposti, M.A.; Johnson, R.S.; Smolak, P.J.; Waugh, J.Y.; Boiani, N.; Timour, M.S.; Gerhart, M.J.; Schooley, K.A.; Smith, C.A.; et al. TRAIL-R2: A novel apoptosis-mediating receptor for TRAIL. EMBO J. 1997, 16, 5386–5397. [Google Scholar] [CrossRef] [PubMed]
- Pitti, R.M.; Marsters, S.A.; Ruppert, S.; Donahue, C.J.; Moore, A.; Ashkenazi, A. Induction of Apoptosis by Apo-2 Ligand, a New Member of the Tumor Necrosis Factor Cytokine Family. J. Biol. Chem. 1996, 271, 12687–12690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.-P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; El-Deiry, W.S. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene 2003, 22, 8628–8633. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, J.P.; Marsters, S.A.; Pitti, R.M.; Gurney, A.; Skubatch, M.; Baldwin, D.; Ramakrishnan, L.; Gray, C.L.; Baker, K.; Wood, W.I.; et al. Control of TRAIL-Induced Apoptosis by a Family of Signaling and Decoy Receptors. Science 1997, 277, 818–821. [Google Scholar] [CrossRef]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor–related apoptosis–inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Pai, R.C.; Fong, S.; Leung, S.; Lawrence, D.A.; Marsters, S.A.; Blackie, C.; Chang, L.; McMurtrey, A.E.; Hebert, A.; et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Investig. 1999, 104, 155–162. [Google Scholar] [CrossRef]
- Alves, C.C.; Terziyska, N.; Grunert, M.; Gündisch, S.; Graubner, U.; Quintanilla-Martinez, L.; Jeremias, I. Leukemia-initiating cells of patient-derived acute lymphoblastic leukemia xenografts are sensitive toward TRAIL. Blood 2012, 119, 4224–4227. [Google Scholar] [CrossRef] [Green Version]
- Kelley, S.K.; Harris, L.A.; Xie, D.; Deforge, L.; Totpal, K.; Bussiere, J.; Fox, J.A. Preclinical studies to predict the disposition of Apo2L/tumor necrosis factor-related apoptosis-inducing ligand in humans: Characterization of in vivo efficacy, pharmacokinetics, and safety. J. Pharmacol. Exp. Ther. 2001, 299, 31–38. [Google Scholar] [PubMed]
- Soria, J.-C.; Márk, Z.; Zatloukal, P.; Szima, B.; Albert, I.; Juhász, E.; Pujol, J.-L.; Kozielski, J.; Baker, N.; Smethurst, D.; et al. Randomized phase ii study of dulanermin in combination with paclitaxel, carboplatin, and bevacizumab in advanced non–small-cell lung cancer. J. Clin. Oncol. 2011, 29, 4442–4451. [Google Scholar] [CrossRef] [PubMed]
- Belada, D.; Mayer, J.; Czuczman, M.S.; Flinn, I.W.; Durbin-Johnson, B.; Bray, G.L. Phase II study of dulanermin plus rituximab in patients with relapsed follicular non-Hodgkin’s lymphoma (NHL). J. Clin. Oncol. 2010, 28, 8104. [Google Scholar] [CrossRef]
- Herbst, R.S.; Eckhardt, S.G.; Kurzrock, R.; Ebbinghaus, S.; O’Dwyer, P.J.; Gordon, M.S.; Novotny, W.; Goldwasser, M.A.; Tohnya, T.M.; Lum, B.L.; et al. Phase I Dose-Escalation Study of Recombinant Human Apo2L/TRAIL, a Dual Proapoptotic Receptor Agonist, in Patients with Advanced Cancer. J. Clin. Oncol. 2010, 28, 2839–2846. [Google Scholar] [CrossRef]
- Emery, J.G.; McDonnell, P.; Burke, M.B.; Deen, K.C.; Lyn, S.; Silverman, C.; Dul, E.; Appelbaum, E.R.; Eichman, C.; DiPrinzio, R.; et al. Osteoprotegerin Is a Receptor for the Cytotoxic Ligand TRAIL. J. Biol. Chem. 1998, 273, 14363–14367. [Google Scholar] [CrossRef] [Green Version]
- Degli-Esposti, M.A.; Dougall, W.C.; Smolak, P.J.; Waugh, J.Y.; Smith, C.A.; Goodwin, R.G. The Novel Receptor TRAIL-R4 Induces NF-κB and Protects against TRAIL-Mediated Apoptosis, yet Retains an Incomplete Death Domain. Immunity 1997, 7, 813–820. [Google Scholar] [CrossRef] [Green Version]
- Stieglmaier, J.; Bremer, E.; Kellner, C.; Liebig, T.M.; Cate, B.T.; Peipp, M.; Schulze-Koops, H.; Pfeiffer, M.; Bühring, H.-J.; Greil, J.; et al. Selective induction of apoptosis in leukemic B-lymphoid cells by a CD19-specific TRAIL fusion protein. Cancer Immunol. Immunother. 2008, 57, 233–246. [Google Scholar] [CrossRef]
- Unverdorben, F.; Richter, F.; Hutt, M.; Seifert, O.; Malinge, P.; Fischer, N.; Kontermann, R.E. Pharmacokinetic properties of IgG and various Fc fusion proteins in mice. mAbs 2016, 8, 120–128. [Google Scholar] [CrossRef] [Green Version]
- Bremer, E.; Kuijlen, J.; Samplonius, D.; Walczak, H.; De Leij, L.; Helfrich, W. Target cell-restricted and -enhanced apoptosis induction by a scFv:sTRAIL fusion protein with specificity for the pancarcinoma-associated antigen EGP2. Int. J. Cancer 2004, 109, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H.; Moosmayer, D.; Wüest, T.; Bartke, T.; Gerlach, E.; Schönherr, U.; Peters, N.; Scheurich, P.; Pfizenmaier, K. Differential activation of TRAIL-R1 and -2 by soluble and membrane TRAIL allows selective surface antigen-directed activation of TRAIL-R2 by a soluble TRAIL derivative. Oncogene 2001, 20, 4101–4106. [Google Scholar] [CrossRef] [Green Version]
- Carter, P.; Presta, L.; Gorman, C.M.; Ridgway, J.B.; Henner, D.; Wong, W.L.; Rowland, A.M.; Kotts, C.; Carver, M.E.; Shepard, H.M. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl. Acad. Sci. USA 1992, 89, 4285–4289. [Google Scholar] [CrossRef] [Green Version]
- Amersdorffer, J.; Steidl, S.; Winderlich, M.; Krohn, A.; Rojkjaer, L. Combination Therapy with an Anti-CD19 Antibody and a Purine Analog. U.S. Patent 20140227277, 14 August 2012. [Google Scholar]
- Gunasekaran, K.; Pentony, M.; Shen, M.; Garrett, L.; Forte, C.; Woodward, A.; Bin Ng, S.; Born, T.; Retter, M.; Manchulenko, K.; et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: Applications to bispecific molecules and monovalent IgG. J. Biol. Chem. 2010, 285, 19637–19646. [Google Scholar] [CrossRef] [Green Version]
- Schneider, B.L.; Munkel, S.; Krippner-Heidenreich, A.; Grunwald, I.; Wels, W.S.; Wajant, H.; Pfizenmaier, K.; Gerspach, J. Potent antitumoral activity of TRAIL through generation of tumor-targeted single-chain fusion proteins. Cell Death Dis. 2010, 1, e68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steger, K.; Brady, J.P.; Wang, W.; Duskin, M.; Donato, K.; Peshwa, M.V. CHO-S antibody titers 1 gram/liter using flow electroporation-mediated transient gene expression followed by rapid migration to high-yield stable cell lines. J. Biomol. Screen. 2015, 20, 545–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Repp, R.; Kellner, C.; Muskulus, A.; Staudinger, M.; Nodehi, S.M.; Glorius, P.; Akramiene, D.; DeChant, M.; Fey, G.H.; Van Berkel, P.H.; et al. Combined Fc-protein- and Fc-glyco-engineering of scFv-Fc fusion proteins synergistically enhances CD16a binding but does not further enhance NK-cell mediated ADCC. J. Immunol. Methods 2011, 373, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Wirt, T.; Rosskopf, S.; Rösner, T.; Eichholz, K.M.; Kahrs, A.; Lutz, S.; Kretschmer, A.; Valerius, T.; Klausz, K.; Otte, A.; et al. An Fc Double-Engineered CD20 Antibody with Enhanced Ability to Trigger Complement-Dependent Cytotoxicity and Antibody-Dependent Cell-Mediated Cytotoxicity. Transfus. Med. Hemother. 2017, 44, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Fischer, U.; Forster, M.; Rinaldi, A.; Risch, T.; Sungalee, S.; Warnatz, H.-J.; Bornhauser, B.; Gombert, M.; Kratsch, C.; Stütz, A.M.; et al. Genomics and drug profiling of fatal TCF3-HLF−positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat. Genet. 2015, 47, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Alsadeq, A.; Lenk, L.; Vadakumchery, A.; Cousins, A.; Vokuhl, C.; Khadour, A.; Vogiatzi, F.; Seyfried, F.; Meyer, L.-H.; Cario, G.; et al. IL7R is associated with CNS infiltration and relapse in pediatric B-cell precursor acute lymphoblastic leukemia. Blood 2018, 132, 1614–1617. [Google Scholar] [CrossRef]
- Vogiatzi, F.; Winterberg, D.; Lenk, L.; Buchmann, S.; Cario, G.; Schrappe, M.; Peipp, M.; Richter-Pechanska, P.; Kulozik, A.E.; Lentes, J.; et al. Daratumumab eradicates minimal residual disease in a preclinical model of pediatric T-cell acute lymphoblastic leukemia. Blood 2019, 134, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Lenk, L.; Carlet, M.; Vogiatzi, F.; Spory, L.; Winterberg, D.; Cousins, A.; Vossen-Gajcy, M.; Ibruli, O.; Vokuhl, C.; Cario, G.; et al. CD79a promotes CNS-infiltration and leukemia engraftment in pediatric B-cell precursor acute lymphoblastic leukemia. Commun. Biol. 2021, 4, 73. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Debatin, K.-M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hockenbery, D.M.; Nuñez, G.; Milliman, C.L.; Schreiber, R.D.; Korsmeyer, S.J. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 1990, 348, 334–336. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Salles, G.; Duell, J.; Barca, E.G.; Tournilhac, O.; Jurczak, W.; Liberati, A.M.; Nagy, Z.; Obr, A.; Gaidano, G.; André, M.; et al. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): A multicentre, prospective, single-arm, phase 2 study. Lancet Oncol. 2020, 21, 978–988. [Google Scholar] [CrossRef]
- Jain, N.; Stock, W.; Zeidan, A.; Atallah, E.; McCloskey, J.; Heffner, L.; Tomlinson, B.; Bhatnagar, B.; Feingold, J.; Ungar, D.; et al. Loncastuximab tesirine, an anti-CD19 antibody-drug conjugate, in relapsed/refractory B-cell acute lymphoblastic leukemia. Blood Adv. 2020, 4, 449–457. [Google Scholar] [CrossRef]
- Bremer, E.; Samplonius, D.F.; Peipp, M.; Van Genne, L.; Kroesen, B.-J.; Fey, G.H.; Gramatzki, M.; De Leij, L.F.; Helfrich, W. Target Cell–Restricted Apoptosis Induction of Acute Leukemic T Cells by a Recombinant Tumor Necrosis Factor–Related Apoptosis-Inducing Ligand Fusion Protein with Specificity for Human CD7. Cancer Res. 2005, 65, 3380–3388. [Google Scholar] [CrossRef] [Green Version]
- Mariani, S.M.; Krammer, P.H. Differential regulation of TRAIL and CD95 ligand in transformed cells of the T and B lymphocyte lineage. Eur. J. Immunol. 1998, 28, 973–982. [Google Scholar] [CrossRef]
- Uckun, F.M.; Myers, D.E.; Qazi, S.; Ozer, Z.; Rose, R.; D’Cruz, O.J.; Ma, H. Recombinant human CD19L-sTRAIL effectively targets B cell precursor acute lymphoblastic leukemia. J. Clin. Investig. 2015, 125, 1006–1018. [Google Scholar] [CrossRef] [Green Version]
- Bossen, C.; Ingold, K.; Tardivel, A.; Bodmer, J.-L.; Gaide, O.; Hertig, S.; Ambrose, C.; Tschopp, J.; Schneider, P. Interactions of Tumor Necrosis Factor (TNF) and TNF Receptor Family Members in the Mouse and Human. J. Biol. Chem. 2006, 281, 13964–13971. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queudeville, M.; Schlegel, P.; Heinz, A.T.; Lenz, T.; Döring, M.; Holzer, U.; Hartmann, U.; Kreyenberg, H.; von Stackelberg, A.; Schrappe, M.; et al. Blinatumomab in pediatric patients with relapsed/refractory B-cell precursor acute lymphoblastic leukemia. Eur. J. Haematol. 2021, 106, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Khaw, S.L.; Suryani, S.; Evans, K.; Richmond, J.; Robbins, A.; Kurmasheva, R.T.; Billups, C.A.; Erickson, S.W.; Guo, Y.; Houghton, P.J.; et al. Venetoclax responses of pediatric ALL xenografts reveal sensitivity of MLL-rearranged leukemia. Blood 2016, 128, 1382–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Bremer, E.; Samplonius, D.; Kroesen, B.-J.; Van Genne, L.; De Leij, L.; Helfrich, W. Exceptionally Potent Anti-Tumor Bystander Activity of an scFv:sTRAIL Fusion Protein with Specificity for EGP2 Toward Target Antigen-Negative Tumor Cells1. Neoplasia 2004, 6, 636–645. [Google Scholar] [CrossRef] [Green Version]
- De Luca, R.; Kachel, P.; Kropivsek, K.; Snijder, B.; Manz, M.G.; Neri, D. A novel dual-cytokine–antibody fusion protein for the treatment of CD38-positive malignancies. Protein Eng. Des. Sel. 2018, 31, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Li, S.; Li, Z.; Peng, H.; Yuan, X.; Jiang, L.; Zhang, Y.; Fan, N.; Hu, X.; Yang, M.; et al. Human Umbilical Cord Mesenchymal Stem Cells as Vehicles of CD20-Specific TRAIL Fusion Protein Delivery: A Double-Target Therapy against Non-Hodgkin’s Lymphoma. Mol. Pharm. 2013, 10, 142–151. [Google Scholar] [CrossRef]
- Wei, G.; Wang, J.; Huang, H.; Zhao, Y. Novel immunotherapies for adult patients with B-lineage acute lymphoblastic leukemia. J. Hematol. Oncol. 2017, 10, 150. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Winterberg, D.; Lenk, L.; Oßwald, M.; Vogiatzi, F.; Gehlert, C.L.; Frielitz, F.-S.; Klausz, K.; Rösner, T.; Valerius, T.; Trauzold, A.; et al. Engineering of CD19 Antibodies: A CD19-TRAIL Fusion Construct Specifically Induces Apoptosis in B-Cell Precursor Acute Lymphoblastic Leukemia (BCP-ALL) Cells In Vivo. J. Clin. Med. 2021, 10, 2634. https://doi.org/10.3390/jcm10122634
Winterberg D, Lenk L, Oßwald M, Vogiatzi F, Gehlert CL, Frielitz F-S, Klausz K, Rösner T, Valerius T, Trauzold A, et al. Engineering of CD19 Antibodies: A CD19-TRAIL Fusion Construct Specifically Induces Apoptosis in B-Cell Precursor Acute Lymphoblastic Leukemia (BCP-ALL) Cells In Vivo. Journal of Clinical Medicine. 2021; 10(12):2634. https://doi.org/10.3390/jcm10122634
Chicago/Turabian StyleWinterberg, Dorothee, Lennart Lenk, Maren Oßwald, Fotini Vogiatzi, Carina Lynn Gehlert, Fabian-Simon Frielitz, Katja Klausz, Thies Rösner, Thomas Valerius, Anna Trauzold, and et al. 2021. "Engineering of CD19 Antibodies: A CD19-TRAIL Fusion Construct Specifically Induces Apoptosis in B-Cell Precursor Acute Lymphoblastic Leukemia (BCP-ALL) Cells In Vivo" Journal of Clinical Medicine 10, no. 12: 2634. https://doi.org/10.3390/jcm10122634
APA StyleWinterberg, D., Lenk, L., Oßwald, M., Vogiatzi, F., Gehlert, C. L., Frielitz, F. -S., Klausz, K., Rösner, T., Valerius, T., Trauzold, A., Peipp, M., Kellner, C., & Schewe, D. M. (2021). Engineering of CD19 Antibodies: A CD19-TRAIL Fusion Construct Specifically Induces Apoptosis in B-Cell Precursor Acute Lymphoblastic Leukemia (BCP-ALL) Cells In Vivo. Journal of Clinical Medicine, 10(12), 2634. https://doi.org/10.3390/jcm10122634