
Serum Amyloid Beta42 Is Not Eliminated by the Cirrhotic Liver: A Pilot Study




Abstract
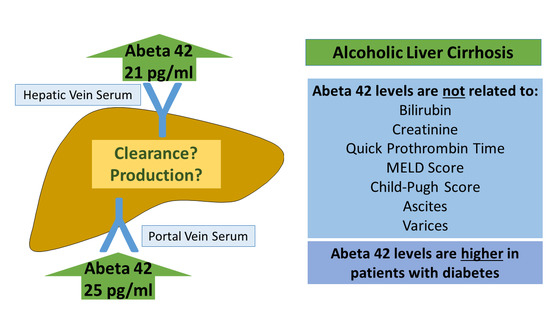
:
1. Introduction
2. Materials and Methods
2.1. Transjugular Intrahepatic Portosystemic Shunt (TIPS)
2.2. ELISA
2.3. Statistics
3. Results
3.1. Aβ42 in Different Blood Compartments of Patients with Liver Cirrhosis
3.2. Aβ42 in Relation to Scores and Measures of Liver Function
3.3. Aβ42 in Patients with Disturbed Glucose Metabolism
3.4. Association of Aβ42 Levels with Markers of Inflammation, Endothelial Function, and Fibrosis
3.5. Aβ42 in Patients with Ascites and Varices
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Nazareth, A.M. Type 2 diabetes mellitus in the pathophysiology of Alzheimer’s disease. Dement. Neuropsychol. 2017, 11, 105–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jellinger, K.A.; Janetzky, B.; Attems, J.; Kienzl, E. Biomarkers for early diagnosis of Alzheimer disease: ‘ALZheimer ASsociated gene’—A new blood biomarker? J. Cell. Mol. Med. 2008, 12, 1094–1117. [Google Scholar] [CrossRef] [Green Version]
- Van Gool, B.; Storck, S.; Reekmans, S.M.; Lechat, B.; Gordts, P.L.S.M.; Pradier, L.; Pietrzik, C.U.; Roebroek, A.J.M. LRP1 Has a Predominant Role in Production over Clearance of Aβ in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 7234–7245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s amyloid-β1-40 peptide from brain by LDL receptor–related protein-1 at the blood-brain barrier. J. Clin. Investig. 2000, 106, 1489–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Bu, X.-L.; Liu, Y.-H.; Zhu, C.; Shen, L.-L.; Jiao, S.-S.; Zhu, X.-Y.; Giunta, B.; Tan, J.; Song, W.-H.; et al. Physiological amyloid-beta clearance in the periphery and its therapeutic potential for Alzheimer’s disease. Acta Neuropathol. 2015, 130, 487–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, D.-Y.; Cheng, Y.; Zhuang, Z.-Q.; He, C.-Y.; Pan, Q.-G.; Tang, M.-Z.; Hu, X.-L.; Shen, Y.-Y.; Wang, Y.-R.; Chen, S.-H.; et al. Physiological clearance of amyloid-beta by the kidney and its therapeutic potential for Alzheimer’s disease. Mol. Psychiatry 2021, 1–9. [Google Scholar] [CrossRef]
- Cheng, Y.; Tian, D.-Y.; Wang, Y.-J. Peripheral clearance of brain-derived Aβ in Alzheimer’s disease: Pathophysiology and therapeutic perspectives. Transl. Neurodegener. 2020, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Joachim, C.L.; Mori, H.; Selkoe, D.J. Amyloid β-protein deposition in tissues other than brain in Alzheimer’s disease. Nat. Cell Biol. 1989, 341, 226–230. [Google Scholar] [CrossRef]
- Chen, S.-H.; Tian, D.-Y.; Shen, Y.-Y.; Cheng, Y.; Fan, D.-Y.; Sun, H.-L.; He, C.-Y.; Sun, P.-Y.; Bu, X.-L.; Zeng, F.; et al. Amyloid-beta uptake by blood monocytes is reduced with ageing and Alzheimer’s disease. Transl. Psychiatry 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Pappolla, M.; Sambamurti, K.; Vidal, R.; Pacheco-Quinto, J.; Poeggeler, B.; Matsubara, E. Evidence for lymphatic Aβ clearance in Alzheimer’s transgenic mice. Neurobiol. Dis. 2014, 71, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Ghiso, J.; Shayo, M.; Calero, M.; Ng, D.; Tomidokoro, Y.; Gandy, S.; Rostagno, A.; Frangione, B. Systemic Catabolism of Alzheimer’s Aβ40 and Aβ42. J. Biol. Chem. 2004, 279, 45897–45908. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-R.; Wang, Q.-H.; Zhang, T.; Liu, Y.-H.; Yao, X.-Q.; Zeng, F.; Li, J.; Zhou, F.-Y.; Wang, L.; Yan, J.-C.; et al. Associations Between Hepatic Functions and Plasma Amyloid-Beta Levels—Implications for the Capacity of Liver in Peripheral Amyloid-Beta Clearance. Mol. Neurobiol. 2017, 54, 2338–2344. [Google Scholar] [CrossRef]
- Buniatian, G.H.; Weiskirchen, R.; Weiss, T.S.; Schwinghammer, U.; Fritz, M.; Seferyan, T.; Proksch, B.; Glaser, M.; Lourhmati, A.; Buadze, M.; et al. Antifibrotic Effects of Amyloid-Beta and Its Loss in Cirrhotic Liver. Cells 2020, 9, 452. [Google Scholar] [CrossRef] [Green Version]
- Roher, A.E.; Esh, C.L.; Kokjohn, T.A.; Castaño, E.M.; Van Vickle, G.D.; Kalback, W.M.; Patton, R.L.; Luehrs, D.C.; Daugs, I.D.; Kuo, Y.-M.; et al. Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer’s disease. Alzheimer’s Dement. 2009, 5, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Molinuevo, J.L.; Ayton, S.; Batrla, R.; Bednar, M.M.; Bittner, T.; Cummings, J.; Fagan, A.M.; Hampel, H.; Mielke, M.; Mikulskis, A.; et al. Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol. 2018, 136, 821–853. [Google Scholar] [CrossRef] [Green Version]
- Bauer, S.; Eisinger, K.; Wiest, R.; Karrasch, T.; Scherer, M.N.; Farkas, S.; Aslanidis, C.; Buechler, C. Connective tissue growth factor level is increased in patients with liver cirrhosis but is not associated with complications or extent of liver injury. Regul. Pept. 2012, 179, 10–14. [Google Scholar] [CrossRef]
- Gressner, A.M.; Yagmur, E.; Lahme, B.; Gressner, O.; Stanzel, S. Connective Tissue Growth Factor in Serum as a New Candidate Test for Assessment of Hepatic Fibrosis. Clin. Chem. 2006, 52, 1815–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gressner, O.A.; Lahme, B.; Demirci, I.; Gressner, A.M.; Weiskirchen, R. Differential effects of TGF-β on connective tissue growth factor (CTGF/CCN2) expression in hepatic stellate cells and hepatocytes. J. Hepatol. 2007, 47, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Laleman, W.; Landeghem, L.; Wilmer, A.; Fevery, J.; Nevens, F. Portal hypertension: From pathophysiology to clinical practice. Liver Int. 2005, 25, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Siramolpiwat, S. Transjugular intrahepatic portosystemic shunts and portal hypertension-related complications. World J. Gastroenterol. 2014, 20, 16996–17010. [Google Scholar] [CrossRef] [PubMed]
- Wiest, R.; Groszmann, R.J. The paradox of nitric oxide in cirrhosis and portal hypertension: Too much, not enough. Hepatology 2002, 35, 478–491. [Google Scholar] [CrossRef]
- Eisinger, K.; Krautbauer, S.; Wiest, R.; Karrasch, T.; Hader, Y.; Scherer, M.N.; Farkas, S.; Aslanidis, C.; Buechler, C. Portal vein omentin is increased in patients with liver cirrhosis but is not associated with complications of portal hypertension. Eur. J. Clin. Investig. 2013, 43, 926–932. [Google Scholar] [CrossRef]
- Lluch, P.; Torondel, B.; Medina, P.; Segarra, G.; Del Olmo, J.A.; Serra, M.A.; Rodrigo, J.M. Plasma concentrations of nitric oxide and asymmetric dimethylarginine in human alcoholic cirrhosis. J. Hepatol. 2004, 41, 55–59. [Google Scholar] [CrossRef]
- Estrada, L.D.; Ahumada, P.; Cabrera, D.; Arab, J.P. Liver Dysfunction as a Novel Player in Alzheimer’s Progression: Looking Outside the Brain. Front. Aging Neurosci. 2019, 11, 174. [Google Scholar] [CrossRef] [Green Version]
- Buechler, C.; Wanninger, J.; Neumeier, M. Adiponectin, a key adipokine in obesity related liver diseases. World J. Gastroenterol. 2011, 17, 2801–2811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.-G.; Krenz, A.; Toussaint, L.E.; Maurer, K.J.; Robinson, S.-A.; Yan, A.; Torres, L.; Bynoe, M.S. Non-alcoholic fatty liver disease induces signs of Alzheimer’s disease (AD) in wild-type mice and accelerates pathological signs of AD in an AD model. J. Neuroinflamm. 2016, 13, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nägga, K.; Gustavsson, A.-M.; Stomrud, E.; Lindqvist, D.; Van Westen, D.; Blennow, K.; Zetterberg, H.; Melander, O.; Hansson, O. Increased midlife triglycerides predict brain β-amyloid and tau pathology 20 years later. Neurology 2017, 90, e73–e81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, B.; Villeneuve, S.; Mack, W.; DeCarli, C.; Chui, H.C.; Jagust, W. Associations Between Serum Cholesterol Levels and Cerebral Amyloidosis. JAMA Neurol. 2014, 71, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Verlinden, W.; Francque, S.; Vonghia, L. Peripheral Venous, Portal Venous, Hepatic Venous, and Arterial and Intrahepatic Cytokine Levels as Biomarkers and Functional Correlations. In Biomarkers in Liver Disease. Biomarkers in Disease: Methods, Discoveries and Applications; Patel, V., Preedy, V., Eds.; Springer: Dordrecht, The Netherlands, 2017. [Google Scholar] [CrossRef]
- Fontana, L.; Eagon, J.C.; Trujillo, M.E.; Scherer, P.E.; Klein, S. Visceral Fat Adipokine Secretion Is Associated with Systemic Inflammation in Obese Humans. Diabetes 2007, 56, 1010–1013. [Google Scholar] [CrossRef] [Green Version]
- Wiest, R.; Weigert, J.; Wanninger, J.; Neumeier, M.; Bauer, S.; Schmidhofer, S.; Farkas, S.; Scherer, M.N.; Schäffler, A.; Schölmerich, J. Impaired hepatic removal of interleukin-6 in patients with liver cirrhosis. Cytokine 2011, 53, 178–183. [Google Scholar] [CrossRef]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buechler, C.; Eisinger, K.; Krautbauer, S. Diagnostic and prognostic potential of the macrophage specific receptor CD163 in inflammatory diseases. Inflamm. Allergy Drug Targets 2013, 12, 391–402. [Google Scholar] [CrossRef]
- Holland-Fischer, P.; Gronbaek, H.; Sandahl, T.D.; Moestrup, S.K.; Riggio, O.; Ridola, L.; Aagaard, N.K.; Møller, H.J.; Vilstrup, H. Kupffer cells are activated in cirrhotic portal hypertension and not normalised by TIPS. Gut 2011, 60, 1389–1393. [Google Scholar] [CrossRef]
- Buechler, C.; Haberl, E.M.; Rein-Fischboeck, L.; Aslanidis, C. Adipokines in Liver Cirrhosis. Int. J. Mol. Sci. 2017, 18, 1392. [Google Scholar] [CrossRef] [Green Version]
- Eisinger, K.; Krautbauer, S.; Wiest, R.; Weiss, T.S.; Buechler, C. Reduced serum chemerin in patients with more severe liver cirrhosis. Exp. Mol. Pathol. 2015, 98, 208–213. [Google Scholar] [CrossRef]
- Krautbauer, S.; Wanninger, J.; Eisinger, K.; Hader, Y.; Beck, M.; Kopp, A.; Schmid, A.; Weiss, T.S.; Dorn, C.; Buechler, C. Chemerin is highly expressed in hepatocytes and is induced in non-alcoholic steatohepatitis liver. Exp. Mol. Pathol. 2013, 95, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Buechler, C.; Feder, S.; Haberl, E.M.; Aslanidis, C. Chemerin Isoforms and Activity in Obesity. Int. J. Mol. Sci. 2019, 20, 1128. [Google Scholar] [CrossRef] [Green Version]
- Horn, P.; Von Loeffelholz, C.; Forkert, F.; Stengel, S.; Reuken, P.; Aschenbach, R.; Stallmach, A.; Bruns, T. Low circulating chemerin levels correlate with hepatic dysfunction and increased mortality in decompensated liver cirrhosis. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef]
- Peschel, G.; Grimm, J.; Gülow, K.; Müller, M.; Buechler, C.; Weigand, K. Chemerin Is a Valuable Biomarker in Patients with HCV Infection and Correlates with Liver Injury. Diagnostics 2020, 10, 974. [Google Scholar] [CrossRef]
- Gong, C.; Wei, D.; Wang, Y.; Ma, J.; Yuan, C.; Zhang, W.; Yu, G.; Zhao, Y. A Meta-Analysis of C-Reactive Protein in Patients With Alzheimer’s Disease. Am. J. Alzheimer’s Dis. Dement. 2016, 31, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Pieri, G.; Agarwal, B.; Burroughs, A.K. C-reactive protein and bacterial infection in cirrhosis. Ann. Gastroenterol. 2014, 27, 113–120. [Google Scholar]
- Hanon, O.; Vidal, J.-S.; Lehmann, S.; Bombois, S.; Allinquant, B.; Tréluyer, J.-M.; Gelé, P.; Delmaire, C.; Blanc, F.; Mangin, J.-F.; et al. Plasma amyloid levels within the Alzheimer’s process and correlations with central biomarkers. Alzheimer’s Dement. 2018, 14, 858–868. [Google Scholar] [CrossRef]
- Roessle, M.; Gerbes, A.L. TIPS for the treatment of refractory ascites, hepatorenal syndrome and hepatic hydrothorax: A critical update. Gut 2010, 59, 988–1000. [Google Scholar] [CrossRef] [Green Version]
- Wiest, R.; Moleda, L.; Farkas, S.; Scherer, M.; Kopp, A.; Wönckhaus, U.; Büchler, C.; Schölmerich, J.; Schäffler, A. Splanchnic concentrations and postprandial release of visceral adipokines. Metabolism 2010, 59, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Schober, P.; Boer, C.; Schwarte, L.A. Correlation Coefficients: Appropriate use and interpretation. Anesth. Analg. 2018, 126, 1763–1768. [Google Scholar] [CrossRef] [PubMed]
- Angermayr, B.; Cejna, M.; Karnel, F.; Gschwantler, M.; Koenig, F.; Pidlich, J.; Mendel, H.; Pichler, L.; Wichlas, M.; Kreil, A.; et al. Child-Pugh versus MELD score in predicting survival in patients undergoing transjugular intrahepatic portosystemic shunt. Gut 2003, 52, 879–885. [Google Scholar] [CrossRef] [Green Version]
- Ott, A.; Stolk, R.; Van Harskamp, F.; Pols, H.A.P.; Hofman, A.; Breteler, M.M. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999, 53, 1937. [Google Scholar] [CrossRef] [PubMed]
- Locascio, J.J.; Fukumoto, H.; Yap, L.; Bottiglieri, T.; Growdon, J.H.; Hyman, B.T.; Irizarry, M.C. Plasma Amyloid β-Protein and C-reactive Protein in Relation to the Rate of Progression of Alzheimer Disease. Arch. Neurol. 2008, 65, 776–785. [Google Scholar] [CrossRef] [Green Version]
- Tejera, D.; Mercan, D.; Sanchez-Caro, J.M.; Hanan, M.; Greenberg, D.; Soreq, H.; Latz, E.; Golenbock, D.; Heneka, M.T. Systemic inflammation impairs microglial Aβ clearance through NLRP 3 inflammasome. EMBO J. 2019, 38, e101064. [Google Scholar] [CrossRef]
- Genc, H.; Dogru, T.; Kara, M.; Tapan, S.; Ercin, C.N.; Acikel, C.; Karslioglu, Y.; Bagci, S. Association of plasma visfatin with hepatic and systemic inflammation in nonalcoholic fatty liver disease. Ann. Hepatol. 2013, 12, 380–387. [Google Scholar] [CrossRef]
- Tilg, H. The Role of Cytokines in Non-Alcoholic Fatty Liver Disease. Dig. Dis. 2010, 28, 179–185. [Google Scholar] [CrossRef]
- Bertolani, C.; Sancho-Bru, P.; Failli, P.; Bataller, R.; Aleffi, S.; DeFranco, R.; Mazzinghi, B.; Romagnani, P.; Milani, S.; Ginés, P.; et al. Resistin as an Intrahepatic Cytokine: Overexpression during Chronic Injury and Induction of Proinflammatory Actions in Hepatic Stellate Cells. Am. J. Pathol. 2006, 169, 2042–2053. [Google Scholar] [CrossRef] [Green Version]
- Siroen, M.P.C.; Wiest, R.; Richir, M.C.; Teerlink, T.; Rauwerda, J.A.; Drescher, F.T.; Zorger, N.; Leeuwen, P.A.M.V. Transjugular intrahepatic portosystemic shunt-placement increases arginine/asymmetric dimethylarginine ratio in cirrhotic patients. World J. Gastroenterol. 2008, 14, 7214–7219. [Google Scholar] [CrossRef]
- Iwakiri, Y. Endothelial dysfunction in the regulation of cirrhosis and portal hypertension. Liver Int. 2011, 32, 199–213. [Google Scholar] [CrossRef] [Green Version]
- Maarouf, C.L.; Walker, J.E.; Sue, L.I.; Dugger, B.N.; Beach, T.G.; Serrano, G.E. Impaired hepatic amyloid-beta degradation in Alzheimer’s disease. PLoS ONE 2018, 13, e0203659. [Google Scholar] [CrossRef]
- Porowski, D.; Wirkowska, A.; Hryniewiecka, E.; Wyzgał, J.; Pacholczyk, M.; Pączek, L. Liver Failure Impairs the Intrahepatic Elimination of Interleukin-6, Tumor Necrosis Factor-Alpha, Hepatocyte Growth Factor, and Transforming Growth Factor-Beta. BioMed Res. Int. 2015, 2015, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Pinçon, A.; De Montgolfier, O.; Akkoyunlu, N.; Daneault, C.; Pouliot, P.; Villeneuve, L.; Lesage, F.; Levy, B.I.; Thorin-Trescases, N.; Thorin, É.; et al. Non-Alcoholic Fatty Liver Disease, and the Underlying Altered Fatty Acid Metabolism, Reveals Brain Hypoperfusion and Contributes to the Cognitive Decline in APP/PS1 Mice. Metabolites 2019, 9, 104. [Google Scholar] [CrossRef] [Green Version]
- Meakin, P.J.; Mezzapesa, A.; Benabou, E.; Haas, M.E.; Bonardo, B.; Grino, M.; Brunel, J.-M.; Desbois-Mouthon, C.; Biddinger, S.B.; Govers, R.; et al. The beta secretase BACE1 regulates the expression of insulin receptor in the liver. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Buechler, C.; Aslanidis, C. Role of lipids in pathophysiology, diagnosis and therapy of hepatocellular carcinoma. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2020, 1865, 158658. [Google Scholar] [CrossRef]
- Lue, L.-F.; Pai, M.-C.; Chen, T.-F.; Hu, C.-J.; Huang, L.-K.; Lin, W.-C.; Wu, C.-C.; Jeng, J.-S.; Blennow, K.; Sabbagh, M.N.; et al. Age-Dependent Relationship Between Plasma Aβ40 and Aβ42 and Total Tau Levels in Cognitively Normal Subjects. Front. Aging Neurosci. 2019, 11, 222. [Google Scholar] [CrossRef] [Green Version]
- Mayeux, R.; Honig, L.S.; Tang, M.-X.; Manly, J.; Stern, Y.; Schupf, N.; Mehta, P.D. Plasma A 40 and A 42 and Alzheimer’s disease: Relation to age, mortality, and risk. Neurology 2003, 61, 1185–1190. [Google Scholar] [CrossRef]
- Meakin, P.J.; Coull, B.M.; Tuharska, Z.; McCaffery, C.; Akoumianakis, I.; Antoniades, C.; Brown, J.; Griffin, K.J.; Platt, F.; Ozber, C.H.; et al. Elevated circulating amyloid concentrations in obesity and diabetes promote vascular dysfunction. J. Clin. Investig. 2020, 130, 4104–4117. [Google Scholar] [CrossRef]
- Ma, F.; Wu, T.; Zhao, J.; Song, A.; Liu, H.; Xu, W.; Huang, G. Folic acid supplementation improves cognitive function by reducing the levels of peripheral inflammatory cytokines in elderly Chinese subjects with MCI. Sci. Rep. 2016, 6, 37486. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cirrhosis Patients | |
|---|---|
| Number | 20 |
| Sex (female/male) | 7/13 |
| MELD score | 9 (6–21) |
| Age (years) | 52 (40–81) |
| Child-Pugh score A/B/C/ | 6/6/8 |
| C-reactive protein (mg/L) | 9.9 (1.0–53.5) |
| Albumin (g/L) | 32.9 (1.6–42.7) |
| Bilirubin (mg/dL) | 1.2 (0.5–4.6) |
| Quick prothrombin time (%) | 71 (28–100) |
| Aspartate aminotransferase (U/L) | 40 (11–82) |
| Alanine aminotransferase (U/L) | 33 (2–68) |
| Creatinine (mg/dL) | 1.1 (0.5–4.5) |
| Ascites: no or little/modest or massive | 8/12 |
| Varices: no or small/large | 7/13 |
| Diabetes no/yes | 12/8 |
| HVS Aβ42 | PVS Aβ42 | |
| SVS Aβ42 | 0.541 (0.014) | 0.463 (0.040) |
| HVS Aβ42 | 0.589 (0.006) | |
| HVS Chemerin | PVS Chemerin | |
| SVS Chemerin | 0.811 (<0.001) | 0.912 (<0.001) |
| HVS Chemerin | 0.838 (<0.001) | |
| HVS IL-6 | PVS IL-6 | |
| SVS IL-6 | 0.950 (<0.001) | 0.689 (0.001) |
| HVS IL-6 | 0.726 (<0.001) |
| CRP | Resistin | Chemerin | IL-6 | Visfatin | Arginine | ADMA | CTGF | |
|---|---|---|---|---|---|---|---|---|
| r | 0.390 | −0.011 | −0.290 | 0.262 | 0.040 | −0.251 | −0.036 | −0.580 |
| p | 0.089 | 0.965 | 0.214 | 0.531 | 0.867 | 0.286 | 0.880 | 0.007 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiest, R.; Weiss, T.S.; Danielyan, L.; Buechler, C. Serum Amyloid Beta42 Is Not Eliminated by the Cirrhotic Liver: A Pilot Study. J. Clin. Med. 2021, 10, 2669. https://doi.org/10.3390/jcm10122669
Wiest R, Weiss TS, Danielyan L, Buechler C. Serum Amyloid Beta42 Is Not Eliminated by the Cirrhotic Liver: A Pilot Study. Journal of Clinical Medicine. 2021; 10(12):2669. https://doi.org/10.3390/jcm10122669
Chicago/Turabian StyleWiest, Reiner, Thomas S. Weiss, Lusine Danielyan, and Christa Buechler. 2021. "Serum Amyloid Beta42 Is Not Eliminated by the Cirrhotic Liver: A Pilot Study" Journal of Clinical Medicine 10, no. 12: 2669. https://doi.org/10.3390/jcm10122669
APA StyleWiest, R., Weiss, T. S., Danielyan, L., & Buechler, C. (2021). Serum Amyloid Beta42 Is Not Eliminated by the Cirrhotic Liver: A Pilot Study. Journal of Clinical Medicine, 10(12), 2669. https://doi.org/10.3390/jcm10122669