
Methylation Status of Corticotropin-Releasing Factor (CRF) Receptor Genes in Colorectal Cancer



 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset for the In Silico Analysis
2.2. Data Preprocessing and DNA Methylation Analysis
2.3. In Silico Determination of Transcription Factor (TF) Binding
2.4. Clinical Samples
2.5. Extraction of ccfDNA
2.6. Sodium Bisulfite Conversion of ccfDNA
2.7. Quantitative Methylation-Specific PCR (qMSP)
2.8. Statistical Analysis
2.9. Automated Machine Learning Analysis
3. Results
3.1. In Silico Analysis of CRFR1 and CRFR2 Methylation in CRC and CD
3.1.1. Analysis in CRC and CD Tissue-Derived Datasets
3.1.2. Analysis of CRC-Derived ccfDNA Data
3.2. In Silico Analysis of TFs Binding in CRFR1 and CRFR2 Promoters
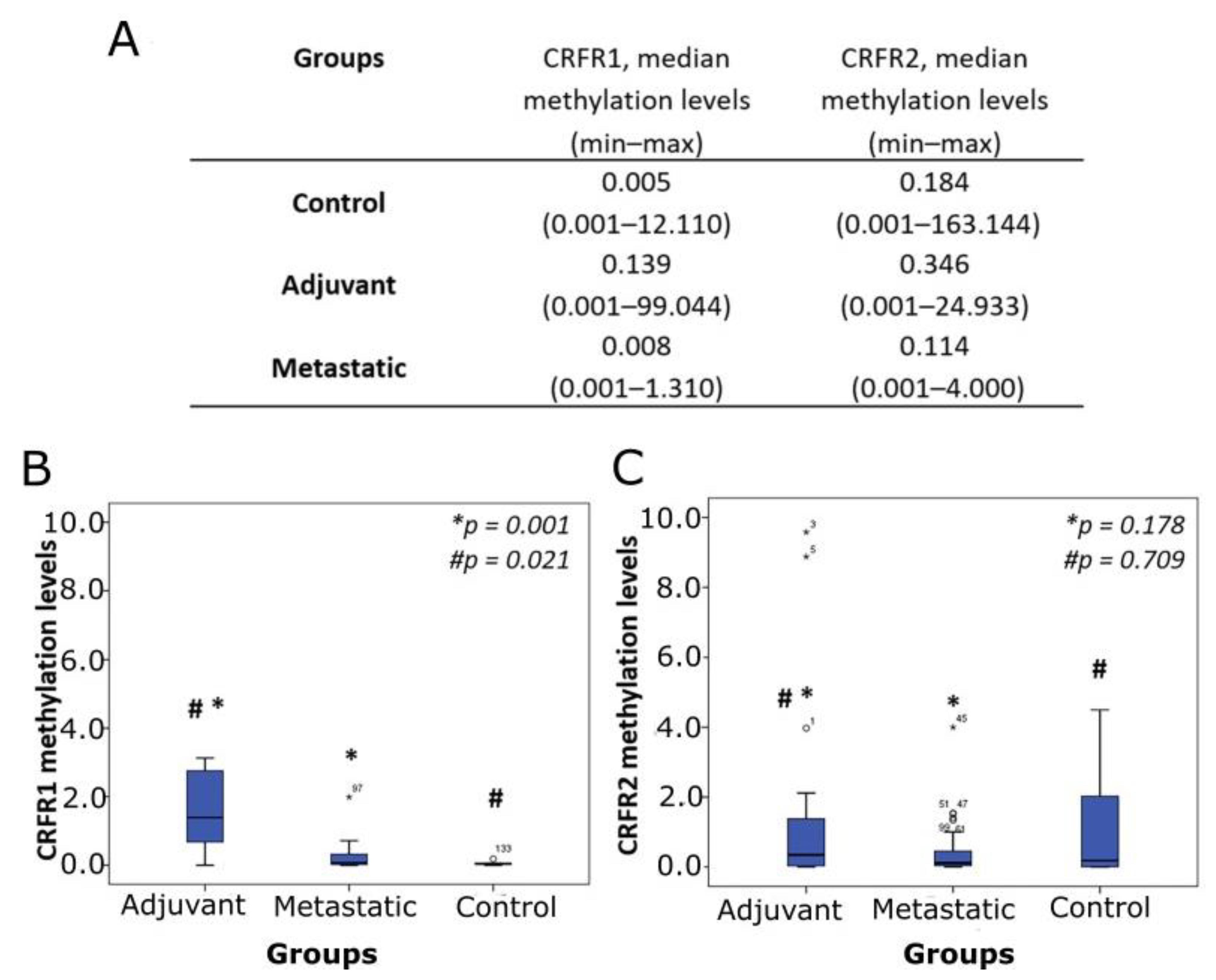
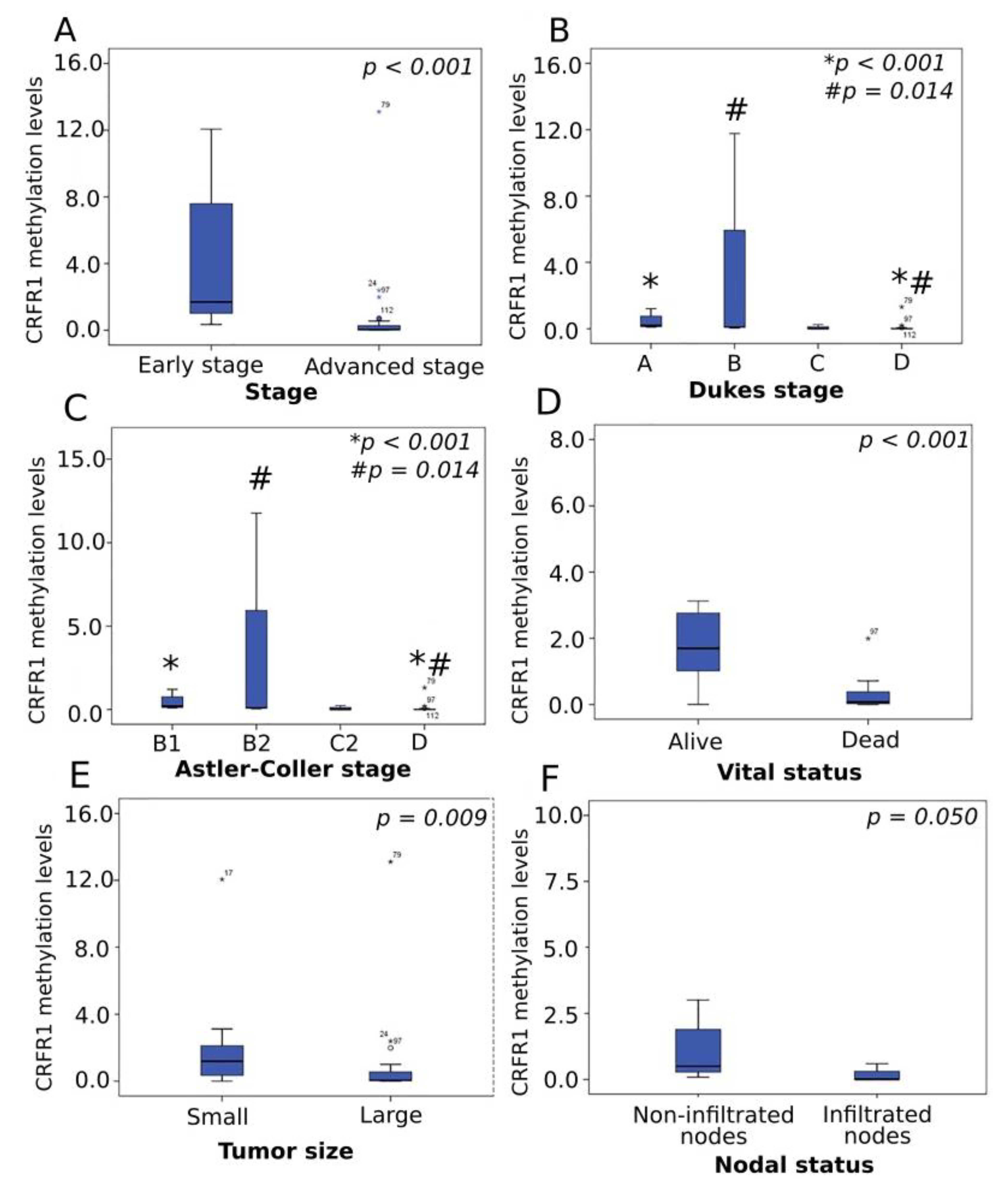
3.3. Methylation Analysis of CRFR1 and CRFR2 in CRC-Derived ccfDNA Clinical Samples
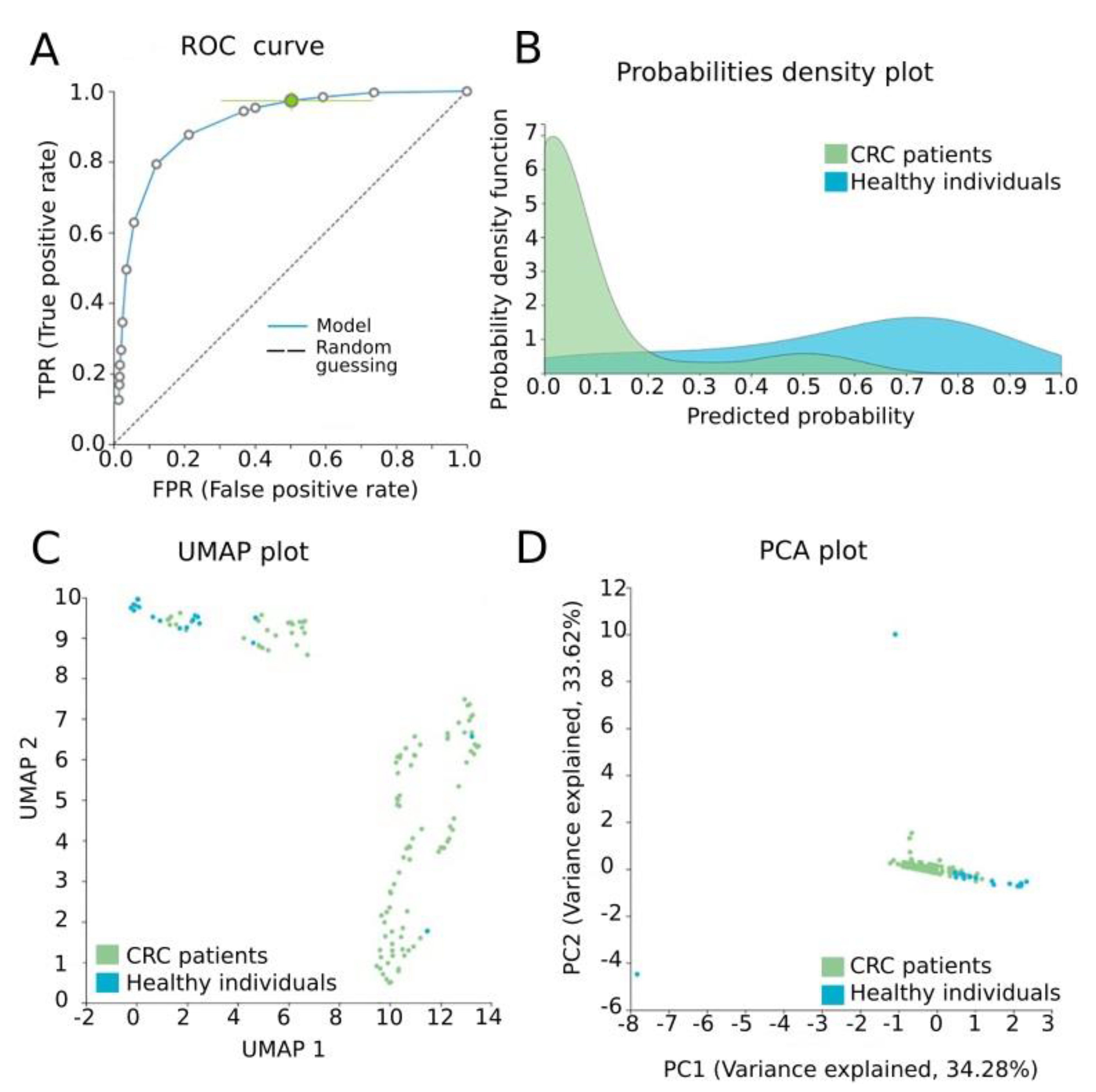
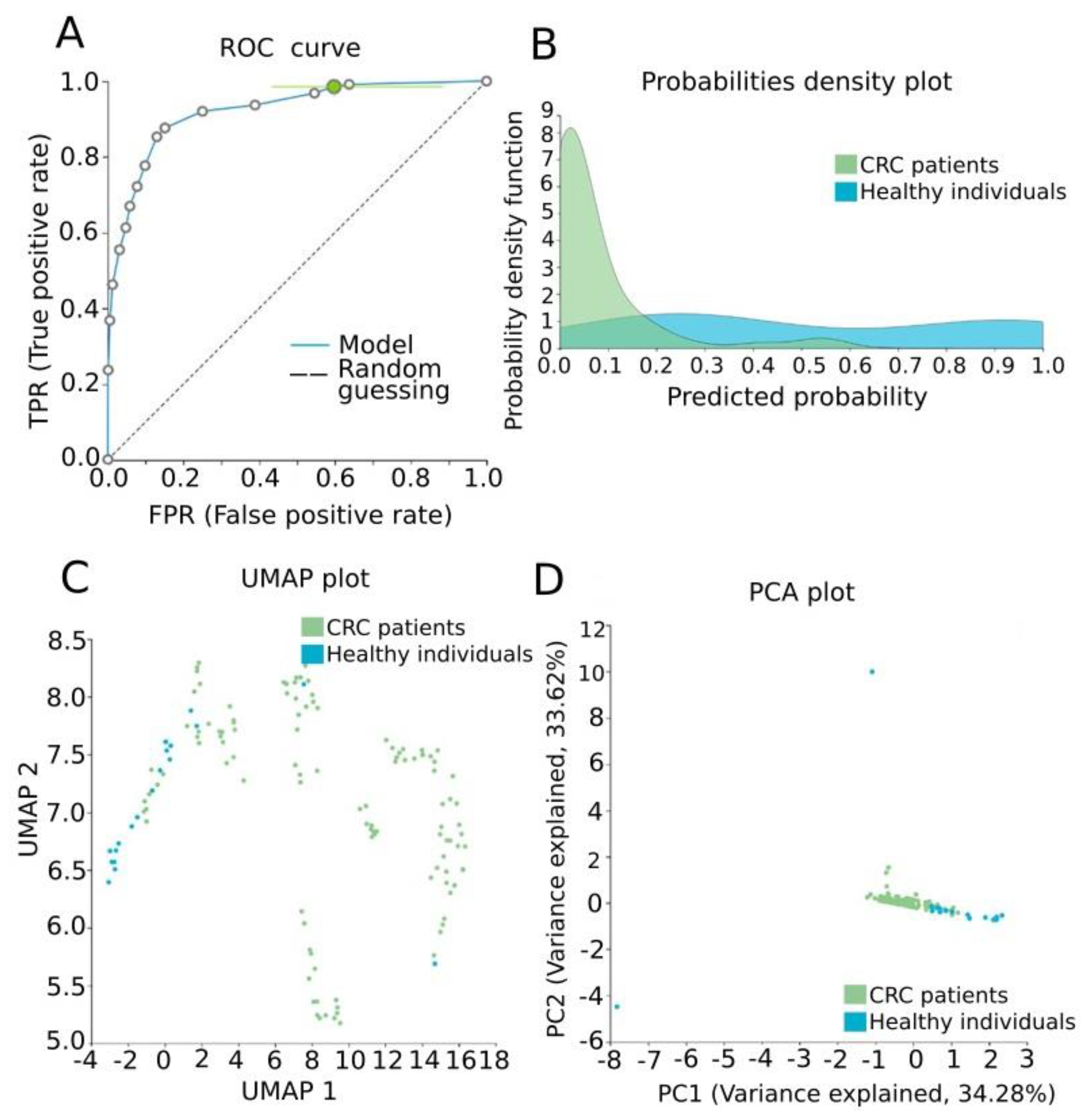
3.4. Automated Machine Learning Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellino, G.; Gallo, G.; Pallante, P.; Capasso, R.; De Stefano, A.; Maretto, I.; Malapelle, U.; Qiu, S.; Nikolaou, S.; Barina, A.; et al. Noninvasive Biomarkers of Colorectal Cancer: Role in Diagnosis and Personalised Treatment Perspectives. Gastroenterol. Res. Pract. 2018, 2018, 2397863. [Google Scholar] [CrossRef] [PubMed]
- Sammarco, G.; Gallo, G.; Vescio, G.; Picciariello, A.; De Paola, G.; Trompetto, M.; Currò, G.; Ammendola, M. Mast Cells, microRNAs and Others: The Role of Translational Research on Colorectal Cancer in the Forthcoming Era of Precision Medicine. J. Clin. Med. 2020, 9, 2852. [Google Scholar] [CrossRef] [PubMed]
- Garborg, K.; Holme, Ø.; Løberg, M.; Kalager, M.; Adami, H.O.; Bretthauer, M. Current status of screening for colorectal cancer. Ann. Oncol. 2013, 24, 1963–1972. [Google Scholar] [CrossRef]
- Kaminski, M.F.; Regula, J.; Kraszewska, E.; Polkowski, M.; Wojciechowska, U.; Didkowska, J.; Zwierko, M.; Rupinski, M.; Nowacki, M.P.; Butruk, E. Quality indicators for colonoscopy and the risk of interval cancer. N. Engl. J. Med. 2010, 362, 1795–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabeneck, L.; Paszat, L.F.; Saskin, R. Endoscopist specialty is associated with incident colorectal cancer after a negative colonoscopy. Clin. Gastroenterol. Hepatol. 2010, 8, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Brenner, H.; Stock, C.; Hoffmeister, M. Effect of screening sigmoidoscopy and screening colonoscopy on colorectal cancer incidence and mortality: Systematic review and meta-analysis of randomised controlled trials and observational studies. BMJ Clin. Res. Ed. 2014, 348, g2467. [Google Scholar] [CrossRef] [Green Version]
- Martín-López, J.E.; Beltrán-Calvo, C.; Rodríguez-López, R.; Molina-López, T. Comparison of the accuracy of CT colonography and colonoscopy in the diagnosis of colorectal cancer. Colorectal Dis. 2014, 16, O82–O89. [Google Scholar] [CrossRef]
- Lin, O.S.-T. Computed tomographic colonography: Hope or hype? World J. Gastroenterol. 2010, 16, 915–920. [Google Scholar] [CrossRef]
- Lansdorp-Vogelaar, I.; van Ballegooijen, M.; Boer, R.; Zauber, A.; Habbema, J.D. A novel hypothesis on the sensitivity of the fecal occult blood test: Results of a joint analysis of 3 randomized controlled trials. Cancer 2009, 115, 2410–2419. [Google Scholar] [CrossRef] [Green Version]
- Imperiale, T.F.; Ransohoff, D.F.; Itzkowitz, S.H.; Levin, T.R.; Lavin, P.; Lidgard, G.P.; Ahlquist, D.A.; Berger, B.M. Multitarget stool DNA testing for colorectal-cancer screening. N. Engl. J. Med. 2014, 370, 1287–1297. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, M.; Terzer, T.; Schrotz-King, P.; Brenner, H. Comparison of Proteomic Technologies for Blood-Based Detection of Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 1189. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C. Real-time liquid biopsy in cancer patients: Fact or fiction? Cancer Res. 2013, 73, 6384–6388. [Google Scholar] [CrossRef] [Green Version]
- Alix-Panabières, C.; Pantel, K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. 2016, 6, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Panagopoulou, M.; Esteller, M.; Chatzaki, E. Circulating Cell-Free DNA in Breast Cancer: Searching for Hidden Information towards Precision Medicine. Cancers 2021, 13, 728. [Google Scholar] [CrossRef]
- Panagopoulou, M.; Karaglani, M.; Balgkouranidou, I.; Biziota, E.; Koukaki, T.; Karamitrousis, E.; Nena, E.; Tsamardinos, I.; Kolios, G. Circulating cell-free DNA in breast cancer: Size profiling, levels, and methylation patterns lead to prognostic and predictive classifiers. Oncogene 2019, 38, 3387–3401. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulou, M.; Karaglani, M.; Balgkouranidou, I.; Pantazi, C.; Kolios, G.; Kakolyris, S.; Chatzaki, E. Circulating cell-free DNA release in vitro: Kinetics, size profiling, and cancer-related gene methylation. Cell. Physiol. 2019, 234, 14079–14089. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Tiirikainen, M. Diagnostic Power of DNA Methylation Classifiers for Early Detection of Cancer. Trends Cancer 2020, 6, 78–81. [Google Scholar] [CrossRef]
- Chatzaki, E.; Crowe, P.D.; Wang, L.; Million, M.; Taché, Y.; Grigoriadis, D.E. CRF receptor type 1 and 2 expression and anatomical distribution in the rat colon. J. Neurochem. 2004, 90, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Chatzaki, E.; Lambropoulou, M.; Constantinidis, T.C.; Papadopoulos, N.; Taché, Y.; Minopoulos, G.; Grigoriadis, D.E. Corticotropin-releasing factor (CRF) receptor type 2 in the human stomach: Protective biological role by inhibition of apoptosis. J. Cell. Physiol. 2006, 209, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Larauche, M.; Kiank, C.; Tache, Y. Corticotropin releasing factor signaling in colon and ileum: Regulation by stress and pathophysiological implications. J. Physiol. Pharmacol. 2009, 60 (Suppl. 7), 33–46. [Google Scholar]
- Chatzaki, E.; Murphy, B.J.; Wang, L.; Million, M.; Ohning, G.V.; Crowe, P.D.; Petroski, R.; Taché, Y.; Grigoriadis, D.E. Differential profile of CRF receptor distribution in the rat stomach and duodenum assessed by newly developed CRF receptor antibodies. J. Neurochem. 2004, 88, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chatzaki, E.; Charalampopoulos, I.; Leontidis, C.; Mouzas, I.A.; Tzardi, M.; Tsatsanis, C.; Margioris, A.N.; Gravanis, A. Urocortin in human gastric mucosa: Relationship to inflammatory activity. J. Clin. Endocrinol. Metab. 2003, 88, 478–483. [Google Scholar] [CrossRef] [Green Version]
- Baritaki, S.; de Bree, E.; Chatzaki, E.; Pothoulakis, C. Chronic Stress, Inflammation, and Colon Cancer: A CRH System-Driven Molecular Crosstalk. J. Clin. Med. 2019, 8, 1669. [Google Scholar] [CrossRef] [Green Version]
- Paschos, K.A.; Kolios, G.; Chatzaki, E. The corticotropin-releasing factor system in inflammatory bowel disease: Prospects for new therapeutic approaches. Drug Discov. Today 2009, 14, 713–720. [Google Scholar] [CrossRef]
- Yuan, P.Q.; Wu, S.V.; Elliott, J.; Anton, P.A.; Chatzaki, E.; Million, M.; Taché, Y. Expression of corticotropin releasing factor receptor type 1 (CRF1) in the human gastrointestinal tract and upregulation in the colonic mucosa in patients with ulcerative colitis. Peptides 2012, 38, 62–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatzaki, E.; Anton, P.A.; Million, M.; Lambropoulou, M.; Constantinidis, T.; Kolios, G.; Taché, Y.; Grigoriadis, D.E. Corticotropin-releasing factor receptor subtype 2 in human colonic mucosa: Down-regulation in ulcerative colitis. World J. Gastroenterol. 2013, 19, 1416–1423. [Google Scholar] [CrossRef]
- Chatoo, M.; Li, Y.; Ma, Z.; Coote, J.; Du, J.; Chen, X. Involvement of Corticotropin-Releasing Factor and Receptors in Immune Cells in Irritable Bowel Syndrome. Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Hong, Y.; Dai, L.; Qian, Y.; Zhu, C.; Wu, B.; Li, S. CRH promotes human colon cancer cell proliferation via IL-6/JAK2/STAT3 signaling pathway and VEGF-induced tumor angiogenesis. Mol. Carcinog. 2017, 56, 2434–2445. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Huerta-Yepez, S.; Law, I.K.; Baay-Guzman, G.J.; Tirado-Rodriguez, B.; Hoffman, J.M.; Iliopoulos, D.; Hommes, D.W.; Verspaget, H.W.; Chang, L.; et al. Diminished expression of CRHR2 in human colon cancer promotes tumor growth and EMT via persistent IL-6/Stat3 signaling. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 610–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaprara, A.; Pazaitou-Panayiotou, K.; Kortsaris, A.; Chatzaki, E. The corticotropin releasing factor system in cancer: Expression and pathophysiological implications. Cell. Mol. Life Sci. 2010, 67, 1293–1306. [Google Scholar] [CrossRef]
- Pape, J.C.; Carrillo-Roa, T.; Rothbaum, B.O.; Nemeroff, C.B.; Czamara, D.; Zannas, A.S.; Iosifescu, D.; Mathew, S.J.; Neylan, T.C.; Mayberg, H.S.; et al. DNA methylation levels are associated with CRF(1) receptor antagonist treatment outcome in women with post-traumatic stress disorder. Clin. Epigenetics 2018, 10, 136. [Google Scholar] [CrossRef]
- Schartner, C.; Ziegler, C.; Schiele, M.A.; Kollert, L.; Weber, H.; Zwanzger, P.; Arolt, V.; Pauli, P.; Deckert, J.; Reif, A.; et al. CRHR1 promoter hypomethylation: An epigenetic readout of panic disorder? Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2017, 27, 360–371. [Google Scholar] [CrossRef]
- Koureta, M.; Karaglani, M.; Panagopoulou, M.; Balgkouranidou, I.; Papadaki-Anastasopoulou, A.; Zarouchlioti, C.; Dekavallas, S.; Kolios, G.; Lambropoulou, M.; Baritaki, S.; et al. Corticotropin Releasing Factor Receptors in breast cancer: Expression and activity in hormone-dependent growth in vitro. Peptides 2020, 129, 170316. [Google Scholar] [CrossRef]
- Kobayashi, M.; Matsubara, N.; Nakachi, Y.; Okazaki, Y.; Uchino, M.; Ikeuchi, H.; Song, J.; Kimura, K.; Yasuhara, M.; Babaya, A.; et al. Hypermethylation of Corticotropin Releasing Hormone Receptor-2 Gene in Ulcerative Colitis Associated Colorectal Cancer. In Vivo 2020, 34, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Pothoulakis, C.; Torre-Rojas, M.; Duran-Padilla, M.A.; Gevorkian, J.; Zoras, O.; Chrysos, E.; Chalkiadakis, G.; Baritaki, S. CRHR2/Ucn2 signaling is a novel regulator of miR-7/YY1/Fas circuitry contributing to reversal of colorectal cancer cell resistance to Fas-mediated apoptosis. Int. J. Cancer 2018, 142, 334–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li Yim, A.Y.F.; de Bruyn, J.R.; Duijvis, N.W.; Sharp, C.; Ferrero, E.; de Jonge, W.J.; Wildenberg, M.E.; Mannens, M.; Buskens, C.J.; D’Haens, G.R.; et al. A distinct epigenetic profile distinguishes stenotic from non-inflamed fibroblasts in the ileal mucosa of Crohn’s disease patients. PLoS ONE 2018, 13, e0209656. [Google Scholar] [CrossRef]
- Ishak, M.; Baharudin, R.; Rose, I.M.; Sagap, I.; Mazlan, L.; Azman, Z.A.M.; Abu, N.; Jamal, R.; Lee, L.H.; Mutalib, N.S.A. Genome-Wide Open Chromatin Methylome Profiles in Colorectal Cancer. Biomolecules 2020, 10, 719. [Google Scholar] [CrossRef]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.O.; Park, D.I.; Han, Y.K.; Kang, K.; Park, S.G.; Park, H.R.; Yi, J.M. Genome-Wide Analysis of the DNA Methylation Profile Identifies the Fragile Histidine Triad (FHIT) Gene as a New Promising Biomarker of Crohn’s Disease. J. Clin. Med. 2020, 9, 1338. [Google Scholar] [CrossRef] [PubMed]
- Müller, F.; Scherer, M.; Assenov, Y.; Lutsik, P.; Walter, J.; Lengauer, T.; Bock, C. RnBeads 2.0: Comprehensive analysis of DNA methylation data. Genome Biol. 2019, 20, 55. [Google Scholar] [CrossRef] [Green Version]
- Triche, T.J., Jr.; Weisenberger, D.J.; Van Den Berg, D.; Laird, P.W.; Siegmund, K.D. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 2013, 41, e90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teschendorff, A.E.; Marabita, F.; Lechner, M.; Bartlett, T.; Tegner, J.; Gomez-Cabrero, D.; Beck, S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 2013, 29, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.A.; Lemire, M.; Choufani, S.; Butcher, D.T.; Grafodatskaya, D.; Zanke, B.W.; Gallinger, S.; Hudson, T.J.; Weksberg, R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013, 8, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Du, P.; Zhang, X.; Huang, C.C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef] [Green Version]
- Assenov, Y.; Müller, F.; Lutsik, P.; Walter, J.; Lengauer, T.; Bock, C. Comprehensive analysis of DNA methylation data with RnBeads. Nat. Methods 2014, 11, 1138–1140. [Google Scholar] [CrossRef] [Green Version]
- Li, L.-C.; Dahiya, R. MethPrimer: Designing primers for methylation PCRs. Bioinformatics 2002, 18, 1427–1431. [Google Scholar] [CrossRef] [Green Version]
- Messeguer, X.; Escudero, R.; Farré, D.; Núñez, O.; Martínez, J.; Albà, M.M. PROMO: Detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 2002, 18, 333–334. [Google Scholar] [CrossRef]
- Panagopoulou, M.; Lambropoulou, M.; Balgkouranidou, I.; Nena, E.; Karaglani, M.; Nicolaidou, C.; Asimaki, A.; Konstantinidis, T.; Constantinidis, T.C.; Kolios, G.; et al. Gene promoter methylation and protein expression of BRMS1 in uterine cervix in relation to high-risk human papilloma virus infection and cancer. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2017, 39, 1010428317697557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Tsamardinos, I.; Charonyktakis, P.; Lakiotaki, K.; Borboudakis, G.; Zenklusen, J.C.; Juhl, H.; Chatzaki, E.; Lagani, V. Just Add Data: Automated Predictive Modeling and BioSignature Discovery. bioRxiv 2020. [Google Scholar] [CrossRef]
- Tsamardinos, I.; Greasidou, E.; Borboudakis, G. Bootstrapping the out-of-sample predictions for efficient and accurate cross-validation. Mach. Learn. 2018, 107, 1895–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boser, B.E.; Guyon, I.M.; Vapnik, V.N. A training algorithm for optimal margin classifiers. In Proceedings of the Fifth Annual Workshop on Computational Learning Theory, Pittsburgh, PA, USA, 27–29 July 1992; pp. 144–152. [Google Scholar]
- Hoerl, A.E.; Kennard, R.W. Ridge Regression: Biased Estimation for Nonorthogonal Problems. Technometrics 1970, 12, 55–67. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Moore II, D.H. Classification and Regression Trees; Breiman, L., Friedman, J.H., Olshen, R.A., Stone, C.J., Eds.; Brooks/Cole Publishing: Monterey, CA, USA, 1987; Volume 8, pp. 534–535. [Google Scholar] [CrossRef]
- Brenet, F.; Moh, M.; Funk, P.; Feierstein, E.; Viale, A.J.; Socci, N.D.; Scandura, J.M. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS ONE 2011, 6, e14524. [Google Scholar] [CrossRef]
- Wagner, J.R.; Busche, S.; Ge, B.; Kwan, T.; Pastinen, T.; Blanchette, M. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol. 2014, 15, R37. [Google Scholar] [CrossRef] [Green Version]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [Green Version]
- Tate, P.H.; Bird, A.P. Effects of DNA methylation on DNA-binding proteins and gene expression. Curr. Opin. Genet. Dev. 1993, 3, 226–231. [Google Scholar] [CrossRef]
- Liu, Y.; Fang, X.; Yuan, J.; Sun, Z.; Li, C.; Li, R.; Li, L.; Zhu, C.; Wan, R.; Guo, R.; et al. The role of corticotropin-releasing hormone receptor 1 in the development of colitis-associated cancer in mouse model. Endocr. Relat. Cancer 2014, 21, 639–651. [Google Scholar] [CrossRef] [Green Version]
- Jjingo, D.; Conley, A.B.; Yi, S.V.; Lunyak, V.V.; Jordan, I.K. On the presence and role of human gene-body DNA methylation. Oncotarget 2012, 3, 462–474. [Google Scholar] [CrossRef] [Green Version]
- Aran, D.; Toperoff, G.; Rosenberg, M.; Hellman, A. Replication timing-related and gene body-specific methylation of active human genes. Hum. Mol. Genet. 2011, 20, 670–680. [Google Scholar] [CrossRef]
- Jones, P.A. The DNA methylation paradox. Trends Genet. TIG 1999, 15, 34–37. [Google Scholar] [CrossRef]
- Kulis, M.; Heath, S.; Bibikova, M.; Queirós, A.C.; Navarro, A.; Clot, G.; Martínez-Trillos, A.; Castellano, G.; Brun-Heath, I.; Pinyol, M.; et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat. Genet. 2012, 44, 1236–1242. [Google Scholar] [CrossRef]
- Héberlé, É.; Bardet, A.F. Sensitivity of transcription factors to DNA methylation. Essays Biochem. 2019, 63, 727–741. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Wang, L.; Liu, X.; Wu, Q.; Zhang, H.; Hu, F.; Sun, X. Human corticotrophin releasing factor inhibits cell proliferation and promotes apoptosis through upregulation of tumor protein p53 in human glioma. Oncol. Lett. 2018, 15, 8378–8386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourou, K.; Exarchos, T.P.; Exarchos, K.P.; Karamouzis, M.V.; Fotiadis, D.I. Machine learning applications in cancer prognosis and prediction. Comput. Struct. Biotechnol. J. 2015, 13, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatzaki, E.; Tsamardinos, I. Somatic copy number aberrations detected in circulating tumor DNA can hold diagnostic value for early detection of hepatocellular carcinoma. EBioMedicine 2020, 57, 102851. [Google Scholar] [CrossRef] [PubMed]
- Markaki, M.; Tsamardinos, I.; Langhammer, A.; Lagani, V.; Hveem, K.; Røe, O.D. A Validated Clinical Risk Prediction Model for Lung Cancer in Smokers of All Ages and Exposure Types: A HUNT Study. EBioMedicine 2018, 31, 36–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamou, M.; Antoniou, G.; Greasidou, E.; Lagani, V.; Charonyktakis, P.; Tsamardinos, I.; Doyle, M. Toward Automatic Risk Assessment to Support Suicide Prevention. Crisis 2019, 40, 249–256. [Google Scholar] [CrossRef] [Green Version]
- Karaglani, M.; Gourlia, K.; Tsamardinos, I.; Chatzaki, E. Accurate Blood-Based Diagnostic Biosignatures for Alzheimer’s Disease via Automated Machine Learning. J. Clin. Med. 2020, 9, 3016. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulou, M.; Karaglani, M.; Manolopoulos, V.G.; Iliopoulos, I.; Tsamardinos, I.; Chatzaki, E. Deciphering the Methylation Landscape in Breast Cancer: Diagnostic and Prognostic Biosignatures through Automated Machine Learning. Cancers 2021, 13, 1677. [Google Scholar] [CrossRef] [PubMed]
- Wan, N.; Weinberg, D.; Liu, T.-Y.; Niehaus, K.; Ariazi, E.A.; Delubac, D.; Kannan, A.; White, B.; Bailey, M.; Bertin, M.; et al. Machine learning enables detection of early-stage colorectal cancer by whole-genome sequencing of plasma cell-free DNA. BMC Cancer 2019, 19, 832. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Zhao, Q.; Wei, W.; Zheng, L.; Yi, S.; Li, G.; Wang, W.; Sheng, H.; Pu, H.; Mo, H.; et al. Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer. J. Sci. Transl. Med. 2020, 12, eaax7533. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Platform | Correlated Groups | References |
|---|---|---|---|
| GSE149282 | EPIC | Twelve CRC vs. 12 adjacent colon tissues | [35] |
| GSE122126 | 450k | Four CRC ccfDNAs vs. four healthy ccfDNA | [36] |
| GSE105798 | 450k | Three CD vs. eight normal colon tissues | [37] |
| GSE99788 | EPIC | Thirteen CD vs. five normal colon fibroblasts | [34] |
| Clinical Parameter | Total (n = 113) (%) | Adjuvant Group (n = 42) (%) | Metastatic Group (n = 71) (%) | Normal (n = 20) (%) |
|---|---|---|---|---|
| Age (years) | ||||
| Mean ± SD | 67.0 ± 9.7 | 69.5 ± 8.7 | 65.5 ± 10 | 58.9 (±9.0) |
| Median, range | 68, 44–87 | 70.5, 47–87 | 67, 44–85 | 59, 43–76 |
| Gender | ||||
| Male | 78 (69%) | 30 (71.4%) | 48 (67.6%) | 11 (55%) |
| Female | 35 (31%) | 12 (28.6%) | 23 (32.4%) | 9 (45%) |
| BMI | ||||
| <18.5 | 2 (1.8%) | 0 | 2 (2.8%) | 0 |
| 18.5–24.9 | 12 (10.6%) | 6 (14.3%) | 6 (8.5%) | 8 (40%) |
| 25–29.9 | 35 (31%) | 24 (57.1%) | 11 (15.5%) | 6 (30%) |
| ≥30 | 13 (11.5%) | 11 (26.2%) | 2 (2.8%) | 3 (15%) |
| Not available | 51 (45.1%) | 1 (2.4%) | 50 (70.4%) | 3 (15%) |
| Cancer location | ||||
| R | 19 (16.8%) | 13 (31%) | 6 (8.5%) | |
| A | 7 (6.2%) | 4 (9.5%) | 3 (4.2%) | |
| S | 28 (24.8%) | 18 (42.9%) | 10 (14.1%) | |
| T | 2 (1.8%) | 2 (4.8%) | 0 | |
| D | 3 (2.7%) | 3 (7.1%) | 0 | |
| C | 4 (3.5%) | 2 (4.8%) | 2 (2.8%) | |
| Νot available | 50 (44.2%) | 0 | 50 (70.4%) | |
| Dukes classification | ||||
| A | 14 (12.4%) | 14 (33.3%) | 0 | |
| B | 14 (12.4%) | 13 (31%) | 0 | |
| C | 13 (11.5%) | 13 (31%) | 0 | |
| D | 69 (61%) | 0 | 71 (100%) | |
| Not available | 3 (2.7%) | 2 (4.8%) | 0 | |
| Astler–Coller classification | ||||
| A | 2 (1.8%) | 2 (4.8%) | 0 | |
| B1 | 12 (10.6%) | 12 (28.6%) | 0 | |
| B2 | 13 (11.5%) | 13 (31%) | 0 | |
| B3 | 0 | 0 | 0 | |
| C1 | 1 (0.9%) | 1 (2.4%) | 0 | |
| C2 | 12 (10.6%) | 12 (28.6%) | 0 | |
| C3 | 0 | 0 | 0 | |
| D | 71 (62.8%) | 0 | 71(100%) | |
| Not available | 2 (1.8%) | 2 (4.8%) | 0 | |
| Stage | ||||
| Ι | 15 (13.3%) | 15 (35.7%) | 0 | |
| ΙΙ | 13 (11.5%) | 13 (31%) | 0 | |
| ΙΙΙ | 13 (11.5%) | 13 (31%) | 0 | |
| IV | 71 (62.8%) | 0 | 71(100%) | |
| Not available | 1 (0.9%) | 1 (2.4%) | 0 | |
| Grade | ||||
| 1 | 41 (36.3%) | 18 (42.9%) | 14 (19.7%) | |
| 2 | 50 (44.2%) | 18 (42.9%) | 41 (57.7%) | |
| 3 | 12 (10.6%) | 5 (11.9%) | 7 (9.9%) | |
| Not available | 10 (8.9%) | 1 (2.4%) | 9 (12.7%) | |
| Tumor size | ||||
| T1 | 3 (2.7%) | 3 (7.15%) | 0 | |
| T2 | 24 (21.2%) | 13 (30.95%) | 11 (15.5%) | |
| T3 | 66 (58.4%) | 21 (50%) | 45 (63.4%) | |
| T4 | 12 (10.6%) | 4 (9.5%) | 8 (11.3%) | |
| Not available | 8 (7.1%) | 1 (2.4%) | 7 (9.8%) | |
| LN status | ||||
| N0 | 52 (46%) | 27 (64.3%) | 0 | |
| N1 | 25 (22.1%) | 9 (21.4%) | 28 (39.4%) | |
| N2 | 26 (23%) | 4 (9.5%) | 35 (49.3%) | |
| Not available | 10 (8.9%) | 2 (4.8%) | 8 (11.3%) | |
| Metastatic site | ||||
| Lung | 21 (18.6%) | 0 | 21 (29.6%) | |
| Liver | 55 (48.7%) | 0 | 55 (77.5%) | |
| Pancreas | 1 (0.9%) | 0 | 1 (1.4%) | |
| Bone | 1 (0.9%) | 0 | 1 (1.4%) | |
| Peritoneum | 10 (8.9%) | 0 | 10 (14%) | |
| Brain | 2 (1.8%) | 0 | 2 (2.8%) | |
| Testis | 1 (0.9%) | 0 | 1 (1.4%) | |
| Uterus | 1 (0.9%) | 0 | 1 (1.4%) |
| STUDY | CpG ID | Compared Study Groups | Mean β-Value 1 * | Mean β-Value 2 * | Δ β-Value # | Methylation, CRC vs. Normal | Gene Location | Location Relative to CpG | FDR |
|---|---|---|---|---|---|---|---|---|---|
| GSE149 282 | cg08473090 | Adjacent vs. CRC tissue | 0.089 | 0.330 | +0.241 | Up | TSS1500 | Island | 3.174 × 10−3 |
| GSE149 282 | cg08929103 | Adjacent vs. CRC tissue | 0.563 | 0.239 | −0.323 | Down | TSS1500 | N shore | 4.453 × 10−5 |
| GSE149 282 | cg11338426 | Adjacent vs. CRC tissue | 0.103 | 0.269 | +0.166 | Up | First Exon | Island | 1.786 × 10−2 |
| GSE149 282 | cg12577105 | Adjacent vs. CRC tissue | 0.047 | 0.163 | +0.116 | Up | TSS1500 | Island | 8.456 × 10−3 |
| GSE149 282 | cg13521908 | Adjacent vs. CRC tissue | 0.077 | 0.249 | +0.172 | Up | First Exon | Island | 3.136 × 10−3 |
| GSE149 282 | cg18757974 | Adjacent vs. CRC tissue | 0.062 | 0.218 | +0.156 | Up | TSS1500 | Island | 7.654 × 10−3 |
| GSE122 2126 | cg08929103 | Healthy vs. CRC ccfDNA | 0.767 | 0.477 | −0.291 | Down | TSS1500 | N shore | 1.261 × 10−3 |
| GSE122 2126 | cg13521908 | Healthy vs. CRC ccfDNA | 0.117 | 0.213 | +0.096 | Up | First Exon | Island | 2.545 × 10−2 |
| STUDY | CpG ID | Compared Study Groups | Mean β-Value 1 * | Mean β-Value 2 * | Δ β-Value # | Methylation, Diseased vs. Normal | Gene Location | Location Relative to CpG | FDR |
|---|---|---|---|---|---|---|---|---|---|
| GSE149 282 | cg01718447 | Adjacent vs. CRC tissue | 0.126 | 0.536 | +0.410 | Up | TSS200 | Island | 2.375 × 10−3 |
| GSE149 282 | cg02712145 | Adjacent vs. CRC tissue | 0.166 | 0.471 | +0.305 | Up | TSS1500 | Island | 2.785 × 10−2 |
| GSE149 282 | cg04922810 | Adjacent vs. CRC tissue | 0.077 | 0.435 | +0.358 | Up | First Exon | Island | 7.024 × 10−3 |
| GSE149 282 | cg07658503 | Adjacent vs. CRC tissue | 0.051 | 0.313 | +0.262 | Up | TSS200 | Island | 3.165 × 10−2 |
| GSE149 282 | cg13094036 | Adjacent vs. CRC tissue | 0.089 | 0.343 | +0.254 | Up | TSS1500 | Island | 3.663 × 10−3 |
| GSE149 282 | cg14896516 | Adjacent vs. CRC tissue | 0.106 | 0.352 | +0.246 | Up | TSS1500 | Island | 1.861 × 10−4 |
| GSE149 282 | cg18266052 | Adjacent vs. CRC tissue | 0.100 | 0.418 | +0.318 | Up | First Exon | Island | 4.872 × 10−2 |
| GSE149 282 | cg21773872 | Adjacent vs. CRC tissue | 0.146 | 0.655 | +0.509 | Up | TSS200 | Island | 1.242 × 10−6 |
| GSE149 282 | cg24214442 | Adjacent vs. CRC tissue | 0.123 | 0.463 | +0.340 | Up | First Exon | Island | 3.225 × 10−6 |
| GSE149 282 | cg24610236 | Adjacent vs. CRC tissue | 0.074 | 0.451 | +0.378 | Up | First Exon | Island | 1.568 × 10−5 |
| GSE149 282 | cg27430726 | Adjacent vs. CRC tissue | 0.133 | 0.457 | +0.325 | Up | First Exon | Island | 7.102 × 10−5 |
| GSE1222126 | cg04863452 | Healthy vs. CRC ccfDNA | 0.053 | 0.110 | +0.057 | Up | TSS200 | Island | 1.065 × 10−3 |
| GSE1222126 | cg04923928 | Healthy vs. CRC ccfDNA | 0.045 | 0.143 | +0.098 | Up | First Exon | Island | 1.665 × 10−4 |
| GSE1222126 | cg15615793 | Healthy vs. CRC ccfDNA | 0.486 | 0.629 | +0.142 | Up | TSS1500 | S_Shore | 9.913 × 10−3 |
| GSE1222126 | cg18351440 | Healthy vs. CRC ccfDNA | 0.885 | 0.806 | −0.079 | Down | TSS1500 | N_Shelf | 3.929 × 10−5 |
| GSE105799 | cg21773872 | Normal vs. CD | 0.089 | 0.041 | −0.049 | Down | TSS200 | Island | 2.386 × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panagopoulou, M.; Cheretaki, A.; Karaglani, M.; Balgkouranidou, I.; Biziota, E.; Amarantidis, K.; Xenidis, N.; Kakolyris, S.; Baritaki, S.; Chatzaki, E. Methylation Status of Corticotropin-Releasing Factor (CRF) Receptor Genes in Colorectal Cancer. J. Clin. Med. 2021, 10, 2680. https://doi.org/10.3390/jcm10122680
Panagopoulou M, Cheretaki A, Karaglani M, Balgkouranidou I, Biziota E, Amarantidis K, Xenidis N, Kakolyris S, Baritaki S, Chatzaki E. Methylation Status of Corticotropin-Releasing Factor (CRF) Receptor Genes in Colorectal Cancer. Journal of Clinical Medicine. 2021; 10(12):2680. https://doi.org/10.3390/jcm10122680
Chicago/Turabian StylePanagopoulou, Maria, Antonia Cheretaki, Makrina Karaglani, Ioanna Balgkouranidou, Eirini Biziota, Kyriakos Amarantidis, Nikolaos Xenidis, Stylianos Kakolyris, Stavroula Baritaki, and Ekaterini Chatzaki. 2021. "Methylation Status of Corticotropin-Releasing Factor (CRF) Receptor Genes in Colorectal Cancer" Journal of Clinical Medicine 10, no. 12: 2680. https://doi.org/10.3390/jcm10122680
APA StylePanagopoulou, M., Cheretaki, A., Karaglani, M., Balgkouranidou, I., Biziota, E., Amarantidis, K., Xenidis, N., Kakolyris, S., Baritaki, S., & Chatzaki, E. (2021). Methylation Status of Corticotropin-Releasing Factor (CRF) Receptor Genes in Colorectal Cancer. Journal of Clinical Medicine, 10(12), 2680. https://doi.org/10.3390/jcm10122680