Severe COVID-19 Lung Infection in Older People and Periodontitis


Abstract
:1. Introduction
2. Pathogenesis of COVID-19
3. Predictors of Severe Lung COVID-19 Infection
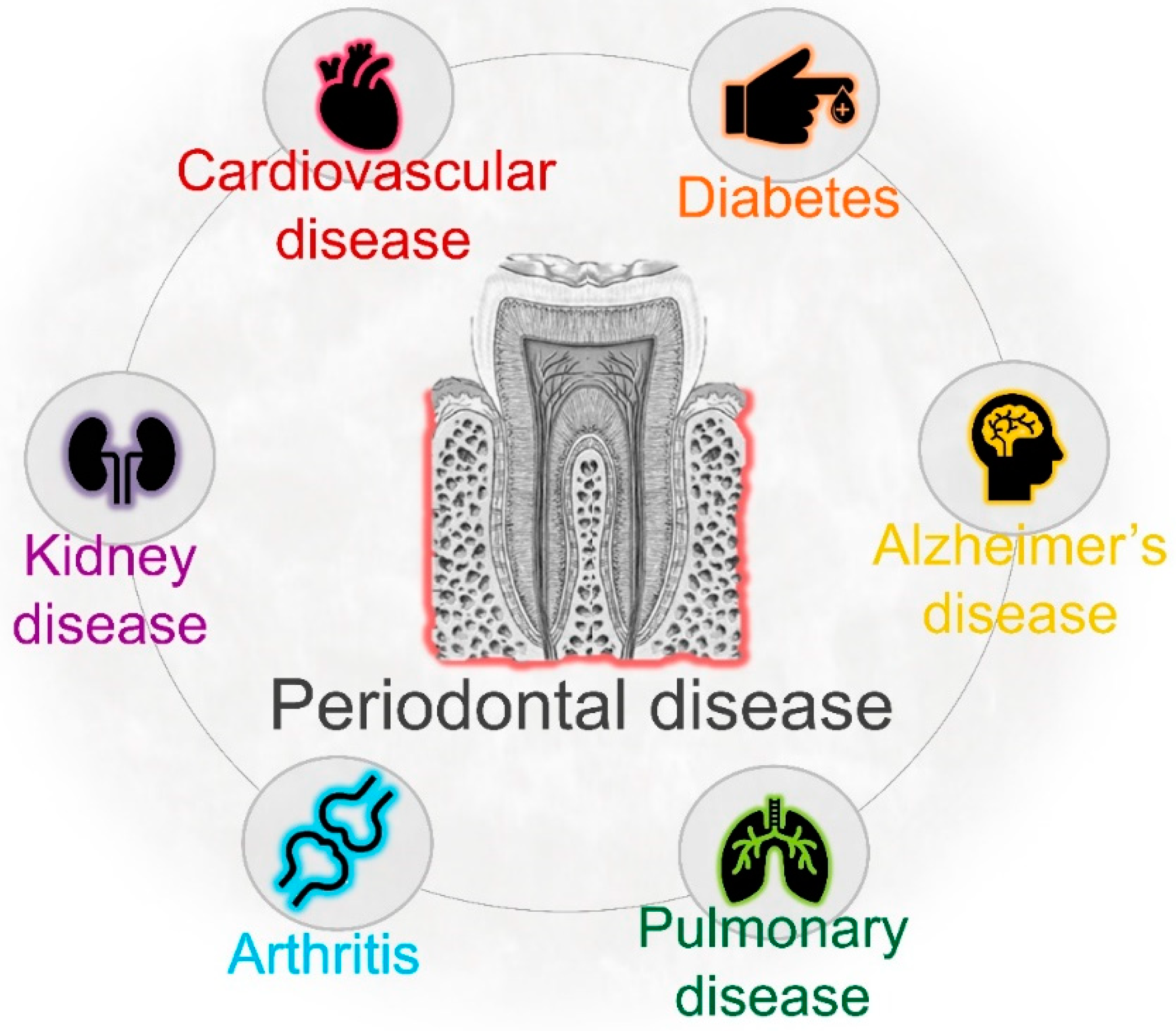
4. Periodontal Infection and Systemic Health
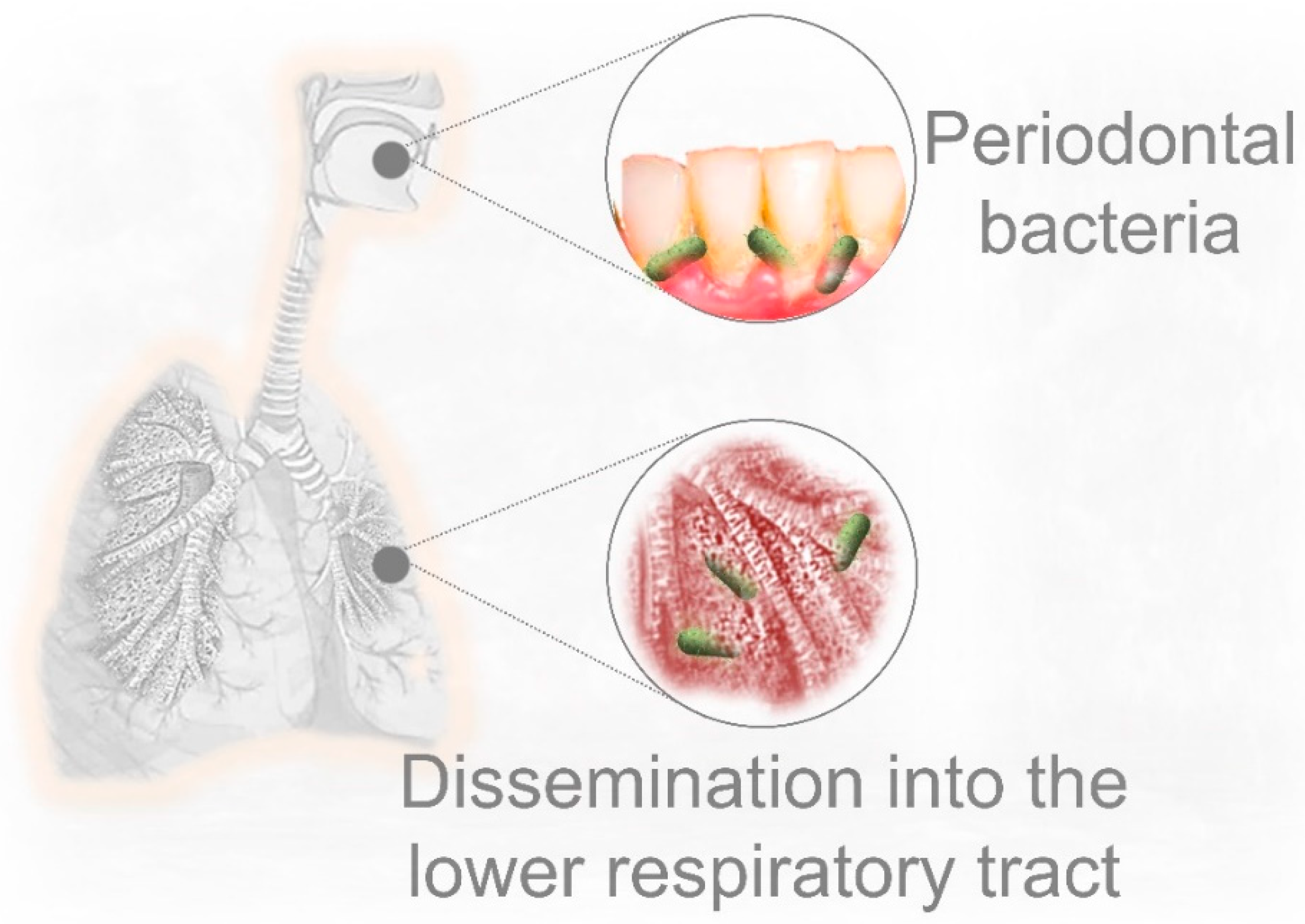
5. Pre-Existing Periodontal Bacterial Infection and Respiratory Disease
6. Could Periodontal Bacteria Dissemination into the Lower Respiratory Tract Contribute to Severe COVID-19 Lung Infection?
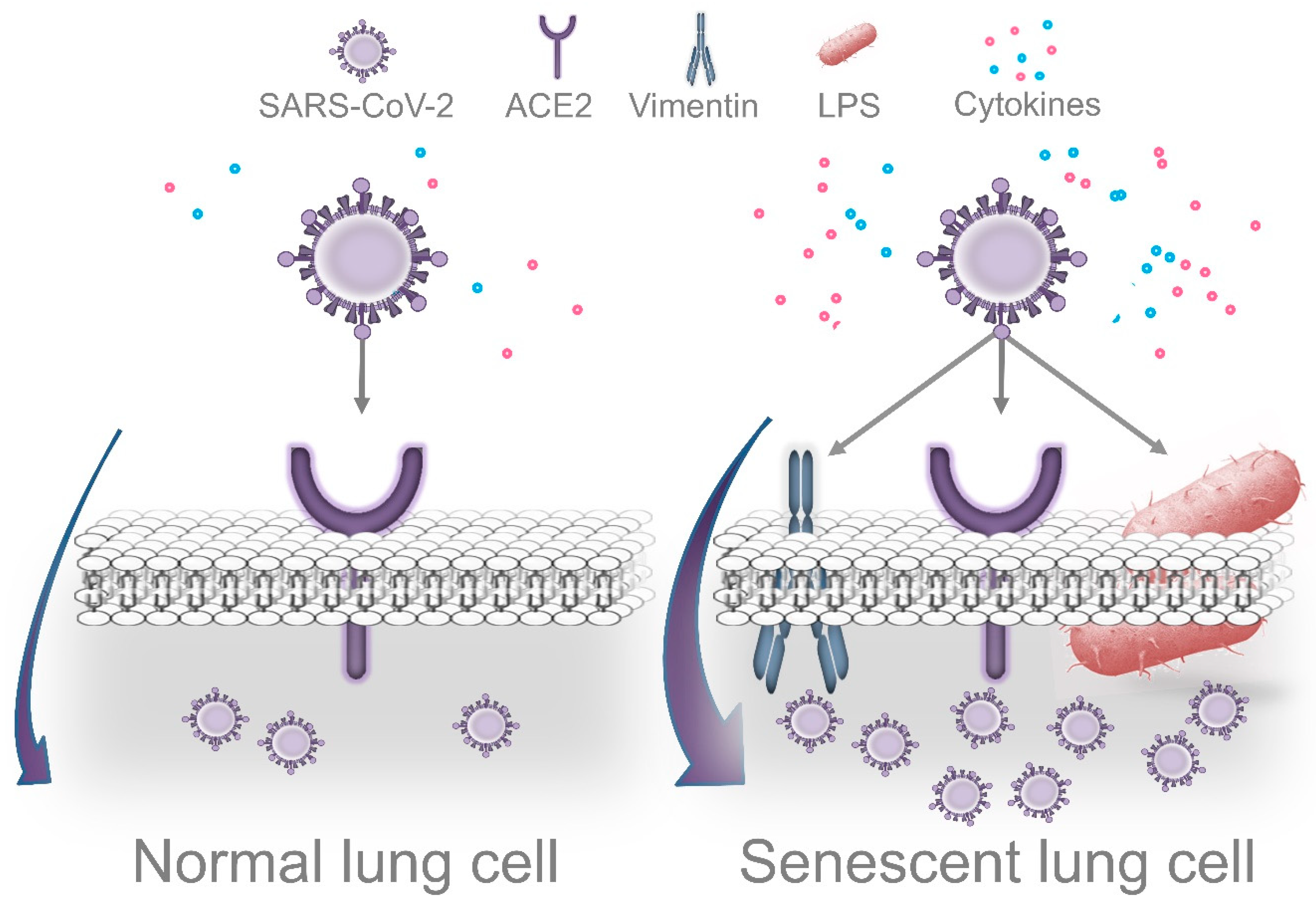
6.1. LPS-Induced Senescence and SARS-CoV-2 Replication
6.2. Periodontal Bacteria and Exacerbation of Lung Inflammation in Response to SARS-CoV-2
6.3. Impaired Immune Surveillance Caused by Periodontal Bacteria and SARS-CoV-2 Replication
7. Periodontal Status and Ventilator-Associated Pneumonia (VAP)
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chakraborty, I.; Maity, P. COVID-19 outbreak: Migration, effects on society, global environment and prevention. Sci. Total Environ. 2020, 728, 138882. [Google Scholar] [CrossRef] [PubMed]
- Mueller, A.L.; Mcnamara, M.S.; Sinclair, D.A. Why does COVID-19 disproportionately affect older people? Aging 2020, 12, 9959–9981. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.; Hirsch, J.S.; Narasimhan, M.; Crawford, J.M.; McGinn, T.; Davidson, K.W.; Barnaby, D.P.; Becker, L.B.; Chelico, J.D.; Cohen, S.L.; et al. Presenting Characteristics, Comorbidities, and Outcomes Among 5700 Patients Hospitalized With COVID-19 in the New York City Area. JAMA 2020, 323, 2052–2059. [Google Scholar] [CrossRef] [PubMed]
- Sanyaolu, A.; Okorie, C.; Marinkovic, A.; Patidar, R.; Younis, K.; Desai, P.; Hosein, Z.; Padda, I.; Mangat, J.; Altaf, M. Comorbidity and its Impact on Patients with COVID-19. SN Compr. Clin. Med. 2020, 2, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Huh, K.; Kang, M.; Hong, J.; Bae, G.H.; Lee, R.; Na, Y.; Choi, H.; Gong, S.Y.; Choi, Y.H.; et al. Effect of underlying comorbidities on the infection and severity of COVID-19 in Korea: A nationwide case-control study. J. Korean Med. Sci. 2020. [Google Scholar] [CrossRef]
- Callender, L.A.; Curran, M.; Bates, S.M.; Mairesse, M.; Weigandt, J.; Betts, C.J. The Impact of Pre-existing Comorbidities and Therapeutic Interventions on COVID-19. Front. Immunol. 2020, 11, 1991. [Google Scholar] [CrossRef]
- Du, Y.; Tu, L.; Zhu, P.; Mu, M.; Wang, R.; Yang, P.; Wang, X.; Hu, C.; Ping, R.; Hu, P.; et al. Clinical features of 85 fatal cases of COVID-19 from Wuhan: A retrospective observational study. Am. J. Respir. Crit. Care Med. 2020, 201, 1372–1379. [Google Scholar] [CrossRef] [Green Version]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Sone, H.; Kagawa, Y. Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia 2005. [Google Scholar] [CrossRef] [PubMed]
- Erusalimsky, J.D.; Kurz, D.J. Cellular senescence in vivo: Its relevance in ageing and cardiovascular disease. Exp. Gerontol. 2005. [Google Scholar] [CrossRef] [PubMed]
- Chilosi, M.; Carloni, A.; Rossi, A.; Poletti, V. Premature lung aging and cellular senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema. Transl. Res. 2013, 162, 156–173. [Google Scholar] [CrossRef] [PubMed]
- Fulop, G.A.; Kiss, T.; Tarantini, S.; Balasubramanian, P.; Yabluchanskiy, A.; Farkas, E.; Bari, F.; Ungvari, Z.; Csiszar, A. Nrf2 deficiency in aged mice exacerbates cellular senescence promoting cerebrovascular inflammation. GeroScience 2018. [Google Scholar] [CrossRef]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef]
- McCulloch, K.; Litherland, G.J.; Rai, T.S. Cellular senescence in osteoarthritis pathology. Aging Cell 2017, 16, 210–218. [Google Scholar] [CrossRef]
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Toussaint, O.; Medrano, E.E.; Von Zglinicki, T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp. Gerontol. 2000, 35, 927–945. [Google Scholar] [CrossRef]
- Feng, X.; Feng, G.; Xing, J.; Shen, B.; Tan, W.; Huang, D.; Lu, X.; Tao, T.; Zhang, J.; Li, L.; et al. Repeated lipopolysaccharide stimulation promotes cellular senescence in human dental pulp stem cells (DPSCs). Cell Tissue Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Nyunoya, T.; Monick, M.M.; Klingelhutz, A.; Yarovinsky, T.O.; Cagley, J.R.; Hunninghake, G.W. Cigarette smoke induces cellular senescence. Am. J. Respir. Cell Mol. Biol. 2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blazkova, H.; Krejcikova, K.; Moudry, P.; Frisan, T.; Hodny, Z.; Bartek, J. Bacterial intoxication evokes cellular senescence with persistent DNA damage and cytokine signalling. J. Cell. Mol. Med. 2010, 14, 357–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasso, F.; Frisan, T. Bacterial Genotoxins: Merging the DNA Damage Response into Infection Biology. Biomolecules 2015, 5, 1762–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, R.C.; Schroeder, H.E. Pathogenesis of inflammatory periodontal disease: A summary of current work. Lab. Investig. 1976, 34, 235–249. [Google Scholar]
- Tonetti, M.S.; Jepsen, S.; Jin, L.; Otomo-Corgel, J. Impact of the global burden of periodontal diseases on health, nutrition and wellbeing of mankind: A call for global action. J. Clin. Periodontol. 2017, 44, 456–462. [Google Scholar] [CrossRef] [Green Version]
- Kassebaum, N.J.; Smith, A.G.C.; Bernabé, E.; Fleming, T.D.; Reynolds, A.E.; Vos, T.; Murray, C.J.L.; Marcenes, W. Global, Regional, and National Prevalence, Incidence, and Disability-Adjusted Life Years for Oral Conditions for 195 Countries, 1990–2015: A Systematic Analysis for the Global Burden of Diseases, Injuries, and Risk Factors. J. Dent. Res. 2017. [Google Scholar] [CrossRef]
- Eke, P.I.; Dye, B.A.; Wei, L.; Thornton-Evans, G.O.; Genco, R.J. Prevalence of periodontitis in adults in the united states: 2009 and 2010. J. Dent. Res. 2012, 91, 914–920. [Google Scholar] [CrossRef]
- Eke, P.I.; Dye, B.A.; Wei, L.; Slade, G.D.; Thornton-Evans, G.O.; Borgnakke, W.S.; Taylor, G.W.; Page, R.C.; Beck, J.D.; Genco, R.J. Update on Prevalence of Periodontitis in Adults in the United States: NHANES 2009 to 2012. J. Periodontol. 2015. [Google Scholar] [CrossRef] [Green Version]
- Garlet, G.P. Critical reviews in oral biology & medicine: Destructive and protective roles of cytokines in periodontitis: A re-appraisal from host defense and tissue destruction viewpoints. J. Dent. Res. 2010, 89, 1349–1363. [Google Scholar] [CrossRef]
- Pan, W.; Wang, Q.; Chen, Q. The cytokine network involved in the host immune response to periodontitis. Int. J. Oral Sci. 2019, 11, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Kolltveit, K.M.; Tronstad, L.; Olsen, I. Systemic diseases caused by oral infection. Clin. Microbiol. Rev. 2000, 13, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, G.; Guiglia, R.; Russo, L.L.; Campisi, G. Dentistry and internal medicine: From the focal infection theory to the periodontal medicine concept. Eur. J. Intern. Med. 2010, 21, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.S. From focal sepsis to periodontal medicine: A century of exploring the role of the oral microbiome in systemic disease. J. Physiol. 2017, 595, 465–476. [Google Scholar] [CrossRef]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019. [Google Scholar] [CrossRef] [Green Version]
- Scannapieco, F.A.; Bush, R.B.; Paju, S. Associations between periodontal disease and risk for nosocomial bacterial pneumonia and chronic obstructive pulmonary disease. A systematic review. Ann. Periodontol. 2003, 8, 54–69. [Google Scholar] [CrossRef] [Green Version]
- Beck, J.; Garcia, R.; Heiss, G.; Vokonas, P.S.; Offenbacher, S. Periodontal Disease and Cardiovascular Disease. J. Periodontol. 1996. [Google Scholar] [CrossRef]
- Taylor, G.W.; Borgnakke, W.S. Periodontal disease: Associations with diabetes, glycemic control and complications. Oral Dis. 2008, 14, 191–203. [Google Scholar] [CrossRef] [Green Version]
- Sfyroeras, G.S.; Roussas, N.; Saleptsis, V.G.; Argyriou, C.; Giannoukas, A.D. Association between periodontal disease and stroke. J. Vasc. Surg. 2012, 55, 1178–1184. [Google Scholar] [CrossRef] [Green Version]
- Kassebaum, N.J.; Bernabé, E.; Dahiya, M.; Bhandari, B.; Murray, C.J.L.; Marcenes, W. Global burden of severe periodontitis in 1990-2010: A systematic review and meta-regression. J. Dent. Res. 2014, 93, 1045–1053. [Google Scholar] [CrossRef]
- López, R.; Smith, P.C.; Göstemeyer, G.; Schwendicke, F. Ageing, dental caries and periodontal diseases. J. Clin. Periodontol. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haytac, M.C.; Ozcelik, O.; Mariotti, A. Periodontal disease in men. Periodontol. 2000 2013, 61, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Ioannidou, E. The sex and gender intersection in chronic periodontitis. Front. Public Health 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, R.G.; Yip, J.K.; Mijares, D.Q.; Boylan, R.J.; Haffajee, A.D.; Socransky, S.S. Destructive periodontal diseases in minority populations. Dent. Clin. N. Am. 2003, 47, 103–114. [Google Scholar] [CrossRef]
- Weatherspoon, D.J.; Borrell, L.N.; Johnson, C.W.; Mujahid, M.S.; Neighbors, H.W.; Adar, S.D. Racial and ethnic differences in self-reported periodontal disease in the multi-ethnic study of atherosclerosis (MESA). Oral Heal. Prev. Dent. 2016. [Google Scholar] [CrossRef]
- Borrell, L.N.; Beck, J.D.; Heiss, G. Socioeconomic disadvantage and periodontal disease: The dental atherosclerosis risk in communities study. Am. J. Public Health 2006. [Google Scholar] [CrossRef] [PubMed]
- Borrell, L.N.; Crawford, N.D. Socioeconomic position indicators and periodontitis: Examining the evidence. Periodontol. 2000 2012. [Google Scholar] [CrossRef]
- Scannapieco, F.A. Role of Oral Bacteria in Respiratory Infection. J. Periodontol. 1999. [Google Scholar] [CrossRef]
- Barros, S.P.; Suruki, R.; Loewy, Z.G.; Beck, J.D.; Offenbacher, S. A Cohort Study of the Impact of Tooth Loss and Periodontal Disease on Respiratory Events among COPD Subjects: Modulatory Role of Systemic Biomarkers of Inflammation. PLoS ONE 2013, 8, e68592. [Google Scholar] [CrossRef]
- Rabi, F.A.; Al Zoubi, M.S.; Al-Nasser, A.D.; Kasasbeh, G.A.; Salameh, D.M. Sars-cov-2 and coronavirus disease 2019: What we know so far. Pathogens 2020, 9, 231. [Google Scholar] [CrossRef]
- Stadnytskyi, V.; Bax, C.E.; Bax, A.; Anfinrud, P. The airborne lifetime of small speech droplets and their potential importance in SARS-CoV-2 transmission. Proc. Natl. Acad. Sci. USA 2020, 117, 11875–11877. [Google Scholar] [CrossRef] [PubMed]
- Jayaweera, M.; Perera, H.; Gunawardana, B.; Manatunge, J. Transmission of COVID-19 virus by droplets and aerosols: A critical review on the unresolved dichotomy. Environ. Res. 2020, 188, 109819. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.B.; Chow, K.; Mathias, R. Dental procedure aerosols and COVID-19. Lancet Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020. [Google Scholar] [CrossRef]
- Bunyavanich, S.; Do, A.; Vicencio, A. Nasal Gene Expression of Angiotensin-Converting Enzyme 2 in Children and Adults. JAMA J. Am. Med. Assoc. 2020, 323, 2427–2429. [Google Scholar] [CrossRef]
- Patel, A.B.; Verma, A. Nasal ACE2 Levels and COVID-19 in Children. JAMA J. Am. Med. Assoc. 2020. [Google Scholar] [CrossRef]
- Belser, J.A. Assessment of SARS-CoV-2 replication in the context of other respiratory viruses. Lancet Respir. Med. 2020, 8, 651–652. [Google Scholar] [CrossRef]
- Chu, H.; Chan, J.F.W.; Wang, Y.; Yuen, T.T.T.; Chai, Y.; Hou, Y.; Shuai, H.; Yang, D.; Hu, B.; Huang, X.; et al. Comparative replication and immune activation profiles of SARS-CoV-2 and SARS-CoV in human lungs: An ex vivo study with implications for the pathogenesis of COVID-19. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.Q.; Tam, J.P.; Liu, D.X. Cell cycle arrest and apoptosis induced by the coronavirus infectious bronchitis virus in the absence of p53. Virology 2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.H.; Huang, M.; Fang, S.G.; Liu, D.X. Coronavirus infection induces DNA replication stress partly through interaction of its nonstructural protein 13 with the p125 subunit of DNA polymerase δ. J. Biol. Chem. 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagga, S.; Bouchard, M.J. Cell cycle regulation during viral infection. Methods Mol. Biol. 2014. [Google Scholar] [CrossRef]
- Luftig, M.A. Viruses and the DNA Damage Response: Activation and Antagonism. Annu. Rev. Virol. 2014. [Google Scholar] [CrossRef] [Green Version]
- Bouhaddou, M.; Memon, D.; Meyer, B.; White, K.M.; Rezelj, V.V.; Marrero, M.C.; Polacco, B.J.; Melnyk, J.E.; Ulferts, S.; Kaake, R.M.; et al. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell 2020. [Google Scholar] [CrossRef]
- Mogensen, T.H.; Paludan, S.R. Molecular Pathways in Virus-Induced Cytokine Production. Microbiol. Mol. Biol. Rev. 2001. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020. [Google Scholar] [CrossRef]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodríguez, L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020. [Google Scholar] [CrossRef]
- Catanzaro, M.; Fagiani, F.; Racchi, M.; Corsini, E.; Govoni, S.; Lanni, C. Immune response in COVID-19: Addressing a pharmacological challenge by targeting pathways triggered by SARS-CoV-2. Signal. Transduct. Target. Ther. 2020, 5, 84. [Google Scholar] [CrossRef] [PubMed]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the Eye of the Cytokine Storm. Microbiol. Mol. Biol. Rev. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, P.; Li, W.; Xie, J.; Hou, Y.; You, C. Cytokine storm induced by SARS-CoV-2. Clin. Chim. Acta 2020. [Google Scholar] [CrossRef] [PubMed]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Atkins, J.L.; Masoli, J.A.H.; Delgado, J.; Pilling, L.C.; Kuo, C.-L.; Kuchel, G.A.; Melzer, D. Preexisting Comorbidities Predicting COVID-19 and Mortality in the UK Biobank Community Cohort. J. Gerontol. Ser. A 2020. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, Q.; Chi, J.; Dong, B.; Lv, W.; Shen, L.; Wang, Y. Comorbidities and the risk of severe or fatal outcomes associated with coronavirus disease 2019: A systematic review and meta-analysis. Int. J. Infect. Dis. 2020, 99, 47–56. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Jain, V.; Yuan, J.M. Predictive symptoms and comorbidities for severe COVID-19 and intensive care unit admission: A systematic review and meta-analysis. Int. J. Public Health 2020. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated with Acute Respiratory Distress Syndrome and Death in Patients with Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Domingues, R.; Lippi, A.; Setz, C.; Outeiro, T.F.; Krisko, A. SARS-CoV-2, immunosenescence and inflammaging: Partners in the COVID-19 crime. Aging 2020, 12, 18778–18789. [Google Scholar] [CrossRef]
- Tan, L.; Wang, Q.; Zhang, D.; Ding, J.; Huang, Q.; Tang, Y.Q.; Wang, Q.; Miao, H. Lymphopenia predicts disease severity of COVID-19: A descriptive and predictive study. Signal. Transduct. Target. Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bhatraju, P.K.; Ghassemieh, B.J.; Nichols, M.; Kim, R.; Jerome, K.R.; Nalla, A.K.; Greninger, A.L.; Pipavath, S.; Wurfel, M.M.; Evans, L.; et al. COVID-19 in critically ill patients in the Seattle region—Case series. N. Engl. J. Med. 2020, 382, 2012–2022. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cai, H.; Hu, J.; Lian, J.; Gu, J.; Zhang, S.; Ye, C.; Lu, Y.; Jin, C.; Yu, G.; et al. Epidemiological, clinical characteristics of cases of SARS-CoV-2 infection with abnormal imaging findings. Int. J. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bernheim, A.; Mei, X.; Huang, M.; Yang, Y.; Fayad, Z.A.; Zhang, N.; Diao, K.; Lin, B.; Zhu, X.; Li, K.; et al. Chest CT findings in coronavirus disease 2019 (COVID-19): Relationship to duration of infection. Radiology 2020, 295, 685–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, T.; Murakami, M. COVID-19: A New Virus, but a Familiar Receptor and Cytokine Release Syndrome. Immunity 2020. [Google Scholar] [CrossRef]
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A.; et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, L.M.; Wan, L.; Xiang, T.X.; Le, A.; Liu, J.M.; Peiris, M.; Poon, L.L.M.; Zhang, W. Viral dynamics in mild and severe cases of COVID-19. Lancet Infect. Dis. 2020, 20, 656–657. [Google Scholar] [CrossRef] [Green Version]
- Forner, L.; Larsen, T.; Kilian, M.; Holmstrup, P. Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. J. Clin. Periodontol. 2006. [Google Scholar] [CrossRef]
- Kinane, D.F.; Riggio, M.P.; Walker, K.F.; MacKenzie, D.; Shearer, B. Bacteraemia following periodontal procedures. J. Clin. Periodontol. 2005. [Google Scholar] [CrossRef]
- Lockhart, P.B.; Brennan, M.T.; Sasser, H.C.; Fox, P.C.; Paster, B.J.; Bahrani-Mougeot, F.K. Bacteremia associated with toothbrushing and dental extraction. Circulation 2008. [Google Scholar] [CrossRef] [Green Version]
- Bahrani-Mougeot, F.K.; Paster, B.J.; Coleman, S.; Ashar, J.; Barbuto, S.; Lockhart, P.B. Diverse and novel oral bacterial species in blood following dental procedures. J. Clin. Microbiol. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozarov, E.V.; Dorn, B.R.; Shelburne, C.E.; Dunn, W.A.; Progulske-Fox, A. Human atherosclerotic plaque contains viable invasive Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, e17–e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figuero, E.; Sánchez-Beltrán, M.; Cuesta-Frechoso, S.; Tejerina, J.M.; del Castro, J.A.; Gutiérrez, J.M.; Herrera, D.; Sanz, M. Detection of Periodontal Bacteria in Atheromatous Plaque by Nested Polymerase Chain Reaction. J. Periodontol. 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belstrøm, D. The salivary microbiota in health and disease. J. Oral Microbiol. 2020, 12, 1723975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, Y.; Watanabe, N.; Kamio, N.; Kobayashi, R.; Iinuma, T.; Imai, K. Aspiration of periodontopathic bacteria due to poor oral hygiene potentially contributes to the aggravation of COVID-19. J. Oral Sci. 2020. [Google Scholar] [CrossRef]
- Yoon, J.G.; Yoon, J.; Song, J.Y.; Yoon, S.Y.; Lim, C.S.; Seong, H.; Noh, J.Y.; Cheong, H.J.; Kim, W.J. Clinical significance of a high SARS-CoV-2 viral load in the Saliva. J. Korean Med. Sci. 2020. [Google Scholar] [CrossRef]
- Carrouel, F.; Gonçalves, L.S.; Conte, M.P.; Campus, G.; Fisher, J.; Fraticelli, L.; Gadea-Deschamps, E.; Ottolenghi, L.; Bourgeois, D. Antiviral Activity of Reagents in Mouth Rinses against SARS-CoV-2. J. Dent. Res. 2020. [Google Scholar] [CrossRef]
- Bourgeois, D.; Inquimbert, C.; Ottolenghi, L.; Carrouel, F. Periodontal pathogens as risk factors of cardiovascular diseases, diabetes, rheumatoid arthritis, cancer, and chronic obstructive pulmonary disease—Is there cause for consideration? Microorganisms 2019, 7, 424. [Google Scholar] [CrossRef] [Green Version]
- Page, R.C. The pathobiology of periodontal diseases may affect systemic diseases: Inversion of a paradigm. Ann. Periodontol. 1998, 3, 108–120. [Google Scholar] [CrossRef]
- Inoue, K.I.; Takano, H.; Shimada, A.; Yanagisawa, R.; Sakurai, M.; Yoshino, S.; Sato, H.; Yoshikawa, T. Urinary trypsin inhibitor protects against systemic inflammation induced by lipopolysaccharide. Mol. Pharmacol. 2005. [Google Scholar] [CrossRef]
- Slofstra, S.H.; ten Cate, H.; Spek, C.A. Low dose endotoxin priming is accountable for coagulation abnormalities and organ damage observed in the Shwartzman reaction. A comparison between a single-dose endotoxemia model and a double-hit endotoxin-induced Shwartzman reaction. Thromb. J. 2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levi, M.; Keller, T.T.; Van Gorp, E.; Ten Cate, H. Infection and inflammation and the coagulation system. Cardiovasc. Res. 2003, 60, 26–39. [Google Scholar] [CrossRef]
- Poole, S.; Singhrao, S.K.; Kesavalu, L.; Curtis, M.A.; Crean, S.J. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J. Alzheimer’s Dis. 2013. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yu, C.; Zhang, X.; Chen, H.; Dong, J.; Lu, W.; Song, Z.; Zhou, W. Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signaling pathway in C57BL/6 mice. J. Neuroinflammation 2018. [Google Scholar] [CrossRef] [PubMed]
- Bonnington, K.E.; Kuehn, M.J. Protein selection and export via outer membrane vesicles. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 1612–1619. [Google Scholar] [CrossRef] [Green Version]
- Reis, C.; Da Costa, A.V.; Guimarães, J.T.; Tuna, D.; Braga, A.C.; Pacheco, J.J.; Arosa, F.A.; Salazar, F.; Cardoso, E.M. Clinical improvement following therapy for periodontitis: Association with a decrease in IL-1 and IL-6. Exp. Ther. Med. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konkel, J.E.; O’Boyle, C.; Krishnan, S. Distal consequences of oral inflammation. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Mojon, P. Oral health and respiratory infection. J. Can. Dent. Assoc. 2002, 68, 340–345. [Google Scholar]
- Scannapieco, F.A.; Ho, A.W. Potential Associations Between Chronic Respiratory Disease and Periodontal Disease: Analysis of National Health and Nutrition Examination Survey III. J. Periodontol. 2001. [Google Scholar] [CrossRef]
- Savitt, E.D.; Kent, R.L. Distribution of Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis by Subject Age. J. Periodontol. 1991. [Google Scholar] [CrossRef]
- Okuda, K.; Kimizuka, R.; Abe, S.; Kato, T.; Ishihara, K. Involvement of Periodontopathic Anaerobes in Aspiration Pneumonia. J. Periodontol. 2005. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Wang, M.; Bagby, G.J.; Nelson, S. Importance of TLR2 in Early Innate Immune Response to Acute Pulmonary Infection with Porphyromonas gingivalis in Mice. J. Immunol. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldas, R.R.; Le Gall, F.; Revert, K.; Rault, G.; Virmaux, M.; Gouriou, S.; Héry-Arnaud, G.; Barbier, G.; Boisramé, S. Pseudomonas aeruginosa and periodontal pathogens in the oral cavity and lungs of cystic fibrosis patients: A case-control study. J. Clin. Microbiol. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, T.C.; Chang, P.Y.; Lin, C.L.; Chen, C.H.; Tu, C.Y.; Hsia, T.C.; Shih, C.M.; Hsu, W.H.; Sung, F.C.; Kao, C.H. Periodontal Treatment Reduces Risk of Adverse Respiratory Events in Patients with Chronic Obstructive Pulmonary Disease. Medicine 2016, 95, 3735. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Han, J.; Liu, Z.; Song, Y.; Wang, Z.; Sun, Z. Effects of periodontal treatment on lung function and exacerbation frequency in patients with chronic obstructive pulmonary disease and chronic periodontitis: A 2-year pilot randomized controlled trial. J. Clin. Periodontol. 2014. [Google Scholar] [CrossRef]
- Sodhi, C.P.; Wohlford-Lenane, C.; Yamaguchi, Y.; Prindle, T.; Fulton, W.B.; Wang, S.; McCray, P.B.; Chappell, M.; Hackam, D.J.; Jia, H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-arg9 bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, W.; Zhang, J.; Zhou, X.; Zhang, L.; Song, Y.; Wang, Z. Oral hygiene, periodontal health and chronic obstructive pulmonary disease exacerbations. J. Clin. Periodontol. 2012. [Google Scholar] [CrossRef]
- Van Der Maarel-Wierink, C.D.; Vanobbergen, J.N.O.; Bronkhorst, E.M.; Schols, J.M.G.A.; De Baat, C. Oral health care and aspiration pneumonia in frail older people: A systematic literature review. Gerodontology 2013, 30, 3–9. [Google Scholar] [CrossRef]
- Terpenning, M. Geriatric oral health and pneumonia risk. Clin. Infect. Dis. 2005, 40, 1807–1810. [Google Scholar] [CrossRef] [Green Version]
- Benedyk, M.; Mydel, P.M.; Delaleu, N.; Płaza, K.; Gawron, K.; Milewska, A.; Maresz, K.; Koziel, J.; Pyrc, K.; Potempa, J. Gingipains: Critical Factors in the Development of Aspiration Pneumonia Caused by Porphyromonas gingivalis. J. Innate Immun. 2016. [Google Scholar] [CrossRef]
- Shen, Z.; Xiao, Y.; Kang, L.; Ma, W.; Shi, L.; Zhang, L.; Zhou, Z.; Yang, J.; Zhong, J.; Yang, D.; et al. Genomic diversity of SARS-CoV-2 in Coronavirus Disease 2019 patients. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Chen, X. Effect of lipopolysaccharides on adipogenic potential and premature senescence of adipocyte progenitors. Am. J. Physiol. Endocrinol. Metab. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.M.; Zhao, Y.M.; Luo, X.G.; Feng, Y.; Ren, Y.; Shang, H.; He, Z.Y.; Luo, X.M.; Chen, S.D.; Wang, X.Y. Repeated lipopolysaccharide stimulation induces cellular senescence in BV2 cells. Neuroimmunomodulation 2012. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.O.; Huh, A.J.; Han, S.H.; Kim, J.M. Analysis of cellular senescence induced by lipopolysaccharide in pulmonary alveolar epithelial cells. Arch. Gerontol. Geriatr. 2012. [Google Scholar] [CrossRef]
- Aquino-Martinez, R.; Rowsey, J.L.; Fraser, D.G.; Eckhardt, B.A.; Khosla, S.; Farr, J.N.; Monroe, D.G. LPS-induced premature osteocyte senescence: Implications in inflammatory alveolar bone loss and periodontal disease pathogenesis. Bone 2020, 132, 115220. [Google Scholar] [CrossRef]
- Aquino-Martinez, R.; Khosla, S.; Farr, J.N.; Monroe, D.G. Periodontal disease and senescent cells: New players for an old oral health problem? Int. J. Mol. Sci. 2020, 21, 7441. [Google Scholar] [CrossRef]
- Shivshankar, P.; Boyd, A.R.; Le Saux, C.J.; Yeh, I.T.; Orihuela, C.J. Cellular senescence increases expression of bacterial ligands in the lungs and is positively correlated with increased susceptibility to pneumococcal pneumonia. Aging Cell 2011. [Google Scholar] [CrossRef] [Green Version]
- Nishio, K.; Inoue, A.; Qiao, S.; Kondo, H.; Mimura, A. Senescence and cytoskeleton: Overproduction of vimentin induces senescent-like morphology in human fibroblasts. Histochem. Cell Biol. 2001. [Google Scholar] [CrossRef]
- Yu, Y.T.C.; Chien, S.C.; Chen, I.Y.; Lai, C.T.; Tsay, Y.G.; Chang, S.C.; Chang, M.F. Surface vimentin is critical for the cell entry of SARS-CoV. J. Biomed. Sci. 2016. [Google Scholar] [CrossRef] [Green Version]
- Ramos, I.; Stamatakis, K.; Oeste, C.L.; Pérez-Sala, D. Vimentin as a multifaceted player and potential therapeutic target in viral infections. Int. J. Mol. Sci. 2020, 21, 4675. [Google Scholar] [CrossRef]
- Li, Z.; Paulin, D.; Lacolley, P.; Coletti, D.; Agbulut, O. Vimentin as a target for the treatment of COVID-19. BMJ Open Respir. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kara, C.; Çelen, K.; Dede, F.Ö.; Gökmenoğlu, C.; Kara, N.B. Is periodontal disease a risk factor for developing severe Covid-19 infection? The potential role of Galectin-3. Exp. Biol. Med. 2020, 245, 1425–1427. [Google Scholar] [CrossRef] [PubMed]
- Openo, K.P.; Kadrofske, M.M.; Patterson, R.J.; Wang, J.L. Galectin-3 expression and subcellular localization in senescent human fibroblasts. Exp. Cell Res. 2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Xu, X.; Wang, L.; Liu, G.; Li, Y.; Wu, X.; Jing, Y.; Li, H.; Wang, G. Senescent mesenchymal stem cells promote colorectal cancer cells growth via galectin-3 expression. Cell Biosci. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Alvarez, L.; Ortega, E. The Many Roles of Galectin-3, a Multifaceted Molecule, in Innate Immune Responses against Pathogens. Mediat. Inflamm. 2017, 2017, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Lewinska, A.; Wnuk, M. Helicobacter pylori-induced premature senescence of extragastric cells may contribute to chronic skin diseases. Biogerontology 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, M.; Thomas, R.J.; Atherton, J.; Roberts, I.S.; High, N.J. Galectin-3 binds to Helicobacter pylori O-antigen: It is upregulated and rapidly secreted by gastric epithelial cells in response to H. pylori adhesion. Cell. Microbiol. 2006, 8, 44–54. [Google Scholar] [CrossRef]
- Freund, A.; Patil, C.K.; Campisi, J. P38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011. [Google Scholar] [CrossRef] [Green Version]
- Iwasa, H.; Han, J.; Ishikawa, F. Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells 2003, 8, 131–144. [Google Scholar] [CrossRef]
- Kim, J.A.; Seong, R.K.; Shin, O.S. Enhanced viral replication by cellular replicative senescence. Immune Netw. 2016. [Google Scholar] [CrossRef] [Green Version]
- Hamming, I.; Timens, W.; Bulthuis, M.L.C.; Lely, A.T.; Navis, G.J.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, Z.; Wang, Y.; Zhou, Y.; Ma, Y.; Zuo, W. Single-Cell RNA Expression Profiling of ACE2, the Receptor of SARS-CoV-2. Am. J. Respir. Crit. Care Med. 2020, 202, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Calò, L.A.; Rigato, M.; Bertoldi, G. ACE2/Angiotensin 1-7 protective anti-inflammatory and antioxidant role in hyperoxic lung injury: Support from studies in Bartter’s and Gitelman’s syndromes. QJM 2020. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Yu, C.H.; Li, W.; Li, T.; Luo, W.; Huang, S.; Wu, P.S.; Cai, S.X.; Li, X. Angiotensin-converting enzyme 2/angiotensin-(1-7)/mas axis protects against lung fibrosis by inhibiting the MAPK/NF-κB pathway. Am. J. Respir. Cell Mol. Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Gao, F.; Liu, Z. Angiotensin-converting enzyme 2 attenuates inflammatory response and oxidative stress in hyperoxic lung injury by regulating NF-jB and Nrf2 pathways. QJM 2019. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005. [Google Scholar] [CrossRef]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Cao, Y.; Zeng, Z.; Liang, M.; Xue, Y.; Xi, C.; Zhou, M.; Jiang, W. Angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas axis prevents lipopolysaccharide-induced apoptosis of pulmonary microvascular endothelial cells by inhibiting JNK/NF-κB pathways. Sci. Rep. 2015. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zeng, Z.; Cao, Y.; Liu, Y.; Ping, F.; Liang, M.; Xue, Y.; Xi, C.; Zhou, M.; Jiang, W. Angiotensin-converting enzyme 2 prevents lipopolysaccharide-induced rat acute lung injury via suppressing the ERK1/2 and NF-κB signaling pathways. Sci. Rep. 2016. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.H.; Chapin, O.C.; Johnson, M.D. LPS-stimulated cytokine production in Type I cells is modulated by the renin-angiotensin system. Am. J. Respir. Cell Mol. Biol. 2012. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005. [Google Scholar] [CrossRef] [PubMed]
- Petruk, G.; Puthia, M.; Petrlova, J.; Strömdahl, A.-C.; Kjellström, S.; Schmidtchen, A. SARS-CoV-2 Spike protein binds to bacterial lipopolysaccharide and boosts proinflammatory activity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ye, R.; Liu, Z. ACE2 exhibits protective effects against LPS-induced acute lung injury in mice by inhibiting the LPS-TLR4 pathway. Exp. Mol. Pathol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Sahni, V.; Gupta, S. COVID-19 & Periodontitis: The cytokine connection. Med. Hypotheses 2020, 144. [Google Scholar] [CrossRef]
- Pacha, O.; Sallman, M.A.; Evans, S.E. COVID-19: A case for inhibiting IL-17? Nat. Rev. Immunol. 2020, 20, 345–346. [Google Scholar] [CrossRef]
- Takahashi, K.; Azuma, T.; Motohira, H.; Kinane, D.F.; Kitetsu, S. The potential role of interleukin-17 in the immunopathology of periodontal disease. J. Clin. Periodontol. 2005. [Google Scholar] [CrossRef]
- Hajishengallis, G. Periodontitis: From microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 2015, 15, 30–44. [Google Scholar] [CrossRef]
- Darveau, R.P.; Belton, C.M.; Reife, R.A.; Lamont, R.J. Local Chemokine Paralysis, a Novel Pathogenic Mechanism for Porphyromonas gingivalis. Infect. Immun. 1998, 66, 1660–1665. [Google Scholar] [CrossRef] [Green Version]
- Hajishengallis, G. The inflammophilic character of the periodontitis-associated microbiota. Mol. Oral Microbiol. 2014. [Google Scholar] [CrossRef] [Green Version]
- Hajishengallis, G.; Lamont, R.J. Breaking bad: Manipulation of the host response by Porphyromonas gingivalis. Eur. J. Immunol. 2014. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Michel, R.; Cohen, J.; DeCarlo, A.; Kozarov, E. Intracellular survival and vascular cell-to-cell transmission of Porphyromonas gingivalis. BMC Microbiol. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, I.; Progulske-Fox, A. Invasion of Porphyromonas gingivalis strains into vascular cells and tissue. J. Oral Microbiol. 2015, 7, 28788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eick, S.; Pfister, W. Efficacy of Antibiotics Against Periodontopathogenic Bacteria Within Epithelial Cells: An In Vitro Study. J. Periodontol. 2004. [Google Scholar] [CrossRef] [PubMed]
- Sekot, G.; Posch, G.; Messner, P.; Matejka, M.; Rausch-Fan, X.; Andrukhov, O.; Schäffer, C. Potential of the tannerella forsythia S-layer to delay the immune response. J. Dent. Res. 2011. [Google Scholar] [CrossRef] [Green Version]
- Shenker, B.J.; McKay, T.; Datar, S.; Miller, M.; Chowhan, R.; Demuth, D. Actinobacillus actinomycetemcomitans immunosuppressive protein is a member of the family of cytolethal distending toxins capable of causing a G2 arrest in human T cells. J. Immunol. 1999, 162, 4773–4780. [Google Scholar]
- Rabie, G.; Lally, E.T.; Shenker, B.J. Immunosuppressive properties of Actinobacillus actinomycetemcomitants leukotoxin. Infect. Immun. 1988, 56, 122–127. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Ovadya, Y.; Landsberger, T.; Leins, H.; Vadai, E.; Gal, H.; Biran, A.; Yosef, R.; Sagiv, A.; Agrawal, A.; Shapira, A.; et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun. 2018. [Google Scholar] [CrossRef] [Green Version]
- Goyal, P.; Choi, J.J.; Pinheiro, L.C.; Schenck, E.J.; Chen, R.; Jabri, A.; Satlin, M.J.; Campion, T.R.; Nahid, M.; Ringel, J.B.; et al. Clinical characteristics of COVID-19 in New York City. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Carter, C.; Osborn, M.; Agagah, G.; Aedy, H.; Notter, J. COVID-19 disease: Invasive ventilation. Clin. Integr. Care 2020. [Google Scholar] [CrossRef]
- Meng, L.; Qiu, H.; Wan, L.; Ai, Y.; Xue, Z.; Guo, Q.; Deshpande, R.; Zhang, L.; Meng, J.; Tong, C.; et al. Intubation and Ventilation amid the COVID-19 Outbreak: Wuhan’s Experience. Anesthesiology 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alp, E.; Voss, A. Ventilator associated pneumonia and infection control. Ann. Clin. Microbiol. Antimicrob. 2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chastre, J.; Fagon, J. State of the Art Ventilator-associated Pneumonia. Am. J. Respir Crit Care Med. 2002. [Google Scholar] [CrossRef] [PubMed]
- Kollef, M.H. Prevention of hospital-associated pneumonia and ventilator-associated pneumonia. Crit. Care Med. 2004, 32, 1396–1405. [Google Scholar] [CrossRef]
- Thakuria, B.; Singh, P.; Agrawal, S.; Asthana, V. Profile of infective microorganisms causing ventilator-associated pneumonia: A clinical study from resource limited intensive care unit. J. Anaesthesiol. Clin. Pharmacol. 2013. [Google Scholar] [CrossRef]
- Sarda, C.; Fazal, F.; Rello, J. Management of ventilator-associated pneumonia (VAP) caused by resistant gram-negative bacteria: Which is the best strategy to treat? Expert Rev. Respir. Med. 2019, 13, 787–798. [Google Scholar] [CrossRef]
- Estes, R.J.; Meduri, G.U. The pathogenesis of ventilator-associated pneumonia: I. Mechanisms of bacterial transcolonization and airway inoculation. Intensive Care Med. 1995, 21, 365–383. [Google Scholar] [CrossRef]
- Bahrani-Mougeot, F.K.; Paster, B.J.; Coleman, S.; Barbuto, S.; Brennan, M.T.; Noll, J.; Kennedy, T.; Fox, P.C.; Lockhart, P.B. Molecular analysis of oral and respiratory bacterial species associated with ventilator-associated pneumonia. J. Clin. Microbiol. 2007. [Google Scholar] [CrossRef] [Green Version]
- Hayata, M.; Watanabe, N.; Tamura, M.; Kamio, N.; Tanaka, H.; Nodomi, K.; Miya, C.; Nakayama, E.; Ueda, K.; Ogata, Y.; et al. The periodontopathic bacterium Fusobacterium nucleatum induced proinflammatory cytokine production by human respiratory epithelial cell lines and in the lower respiratory organs in mice. Cell. Physiol. Biochem. 2019. [Google Scholar] [CrossRef] [Green Version]
- Shay, K. Infectious complications of dental and periodontal diseases in the elderly population. Clin. Infect. Dis. 2002. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| References | |
|---|---|
| Prevalence and severity increased in elderly people | [28,40,41] |
| Prevalence significantly higher in males | [28,42,43] |
| Highest prevalence in racial and ethnic minorities | [28,44,45] |
| Higher severity associated with lower socioeconomic status and education | [28,46,47] |
| Association with respiratory infection (pneumonia and COPD) | [36,48,49] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aquino-Martinez, R.; Hernández-Vigueras, S. Severe COVID-19 Lung Infection in Older People and Periodontitis. J. Clin. Med. 2021, 10, 279. https://doi.org/10.3390/jcm10020279
Aquino-Martinez R, Hernández-Vigueras S. Severe COVID-19 Lung Infection in Older People and Periodontitis. Journal of Clinical Medicine. 2021; 10(2):279. https://doi.org/10.3390/jcm10020279
Chicago/Turabian StyleAquino-Martinez, Ruben, and Scarlette Hernández-Vigueras. 2021. "Severe COVID-19 Lung Infection in Older People and Periodontitis" Journal of Clinical Medicine 10, no. 2: 279. https://doi.org/10.3390/jcm10020279
APA StyleAquino-Martinez, R., & Hernández-Vigueras, S. (2021). Severe COVID-19 Lung Infection in Older People and Periodontitis. Journal of Clinical Medicine, 10(2), 279. https://doi.org/10.3390/jcm10020279