
Diagnosis and Risk Prediction of Dilated Cardiomyopathy in the Era of Big Data and Genomics

 and
and 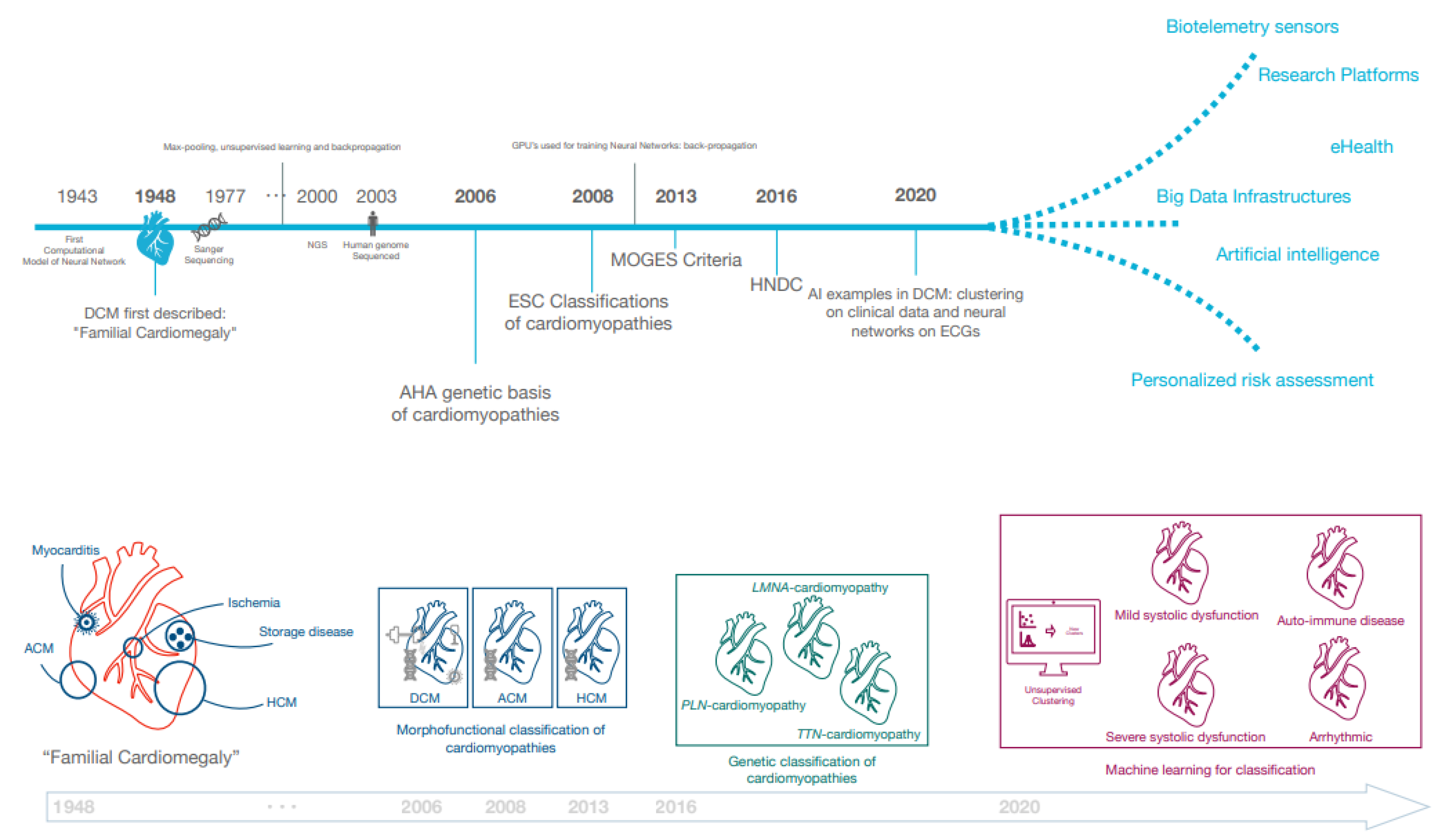{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Defining DCM
2.1. Historic Overview of DCM Definitions
2.2. Diagnosis of DCM and Differential Diagnostic Considerations
3. Classification of DCM in the Era of Genomics
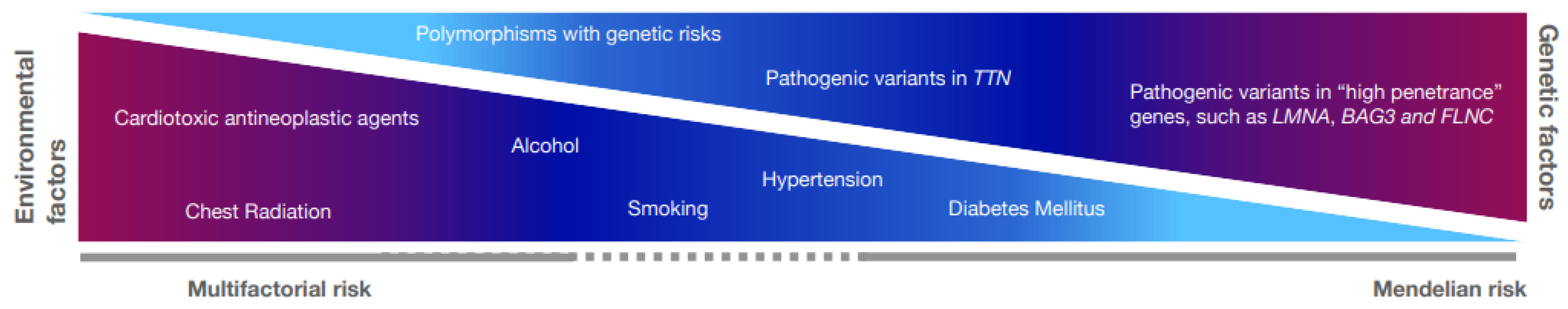
3.1. Genetic Variants in DCM
3.2. Genotype–Phenotype Associations in DCM
3.3. Genome-Wide Association Studies and Genetic Risk Scores in DCM
4. Prognosis of DCM
4.1. Heart Failure and Cardiac Transplantation
4.2. Life-Threatening Ventricular Arrhythmias
5. Big Data Research Opportunities and Artificial Intelligence in DCM
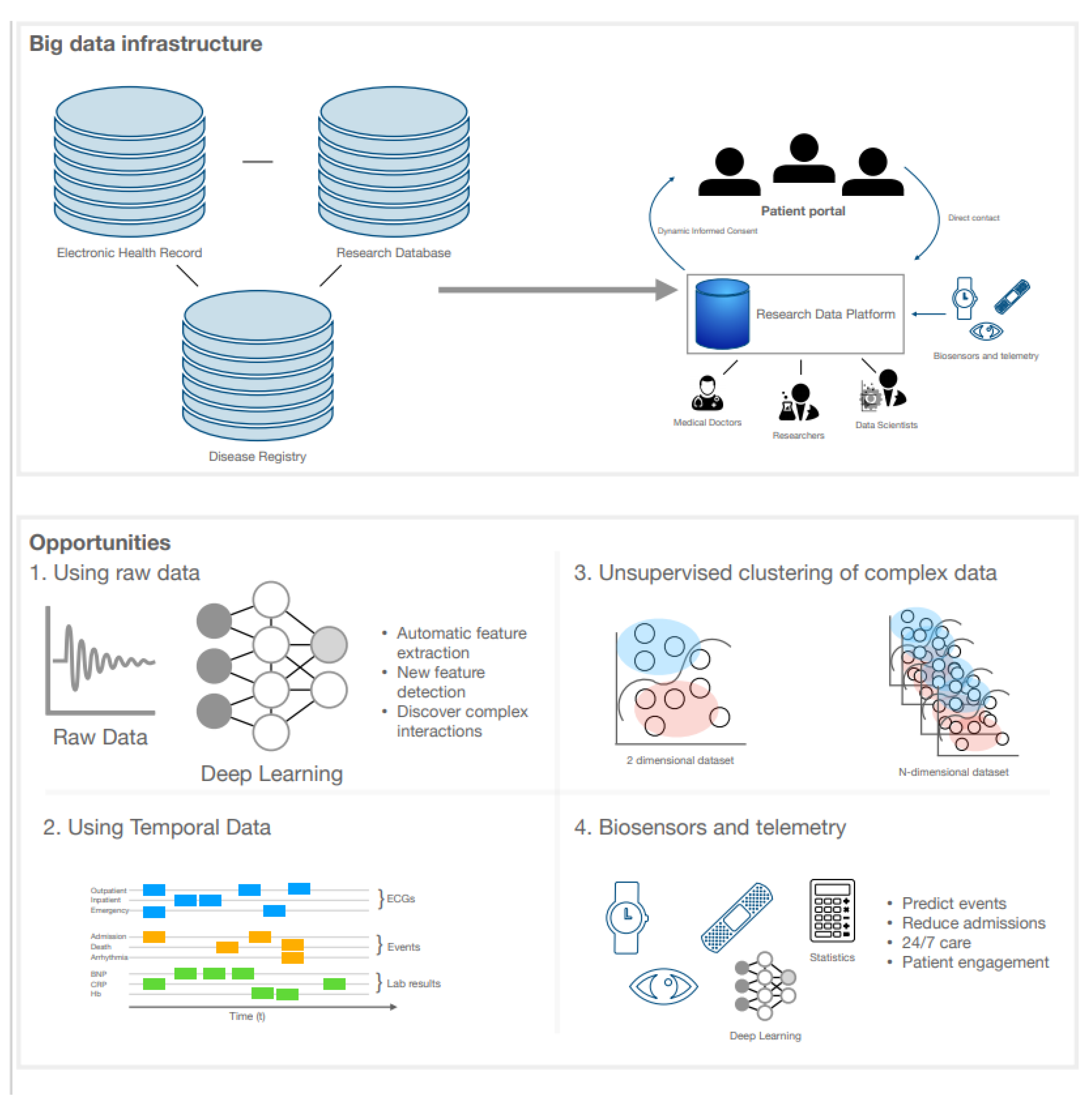
5.1. Big Data Infrastructure
5.2. Clinical Uses of Artificial Intelligence in Cardiomyopathy
5.3. eHealth and Wearables in DCM Management
6. Conclusions
Supplementary Materials
Funding
Conflicts of Interest
References
- McKenna, W.J.; Maron, B.J.; Thiene, G. Classification, Epidemiology, and Global Burden of Cardiomyopathies. Circ. Res. 2017, 121, 722–730. [Google Scholar] [CrossRef] [Green Version]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef]
- Linschoten, M.; Teske, A.J.; Cramer, M.J.; van der Wall, E.; Asselbergs, F.W. Chemotherapy-Related Cardiac Dysfunction: A Systematic Review of Genetic Variants Modulating Individual Risk. Circ. Genom. Precis. Med. 2018, 11, e001753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ware, J.S.; Amor-Salamanca, A.; Tayal, U.; Govind, R.; Serrano, I.; Salazar-Mendiguchía, J.; García-Pinilla, J.M.; Pascual-Figal, D.A.; Nuñez, J.; Guzzo-Merello, G.; et al. Genetic Etiology for Alcohol-Induced Cardiac Toxicity. J. Am. Coll. Cardiol. 2018, 71, 2293–2302. [Google Scholar] [CrossRef]
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Böhm, M.; Duboc, D.; Gimeno, J.; De Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamphuis, J.A.M.; Linschoten, M.; Cramer, M.J.; Doevendans, P.A.; Asselbergs, F.W.; Teske, A.J. Early- and late anthracycline-induced cardiac dysfunction: Echocardiographic characterization and response to heart failure therapy. Cardio-Oncology 2020, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef]
- Sammani, A.; Kayvanpour, E.; Bosman, L.P.; Sedaghat-Hamedani, F.; Proctor, T.; Gi, W.T.; Broezel, A.; Jensen, K.; Katus, H.A.; te Riele, A.S.J.M.; et al. Predicting sustained ventricular arrhythmias in dilated cardiomyopathy: A meta-analysis and systematic review. ESC Heart Fail. 2020, 7, 1430–1441. [Google Scholar] [CrossRef] [Green Version]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.P.J.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed Modification of the Task Force Criteria. Eur. Heart J. 2010, 31, 806–814. [Google Scholar] [CrossRef] [Green Version]
- Jansweijer, J.A.; Hershberger, R.; Van Spaendonck, K.Y. Dilated cardiomyopathy. Clin. Cardiogenetics Second Ed. 2016, 390, 75–89. [Google Scholar] [CrossRef]
- Pugh, T.J.; Kelly, M.A.; Gowrisankar, S.; Hynes, E.; Seidman, M.A.; Baxter, S.M.; Bowser, M.; Harrison, B.; Aaron, D.; Mahanta, L.M.; et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet. Med. 2014, 16, 601–608. [Google Scholar] [CrossRef] [Green Version]
- Begay, R.L.; Graw, S.L.; Sinagra, G.; Asimaki, A.; Rowland, T.J.; Slavov, D.B.; Gowan, K.; Jones, K.L.; Brun, F.; Merlo, M.; et al. Filamin C Truncation Mutations Are Associated With Arrhythmogenic Dilated Cardiomyopathy and Changes in the Cell–Cell Adhesion Structures. JACC Clin. Electrophysiol. 2018, 4, 504–514. [Google Scholar] [CrossRef]
- Kayvanpour, E.; Sedaghat-Hamedani, F.; Amr, A.; Lai, A.; Haas, J.; Holzer, D.B.; Frese, K.S.; Keller, A.; Jensen, K.; Katus, H.A.; et al. Genotype-phenotype associations in dilated cardiomyopathy: Meta-analysis on more than 8000 individuals. Clin. Res. Cardiol. 2017, 106, 127–139. [Google Scholar] [CrossRef]
- Disertori, M.; Gulizia, M.M.; Casolo, G.; Delise, P.; Di Lenarda, A.; Di Tano, G.; Lunati, M.; Mestroni, L.; Salerno-Uriarte, J.; Tavazzi, L. Improving the appropriateness of sudden arrhythmic death primary prevention by implantable cardioverter-defibrillator therapy in patients with low left ventricular ejection fraction. Point of view. J. Cardiovasc. Med. 2016, 17, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Sammani, A.; Jansen, M.; Linschoten, M.; Bagheri, A.; de Jonge, N.; Kirkels, H.; van Laake, L.W.; Vink, A.; van Tintelen, J.P.; Dooijes, D.; et al. UNRAVEL: Big data analytics research data platform to improve care of patients with cardiomyopathies using routine electronic health records and standardised biobanking. Neth. Heart J. 2019, 27, 426–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krittanawong, C.; Zhang, H.J.; Wang, Z.; Aydar, M.; Kitai, T. Artificial Intelligence in Precision Cardiovascular Medicine. J. Am. Coll. Cardiol. 2017, 69, 2657–2664. [Google Scholar] [CrossRef]
- Leur, R.R.; van de Boonstra, M.J.; Bagheri, A.; Roudijk, R.W.; Sammani, A.; Taha, K.; Doevendans, P.A.; Harst, P.; van der Dam, P.M.; van Hassink, R.J.; et al. Big Data and Artificial Intelligence: Opportunities and Threats in Electrophysiology. Arrhythmia Electrophysiol. Rev. 2020, 9, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Alyass, A.; Turcotte, M.; Meyre, D. From big data analysis to personalized medicine for all: Challenges and opportunities. BMC Med. Genom. 2015, 8, 33. [Google Scholar] [CrossRef] [Green Version]
- Obermeyer, Z.; Emanuel, E.J. Predicting the Future—Big Data, Machine Learning, and Clinical Medicine. N. Engl. J. Med. 2016, 375, 1216–1219. [Google Scholar] [CrossRef] [Green Version]
- Evans, W. Familial cardiomegaly. Br. Heart J. 1949, 11, 68–82. [Google Scholar] [CrossRef] [Green Version]
- Schrader, W.H.; Pankey, G.A.; Davis, R.B.; Theologides, A. Familial idiopathic cardiomegaly. Circulation 1961, 24, 599–606. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B. Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functio. Circulation 2006, 113, 1807–1816. [Google Scholar] [CrossRef] [Green Version]
- Thiene, G.; Corrado, D.; Basso, C. Revisiting definition and classification of cardiomyopathies in the era of molecular medicine. Eur. Heart J. 2008, 29, 144–146. [Google Scholar] [CrossRef] [Green Version]
- Arbustini, E.; Narula, N.; Dec, G.W.; Reddy, K.S.; Greenberg, B.; Kushwaha, S.; Marwick, T.; Pinney, S.; Bellazzi, R.; Favalli, V.; et al. The MOGE(S) Classification for a Phenotype–Genotype Nomenclature of Cardiomyopathy. J. Am. Coll. Cardiol. 2013, 62, 2046–2072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubiś, P. The Diagnostic Work-up of Genetic and Inflammatory Dilated Cardiomyopathy. Available online: https://www.escardio.org/Journals/E-Journal-of-Cardiology-Practice/Volume-13/The-diagnostic-work-up-of-genetic-and-inflammatory-dilated-cardiomyopathy (accessed on 15 December 2019).
- Spezzacatene, A.; Sinagra, G.; Merlo, M.; Barbati, G.; Graw, S.L.; Brun, F.; Slavov, D.; Di Lenarda, A.; Salcedo, E.E.; Towbin, J.A.; et al. Arrhythmogenic Phenotype in Dilated Cardiomyopathy: Natural History and Predictors of Life-Threatening Arrhythmias. J. Am. Heart Assoc. 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Zegkos, T.; Panagiotidis, T.; Parcharidou, D.; Efthimiadis, G. Emerging concepts in arrhythmogenic dilated cardiomyopathy. Heart Fail. Rev. 2020. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; Syrris, P.; Prasad, S.K.; Hughes, S.E.; Merrifield, R.; Ward, D.; Pennell, D.J.; McKenna, W.J. Left-Dominant Arrhythmogenic Cardiomyopathy. J. Am. Coll. Cardiol. 2008, 52, 2175–2187. [Google Scholar] [CrossRef] [Green Version]
- Rassi, A.; Rassi, A.; Marin-Neto, J.A. Chagas disease. Lancet 2010, 375, 1388–1402. [Google Scholar] [CrossRef]
- Marques, J.S.; Pinto, F.J. Clinical use of multimodality imaging in the assessment of dilated cardiomyopathy. Heart 2015, 101, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Baughman, K.L. Diagnosis of Myocarditis. Circulation 2006, 113, 593–595. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.T.; Baughman, K.L.; Feldman, A.M.; Frustaci, A.; Jessup, M.; Kuhl, U.; Levine, G.N.; Narula, J.; Starling, R.C.; Towbin, J.; et al. The Role of Endomyocardial Biopsy in the Management of Cardiovascular Disease. J. Am. Coll. Cardiol. 2007, 50, 1914–1931. [Google Scholar] [CrossRef] [Green Version]
- Harakalova, M.; Kummeling, G.; Sammani, A.; Linschoten, M.; Baas, A.F.; Van Der Smagt, J.; Doevendans, P.A.; Van Tintelen, J.P.; Dooijes, D.; Mokry, M.; et al. A systematic analysis of genetic dilated cardiomyopathy reveals numerous ubiquitously expressed and muscle-specific genes. Eur. J. Heart Fail. 2015, 17, 484–493. [Google Scholar] [CrossRef]
- Harakalova, M.; Asselbergs, F.W. Systems analysis of dilated cardiomyopathy in the next generation sequencing era. Wiley Interdiscip. Rev. Syst. Biol. Med. 2018, 10, e1419. [Google Scholar] [CrossRef] [Green Version]
- Milko, L.V.; Funke, B.H.; Hershberger, R.E.; Azzariti, D.R.; Lee, K.; Riggs, E.R.; Rivera-Munoz, E.A.; Weaver, M.A.; Niehaus, A.; Currey, E.L.; et al. Development of Clinical Domain Working Groups for the Clinical Genome Resource (ClinGen): Lessons learned and plans for the future. Genet. Med. 2019, 21, 987–993. [Google Scholar] [CrossRef]
- Morales, A.; Kinnamon, D.D.; Jordan, E.; Platt, J.; Vatta, M.; Dorschner, M.O.; Starkey, C.A.; Mead, J.O.; Ai, T.; Burke, W.; et al. Variant interpretation for dilated cardiomyopathy. Circ. Genom. Precis. Med. 2020, 13, 43–51. [Google Scholar] [CrossRef]
- Asselbergs, F.W.; Sammani, A.; Elliott, P.; Gimeno, J.R.; Tavazzi, L.; Tendera, M.; Kaski, J.P.; Maggioni, A.P.; Rubis, P.P.; Jurcut, R.; et al. Differences between familial and sporadic dilated cardiomyopathy: ESC EORP Cardiomyopathy & Myocarditis registry. ESC Hear. Fail. 2020, ehf2.13100. [Google Scholar] [CrossRef]
- Van Spaendonck-Zwarts, K.Y.; Van Rijsingen, I.A.W.; Van Den Berg, M.P.; Lekanne Deprez, R.H.; Post, J.G.; Van Mil, A.M.; Asselbergs, F.W.; Christiaans, I.; Van Langen, I.M.; Wilde, A.A.M.; et al. Genetic analysis in 418 index patients with idiopathic dilated cardiomyopathy: Overview of 10 years’ experience. Eur. J. Heart Fail. 2013, 15, 628–636. [Google Scholar] [CrossRef]
- Minoche, A.E.; Horvat, C.; Johnson, R.; Gayevskiy, V.; Morton, S.U.; Drew, A.P.; Woo, K.; Statham, A.L.; Lundie, B.; Bagnall, R.D.; et al. Genome sequencing as a first-line genetic test in familial dilated cardiomyopathy. Genet. Med. 2019, 21, 650–662. [Google Scholar] [CrossRef]
- Herman, D.S.; Lam, L.; Taylor, M.R.G.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of Titin Causing Dilated Cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Fatkin, D.; Huttner, I.G. Titin-truncating mutations in dilated cardiomyopathy. Curr. Opin. Cardiol. 2017, 32, 232–238. [Google Scholar] [CrossRef]
- Akinrinade, O.; Koskenvuo, J.W.; Alastalo, T.P. Prevalence of titin truncating variants in general population. PLoS ONE 2015, 10, e0145284. [Google Scholar] [CrossRef] [Green Version]
- Ware, J.S.; Li, J.; Mazaika, E.; Yasso, C.M.; DeSouza, T.; Cappola, T.P.; Tsai, E.J.; Hilfiker-Kleiner, D.; Kamiya, C.A.; Mazzarotto, F.; et al. Shared Genetic Predisposition in Peripartum and Dilated Cardiomyopathies. N. Engl. J. Med. 2016, 374, 233–241. [Google Scholar] [CrossRef]
- Akhtar, M.M.; Lorenzini, M.; Cicerchia, M.; Ochoa, J.P.; Hey, T.M.; Sabater Molina, M.; Restrepo-Cordoba, M.A.; Dal Ferro, M.; Stolfo, D.; Johnson, R.; et al. Clinical phenotypes and prognosis of dilated cardiomyopathy caused by truncating variants in the TTN Gene. Circ. Hear. Fail. 2020, 13, 496–508. [Google Scholar] [CrossRef]
- Jansen, M.; Baas, A.F.; van Spaendonck-Zwarts, K.Y.; Ummels, A.S.; van den Wijngaard, A.; Jongbloed, J.D.H.; van Slegtenhorst, M.A.; Lekanne Deprez, R.H.; Wessels, M.W.; Michels, M.; et al. Mortality Risk Associated With Truncating Founder Mutations in Titin. Circ. Genom. Precis. Med. 2019, 12, e002436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gigli, M.; Merlo, M.; Graw, S.L.; Barbati, G.; Rowland, T.J.; Slavov, D.B.; Stolfo, D.; Haywood, M.E.; Dal Ferro, M.; Altinier, A.; et al. Genetic Risk of Arrhythmic Phenotypes in Patients With Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2019, 74, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- Jansweijer, J.A.; Nieuwhof, K.; Russo, F.; Hoorntje, E.T.; Jongbloed, J.D.H.; Lekanne Deprez, R.H.; Postma, A.V.; Bronk, M.; van Rijsingen, I.A.W.; de Haij, S.; et al. Truncating titin mutations are associated with a mild and treatable form of dilated cardiomyopathy. Eur. J. Heart Fail. 2017, 19, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Corden, B.; Jarman, J.; Whiffin, N.; Tayal, U.; Buchan, R.; Sehmi, J.; Harper, A.; Midwinter, W.; Lascelles, K.; Markides, V.; et al. Association of Titin-Truncating Genetic Variants with Life-threatening Cardiac Arrhythmias in Patients with Dilated Cardiomyopathy and Implanted Defibrillators. JAMA Netw. Open 2019, 2, e196520. [Google Scholar] [CrossRef] [Green Version]
- Franaszczyk, M.; Chmielewski, P.; Truszkowska, G.; Stawinski, P.; Michalak, E.; Rydzanicz, M.; Sobieszczanska-Malek, M.; Pollak, A.; Szczygieł, J.; Kosinska, J.; et al. Titin truncating variants in dilated cardiomyopathy—Prevalence and genotype-phenotype correlations. PLoS ONE 2017, 12, e0169007. [Google Scholar] [CrossRef] [Green Version]
- Hoorntje, E.T.; Bollen, I.A.; Barge-Schaapveld, D.Q.; Van Tienen, F.H.; Te Meerman, G.J.; Jansweijer, J.A.; Van Essen, A.J.; Volders, P.G.; Constantinescu, A.A.; Van Den Akker, P.C.; et al. Lamin A/C -Related Cardiac Disease: Late Onset with a Variable and Mild Phenotype in a Large Cohort of Patients with the Lamin A/C p.(Arg331Gln) Founder Mutation. Circ. Cardiovasc. Genet. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Van Rijsingen, I.A.W.; Van Der Zwaag, P.A.; Groeneweg, J.A.; Nannenberg, E.A.; Jongbloed, J.D.H.; Zwinderman, A.H.; Pinto, Y.M.; Lekanne Dit Deprez, R.H.; Post, J.G.; Tan, H.L.; et al. Outcome in phospholamban R14del carriers results of a large multicentre cohort study. Circ. Cardiovasc. Genet. 2014, 7, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Lennermann, D.; Backs, J.; van den Hoogenhof, M.M.G. New Insights in RBM20 Cardiomyopathy. Curr. Heart Fail. Rep. 2020, 17, 234–246. [Google Scholar] [CrossRef]
- Te Rijdt, W.P.; van Tintelen, J.P.; Vink, A.; van der Wal, A.C.; de Boer, R.A.; van den Berg, M.P.; Suurmeijer, A.J.H. Phospholamban p.Arg14del cardiomyopathy is characterized by phospholamban aggregates, aggresomes, and autophagic degradation. Histopathology 2016, 69, 542–550. [Google Scholar] [CrossRef]
- Wang, X.; Zabell, A.; Koh, W.; Tang, W.H.W. Lamin A/C Cardiomyopathies: Current Understanding and Novel Treatment Strategies. Curr. Treat. Options Cardiovasc. Med. 2017, 19, 21. [Google Scholar] [CrossRef]
- Domínguez, F.; Cuenca, S.; Bilińska, Z.; Toro, R.; Villard, E.; Barriales-Villa, R.; Ochoa, J.P.; Asselbergs, F.; Sammani, A.; Franaszczyk, M.; et al. Dilated Cardiomyopathy Due to BLC2-Associated Athanogene 3 (BAG3) Mutations. J. Am. Coll. Cardiol. 2018, 72, 2471–2481. [Google Scholar] [CrossRef] [PubMed]
- McNally, E.M.; Golbus, J.R.; Puckelwartz, M.J. Genetic mutations and mechanisms in dilated cardiomyopathy. J. Clin. Invest. 2013, 123, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Hershberger, R.E.; Siegfried, J.D. Update 2011: Clinical and genetic issues in familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 2011, 57, 1641–1649. [Google Scholar] [CrossRef] [Green Version]
- Schultheiss, H.P.; Fairweather, D.L.; Caforio, A.L.P.; Escher, F.; Hershberger, R.E.; Lipshultz, S.E.; Liu, P.P.; Matsumori, A.; Mazzanti, A.; McMurray, J.; et al. Dilated cardiomyopathy. Nat. Rev. Dis. Prim. 2019, 5, 32. [Google Scholar] [CrossRef]
- Pirruccello, J.P.; Bick, A.; Wang, M.; Chaffin, M.; Friedman, S.; Yao, J.; Guo, X.; Venkatesh, B.A.; Taylor, K.D.; Post, W.S.; et al. Analysis of cardiac magnetic resonance imaging in 36,000 individuals yields genetic insights into dilated cardiomyopathy. Nat. Commun. 2020, 11, 2254. [Google Scholar] [CrossRef] [PubMed]
- Garnier, S.; Harakalova, M.; Weiss, S.; Mokry, M.; Regitz-Zagrosek, V.; Hengstenberg, C.; Cappola, T.; Isnard, R.; Arbustini, E.; Cook, S.; et al. Genome wide association analysis in dilated cardiomyopathy reveals two new key players in systolic heart failure on chromosome 3p25.1 and 22q11.23. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Meder, B.; Rühle, F.; Weis, T.; Homuth, G.; Keller, A.; Franke, J.; Peil, B.; Bermejo, J.L.; Frese, K.; Huge, A.; et al. A genome-wide association study identifies 6p21 as novel risk locus for dilated cardiomyopathy. Eur. Heart J. 2014, 35, 1069–1077. [Google Scholar] [CrossRef]
- Esslinger, U.; Garnier, S.; Korniat, A.; Proust, C.; Kararigas, G.; Müller-Nurasyid, M.; Empana, J.P.; Morley, M.P.; Perret, C.; Stark, K.; et al. Exome-wide association study reveals novel susceptibility genes to sporadic dilated cardiomyopathy. PLoS ONE 2017, 12, e0172995. [Google Scholar] [CrossRef] [PubMed]
- Villard, E.; Perret, C.; Gary, F.; Proust, C.; Dilanian, G.; Hengstenberg, C.; Ruppert, V.; Arbustini, E.; Wichter, T.; Germain, M.; et al. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur. Heart J. 2011, 32, 1065–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadros, R.; Francis, C.; Xu, X.; Vermeer, A.M.C.; Harper, A.R.; Huurman, R.; Kelu Bisabu, K.; Walsh, R.; Hoorntje, E.T.; te Rijdt, W.P.; et al. Shared genetic pathways contribute to risk of hypertrophic and dilated cardiomyopathies with opposite directions of effect. Nat. Genet. 2021. [Google Scholar] [CrossRef]
- Lewis, C.M.; Vassos, E. Polygenic risk scores: From research tools to clinical instruments. Genome Med. 2020, 12, 44. [Google Scholar] [CrossRef]
- Dec, G.W.; Fuster, V. Idiopathic Dilated Cardiomyopathy. N. Engl. J. Med. 1994, 331, 1564–1575. [Google Scholar] [CrossRef] [PubMed]
- Ushigome, R.; Sakata, Y.; Nochioka, K.; Miyata, S.; Miura, M.; Tadaki, S.; Yamauchi, T.; Sato, K.; Onose, T.; Tsuji, K.; et al. Improved long-term prognosis of dilated cardiomyopathy with implementation of evidenced-based medication: Report from the CHART studies. Circ. J. 2015, 79, 1332–1341. [Google Scholar] [CrossRef] [Green Version]
- Sammani, A.; Wind, A.M.; Kirkels, J.H.; Klöpping, C.; Buijsrogge, M.P.; Ramjakhan, F.Z.; Asselbergs, F.W.; de Jonge, N. Thirty years of heart transplantation at the university medical centre Utrecht. Neth. Hear. J. 2017, 25, 516–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pi, S.-H.; Kim, S.M.; Choi, J.-O.; Kim, E.K.; Chang, S.-A.; Choe, Y.H.; Lee, S.-C.; Jeon, E.-S. Prognostic value of myocardial strain and late gadolinium enhancement on cardiovascular magnetic resonance imaging in patients with idiopathic dilated cardiomyopathy with moderate to severely reduced ejection fraction. J. Cardiovasc. Magn. Reson. 2018, 20, 36. [Google Scholar] [CrossRef]
- Anselmino, M.; De Ferrari, G.M.; Massa, R.; Manca, L.; Tritto, M.; Molon, G.; Curnis, A.; Devecchi, P.; Braga, S.S.; Bartesaghi, G.; et al. Predictors of mortality and hospitalization for cardiac causes in patients with heart failure and nonischemic heart disease: A subanalysis of the ALPHA study. PACE-Pacing Clin. Electrophysiol. 2009, 32, S214–S218. [Google Scholar] [CrossRef]
- Levy, W.C.; Mozaffarian, D.; Linker, D.T.; Sutradhar, S.C.; Anker, S.D.; Cropp, A.B.; Anand, I.; Maggioni, A.; Burton, P.; Sullivan, M.D.; et al. The Seattle Heart Failure Model: Prediction of survival in heart failure. Circulation 2006, 113, 1424–1433. [Google Scholar] [CrossRef]
- Pocock, S.J.; Ariti, C.A.; McMurray, J.J.V.; Maggioni, A.; Køber, L.; Squire, I.B.; Swedberg, K.; Dobson, J.; Poppe, K.K.; Whalley, G.A.; et al. Predicting survival in heart failure: A risk score based on 39 372 patients from 30 studies. Eur. Heart J. 2013, 34, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Lupón, J.; De Antonio, M.; Vila, J.; Peñafiel, J.; Galán, A.; Zamora, E.; Urrutia, A.; Bayes-Genis, A. Development of a novel heart failure risk tool: The Barcelona bio-heart failure risk calculator (BCN bio-HF calculator). PLoS ONE 2014, 9, e85466. [Google Scholar] [CrossRef] [PubMed]
- Dziewięcka, E.; Gliniak, M.; Winiarczyk, M.; Karapetyan, A.; Wiśniowska-Śmiałek, S.; Karabinowska, A.; Dziewięcki, M.; Podolec, P.; Rubiś, P.; Wiśniowska-Śmiałek, S.; et al. Mortality risk in dilated cardiomyopathy: The accuracy of heart failure prognostic models and dilated cardiomyopathy-tailored prognostic model. ESC Hear. Fail. 2020, 7, 2455–2467. [Google Scholar] [CrossRef] [PubMed]
- Tayal, U.; Prasad, S.K. Myocardial remodelling and recovery in dilated cardiomyopathy. JRSM Cardiovasc. Dis. 2017, 6, 204800401773447. [Google Scholar] [CrossRef] [Green Version]
- Tayal, U.; Wage, R.; Newsome, S.; Manivarmane, R.; Izgi, C.; Muthumala, A.; Dungu, J.N.; Assomull, R.; Hatipoglu, S.; Halliday, B.P.; et al. Predictors of left ventricular remodelling in patients with dilated cardiomyopathy—a cardiovascular magnetic resonance study. Eur. J. Heart Fail. 2020, 22, 1160–1170. [Google Scholar] [CrossRef]
- Merlo, M.; Caiffa, T.; Gobbo, M.; Adamo, L.; Sinagra, G. Reverse remodeling in Dilated Cardiomyopathy: Insights and future perspectives. IJC Hear. Vasc. 2018, 18, 52–57. [Google Scholar] [CrossRef]
- Yurista, S.R.; Matsuura, T.R.; Silljé, H.H.W.; Nijholt, K.T.; McDaid, K.S.; Shewale, S.V.; Leone, T.C.; Newman, J.C.; Verdin, E.; van Veldhuisen, D.J.; et al. Ketone Ester Treatment Improves Cardiac Function and Reduces Pathologic Remodeling in Preclinical Models of Heart Failure. Circ. Hear. Fail. 2020. [Google Scholar] [CrossRef]
- Wolff, G.; Lin, Y.; Karathanos, A.; Brockmeyer, M.; Wolters, S.; Nowak, B.; Fürnkranz, A.; Makimoto, H.; Kelm, M.; Schulze, V. Implantable cardioverter/defibrillators for primary prevention in dilated cardiomyopathy post-DANISH: An updated meta-analysis and systematic review of randomized controlled trials. Clin. Res. Cardiol. 2017, 106, 501–513. [Google Scholar] [CrossRef]
- Priori, S.G.; Blomstrom-Lundqvist, C.; Mazzanti, A.; Bloma, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death the Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the Europea. Eur. Heart J. 2015, 36, 2793–2867l. [Google Scholar] [CrossRef] [Green Version]
- Køber, L.; Thune, J.J.; Nielsen, J.C.; Haarbo, J.; Videbk, L.; Korup, E.; Jensen, G.; Hildebrandt, P.; Steffensen, F.H.; Bruun, N.E.; et al. Defibrillator implantation in patients with nonischemic systolic heart failure. N. Engl. J. Med. 2016, 375, 1221–1230. [Google Scholar] [CrossRef] [Green Version]
- Halliday, B.P.; Cleland, J.G.F.; Goldberger, J.J.; Prasad, S.K. Personalizing Risk Stratification for Sudden Death in Dilated Cardiomyopathy: The Past, Present, and Future. Circulation 2017, 136, 215–231. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, M.; Elliott, P.M. Risk Stratification for Sudden Cardiac Death in Non-Ischaemic Dilated Cardiomyopathy. Curr. Cardiol. Rep. 2019, 21, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younis, A.; Goldberger, J.J.; Kutyifa, V.; Zareba, W.; Polonsky, B.; Klein, H.; Aktas, M.K.; Huang, D.; Daubert, J.; Estes, M.; et al. Predicted benefit of an implantable cardioverter-defibrillator: The MADIT-ICD benefit score. Eur. Heart J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Barsheshet, A.; Wang, P.J.; Moss, A.J.; Solomon, S.D.; Al-Ahmad, A.; McNitt, S.; Foster, E.; Huang, D.T.; Klein, H.U.; Zareba, W.; et al. Reverse Remodeling and the Risk of Ventricular Tachyarrhythmias in the MADIT-CRT (Multicenter Automatic Defibrillator Implantation Trial–Cardiac Resynchronization Therapy). J. Am. Coll. Cardiol. 2011, 57, 2416–2423. [Google Scholar] [CrossRef] [PubMed]
- Ellenbogen, K.A.; Levine, J.H.; Berger, R.D.; Daubert, J.P.; Winters, S.L.; Greenstein, E.; Shalaby, A.; Schaechter, A.; Subacius, H.; Kadish, A. Are implantable cardioverter defibrillator shocks a surrogate for sudden cardiac death in patients with nonischemic cardiomyopathy? Circulation 2006, 113, 776–782. [Google Scholar] [CrossRef]
- Hemingway, H.; Asselbergs, F.W.; Danesh, J.; Dobson, R.; Maniadakis, N.; Maggioni, A.; Van Thiel, G.J.M.; Cronin, M.; Brobert, G.; Vardas, P.; et al. Big data from electronic health records for early and late translational cardiovascular research: Challenges and potential. Eur. Heart J. 2018, 39, 1481–1495. [Google Scholar] [CrossRef] [Green Version]
- Bayley, K.B.; Belnap, T.; Savitz, L.; Masica, A.L.; Shah, N.; Fleming, N.S. Challenges in using electronic health record data for CER: Experience of 4 learning organizations and solutions applied. Med. Care 2013. [Google Scholar] [CrossRef]
- Morley, K.I.; Wallace, J.; Denaxas, S.C.; Hunter, R.J.; Patel, R.S.; Perel, P.; Shah, A.D.; Timmis, A.D.; Schilling, R.J.; Hemingway, H. Defining Disease Phenotypes Using National Linked Electronic Health Records: A Case Study of Atrial Fibrillation. PLoS ONE 2014, 9, e110900. [Google Scholar] [CrossRef] [Green Version]
- Miotto, R.; Wang, F.; Wang, S.; Jiang, X.; Dudley, J.T. Deep learning for healthcare: Review, opportunities and challenges. Brief. Bioinform. 2017, 19, 1236–1246. [Google Scholar] [CrossRef]
- Bernard, O.; Lalande, A.; Zotti, C.; Cervenansky, F.; Yang, X.; Heng, P.A.; Cetin, I.; Lekadir, K.; Camara, O.; Gonzalez Ballester, M.A.; et al. Deep Learning Techniques for Automatic MRI Cardiac Multi-Structures Segmentation and Diagnosis: Is the Problem Solved? IEEE Trans. Med. Imaging 2018. [Google Scholar] [CrossRef]
- Ghorbani, A.; Ouyang, D.; Abid, A.; He, B.; Chen, J.H.; Harrington, R.A.; Liang, D.H.; Ashley, E.A.; Zou, J.Y. Deep learning interpretation of echocardiograms. NPJ Digit. Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Blanco, A.; Perez-de-Viñaspre, O.; Pérez, A.; Casillas, A. Boosting ICD multi-label classification of health records with contextual embeddings and label-granularity. Comput. Methods Programs Biomed. 2020, 188, 105264. [Google Scholar] [CrossRef]
- Bagheri, A.; Sammani, A.; van der Heijden, P.G.M.; Asselbergs, F.W.; Oberski, D.L. Automatic ICD-10 classification of diseases from Dutch discharge letters. In Proceedings of the BIOINFORMATICS 2020—11th International Conference on Bioinformatics Models, Methods and Algorithms, Proceedings; Part of 13th International Joint Conference on Biomedical Engineering Systems and Technologies, BIOSTEC, Valletta, Malta, 24 February 2020–26 February 2020; SCITEPRESS—Science and Technology Publications: Valletta, Malta, 2020; Volume 3, pp. 281–289. [Google Scholar]
- Dijk, W.; Van Fiolet, A.; Schuit, E.; Sammani, A.; Groenhof, K.; van der Graaf, R.; de Vries, M.; Alings, M.; Schaap, J.; Asselbergs, F.; et al. Text-Mining in Electronic Healthcare Records for Efficient Recruitment and Data-Collection in Cardiovascular Trials: A Multicenter Validation Study. J. Am. Coll. Cardiol. 2020, 75, 3622. [Google Scholar] [CrossRef]
- Anker, S.; Asselbergs, F.W.; Brobert, G.; Vardas, P.; Grobbee, D.E.; Cronin, M. Big Data in Cardiovascular Disease. Eur. Heart J. 2017, 38, 1863–1865. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.; Kiefer, R.C.; Sharma, D.K.; Prud’hommeaux, E.; Solbrig, H.R. A Consensus-Based Approach for Harmonizing the OHDSI Common Data Model with HL7 FHIR. Stud. Health Technol. Inform. 2017, 245, 887–891. [Google Scholar] [PubMed]
- van de Leur, R.; Taha, K.; Bos, M.N.; van der Heijden, J.F.; Gupta, D.; Cramer, M.J.; Hassink, R.J.; van der Harst, P.; Doevendans, P.A.; Asselbergs, F.W.; et al. Discovering and Visualizing Disease-specific Electrocardiogram Features Using Deep Learning: Proof-of-concept in Phospholamban Gene Mutation Carriers. Circ. Arrhythmia Electrophysiol. 2021, CIRCEP.120.009056. [Google Scholar] [CrossRef]
- Van de Leur, R.R.; Blom, L.J.; Gavves, E.; Hof, I.E.; van der Heijden, J.F.; Clappers, N.C.; Doevendans, P.A.; Hassink, R.J.; van Es, R. Automatic Triage of 12-Lead ECGs Using Deep Convolutional Neural Networks. J. Am. Heart Assoc. 2020, 9, e015138. [Google Scholar] [CrossRef]
- Shah, S.J.; Katz, D.H.; Selvaraj, S.; Burke, M.A.; Yancy, C.W.; Gheorghiade, M.; Bonow, R.O.; Huang, C.C.; Deo, R.C. Phenomapping for novel classification of heart failure with preserved ejection fraction. Circulation 2015, 131, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Verdonschot, J.A.J.; Merlo, M.; Dominguez, F.; Wang, P.; Henkens, M.T.H.M.; Adriaens, M.E.; Hazebroek, M.R.; Masè, M.; Escobar, L.E.; Cobas-Paz, R.; et al. Phenotypic clustering of dilated cardiomyopathy patients highlights important pathophysiological differences. Eur. Heart J. 2020. [Google Scholar] [CrossRef]
- Merlo, M.; Pivetta, A.; Pinamonti, B.; Stolfo, D.; Zecchin, M.; Barbati, G.; Di Lenarda, A.; Sinagra, G. Long-term prognostic impact of therapeutic strategies in patients with idiopathic dilated cardiomyopathy: Changing mortality over the last 30 years. Eur. J. Heart Fail. 2014, 16, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Paldino, A.; De Angelis, G.; Dal Ferro, M.; Faganello, G.; Porcari, A.; Barbati, G.; Korcova, R.; Gentile, P.; Artico, J.; Cannatà, A.; et al. High prevalence of subtle systolic and diastolic dysfunction in genotype-positive phenotype-negative relatives of dilated cardiomyopathy patients. Int. J. Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, Y.; Kerz, M.; Rashid, Z.; Böttcher, S.; Dobson, R.J.; Folarin, A.A. RADAR-base. In Proceedings of the 2018 ACM International Joint Conference and 2018 International Symposium on Pervasive and Ubiquitous Computing and Wearable Computers, Singapore, October 2018; ACM: New York, NY, USA, 2018; pp. 223–226. [Google Scholar]
- Harvard TH Chan School of Public Health FORHEALTH Harvard Application. Available online: https://applab.forhealth.org/#section-8 (accessed on 20 January 2021).
- Barrett, M.; Boyne, J.; Brandts, J.; Brunner-La Rocca, H.-P.; De Maesschalck, L.; De Wit, K.; Dixon, L.; Eurlings, C.; Fitzsimons, D.; Golubnitschaja, O.; et al. Artificial intelligence supported patient self-care in chronic heart failure: A paradigm shift from reactive to predictive, preventive and personalised care. EPMA J. 2019, 10, 445–464. [Google Scholar] [CrossRef] [Green Version]
- Antoniades, C.; Asselbergs, F.W.; Vardas, P. The year in cardiovascular medicine 2020: Digital health and innovation. Eur. Heart J. 2021. [Google Scholar] [CrossRef]
- Yun, J.E.; Park, J.-E.; Park, H.-Y.; Lee, H.-Y.; Park, D.-A. Comparative Effectiveness of Telemonitoring Versus Usual Care for Heart Failure: A Systematic Review and Meta-analysis. J. Card. Fail. 2018, 24, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Perego, G.B.; Landolina, M.; Vergara, G.; Lunati, M.; Zanotto, G.; Pappone, A.; Lonardi, G.; Speca, G.; Iacopino, S.; Varbaro, A.; et al. Implantable CRT device diagnostics identify patients with increased risk for heart failure hospitalization. J. Interv. Card. Electrophysiol. 2008, 23, 235–242. [Google Scholar] [CrossRef]
- Adamson, P.B.; Smith, A.L.; Abraham, W.T.; Kleckner, K.J.; Stadler, R.W.; Shih, A.; Rhodes, M.M. Continuous autonomic assessment in patients with symptomatic heart failure: Prognostic value of heart rate variability measured by an implanted cardiac resynchronization device. Circulation 2004, 110, 2389–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stehlik, J.; Schmalfuss, C.; Bozkurt, B.; Nativi-Nicolau, J.; Wohlfahrt, P.; Wegerich, S.; Rose, K.; Ray, R.; Schofield, R.; Deswal, A.; et al. Continuous Wearable Monitoring Analytics Predict Heart Failure Hospitalization. Circ. Hear. Fail. 2020, 13. [Google Scholar] [CrossRef]
- Singhal, A.; Cowie, M.R. The Role of Wearables in Heart Failure. Curr. Heart Fail. Rep. 2020, 17, 125–132. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sammani, A.; Baas, A.F.; Asselbergs, F.W.; te Riele, A.S.J.M. Diagnosis and Risk Prediction of Dilated Cardiomyopathy in the Era of Big Data and Genomics. J. Clin. Med. 2021, 10, 921. https://doi.org/10.3390/jcm10050921
Sammani A, Baas AF, Asselbergs FW, te Riele ASJM. Diagnosis and Risk Prediction of Dilated Cardiomyopathy in the Era of Big Data and Genomics. Journal of Clinical Medicine. 2021; 10(5):921. https://doi.org/10.3390/jcm10050921
Chicago/Turabian StyleSammani, Arjan, Annette F. Baas, Folkert W. Asselbergs, and Anneline S. J. M. te Riele. 2021. "Diagnosis and Risk Prediction of Dilated Cardiomyopathy in the Era of Big Data and Genomics" Journal of Clinical Medicine 10, no. 5: 921. https://doi.org/10.3390/jcm10050921
APA StyleSammani, A., Baas, A. F., Asselbergs, F. W., & te Riele, A. S. J. M. (2021). Diagnosis and Risk Prediction of Dilated Cardiomyopathy in the Era of Big Data and Genomics. Journal of Clinical Medicine, 10(5), 921. https://doi.org/10.3390/jcm10050921