
Computed Tomographic Changes in Patients with Cystic Fibrosis Treated by Combination Therapy with Lumacaftor and Ivacaftor

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Clinical Data Collection
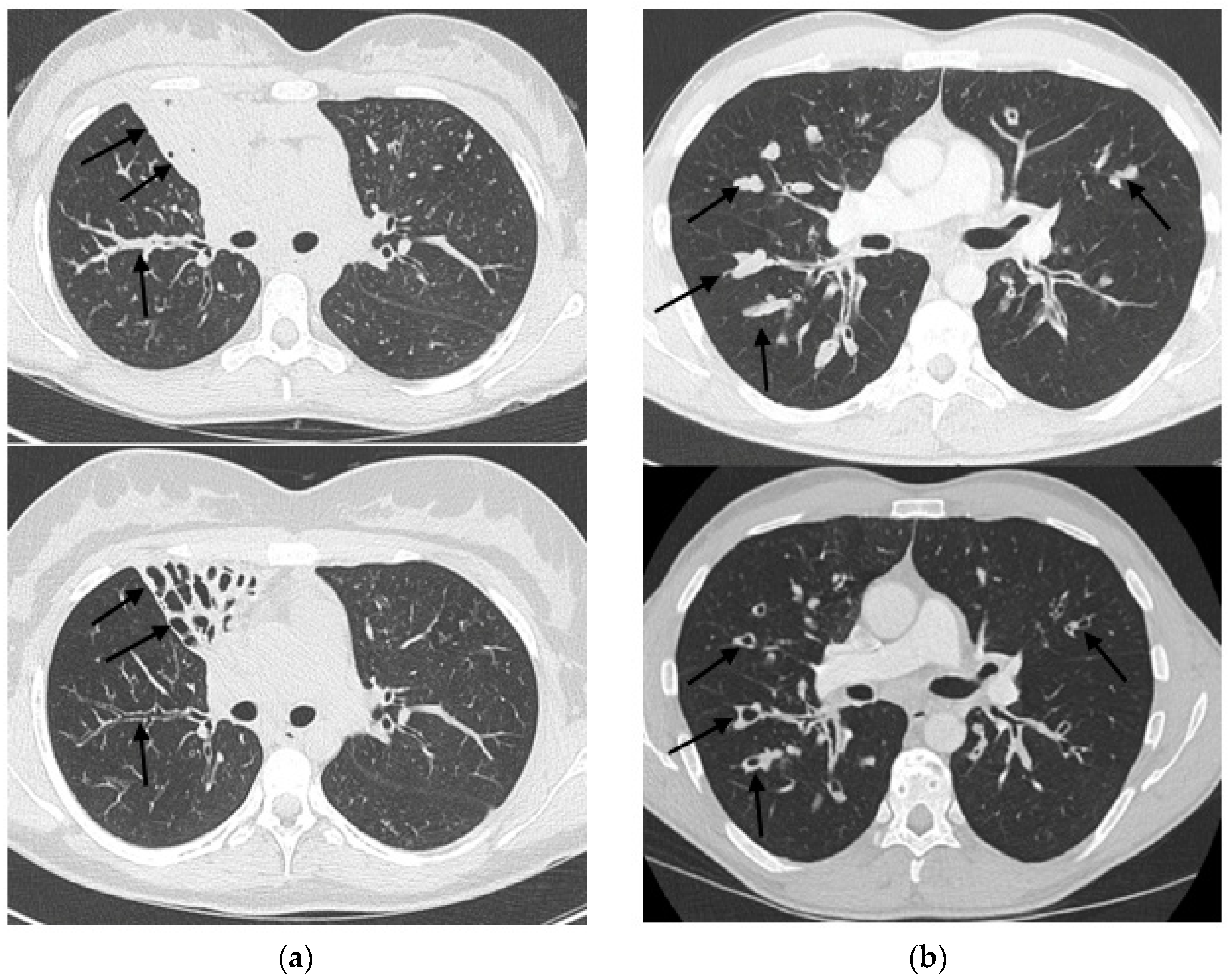
2.3. HRCT Imaging
2.4. Modified Brody Scoring Method
2.5. Statistical Analysis
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. HRCT Scoring System (Modified Brody Score)
| Bronchiectasis Score (range, 0–12) = | Extent of Bronchiectasis in Central Lung 0 = None 1 = 1/3 of Lobe 2 = 1/3–2/3 of Lobe 3 ≥ 2/3 of Lobe | + Extent of Bronchiectasis in Peripheral Lung 0 = None 1 = 1/3 of Lobe 2 = 1/3–2/3 of Lobe 3 ≥ 2/3 of Lobe | × Average Bronchiectasis Size Multiplier Average Multiplier Size 0.5 = 0 1 = 1 1.5 = 1.25 2.0 = 1.5 2.5 = 1.75 3 = 2 |
| Average bronchiectasis size multiplier = | Size of largest dilated bronchus 1 ≤ 2× 2 = 2× –3× 3 ≥ 3× | + Average size of dilated bronchi 1 ≤ 2× 2 = 2× –3× 3 ≥ 3× | ÷ 2 |
| Mucous plugging score (range, 0–6) = | Extent of mucous plugging in central lung 0 = none 1 = 1/3 of lobe 2 = 1/3–2/3 of lobe 3 ≥ 2/3 of lobe | + Extent of mucous plugging in peripheral lung 0 = none 1 = 1/3 of lobe 2 = 1/3–2/3 of lobe 3 ≥ 2/3 of lobe | |
| Peribronchial thickening score (range, 0–9) = | Extent of peribronchial thickening in central lung 0 = none 1 = 1/3 of lobe 2 = 1/3–2/3 of lobe 3 ≥ 2/3 of lobe | + Extent of peribronchial thickening in peripheral lung 0 = none 1 = 1/3 of lobe 2 = 1/3–2/3 of lobe 3 ≥ 2/3 of lobe | × Severity of peribronchial thickening 1 = mild 1.25 = moderate 1.5 = severe |
| Parenchyma score (range, 0–9) = | Extent of dense parenchyma opacity 0 = none 1 = 1/3 of lobe 2 = 1/3–2/3 of lobe 3 ≥ 2/3 of lobe | + Extent of ground glass opacity 0 = none 1 = 1/3 of lobe 2 = 1/3–2/3 of lobe 3 ≥ 2/3 of lobe | + Extent of cysts or bullae 0 = none 1 = 1/3 of lobe 2 = 1/3–2/3 of lobe 3 ≥ 2/3 of lobe |
| Hyperinflation score (range, 0–4,5) | Extent of air trapping 0 = none 1 = 1/3 of lobe 2 = 1/3–2/3 of lobe 3 ≥ 2/3 of lobe | × Appearance of air trapping 1 = subsegmental 1.5 = segmental or larger |
References
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; de Boeck, K.; Flume, P.A.; et al. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elborn, J.S.; Ramsey, B.W.; Boyle, M.P.; Konstan, M.W.; Huang, X.; Marigowda, G.; Waltz, D.; Wainwright, C.E.; VX-809 TRAFFIC and TRANSPORT Study Groups. Efficacy and safety of lumacaftor/ivacaftor combination therapy in patients with cystic fibrosis homozygous for Phe508del CFTR by pulmonary function subgroup: A pooled analysis. Lancet Respir. Med. 2016, 4, 617–626. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.M.; Barry, P.J. Lumacaftor/ivacaftor for patients homozygous for Phe508del-CFTR: Should we curb our enthusiasm? Thorax 2015, 70, 615–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elborn, J.S.; Ramsey, B.; Wainwright, C.; Boyle, M. Response to: ‘Lumacaftor/ivacaftor for patients homozygous for Phe508del-CFTR: Should we curb our enthusiasm?’ by Jones and Barry. Thorax 2016, 71, 185–186. [Google Scholar] [CrossRef] [Green Version]
- Masson, A.; Schneider-Futschik, E.K.; Baatallah, N.; Nguyen-Khoa, T.; Girodon, E.; Hatton, A.; Flament, T.; le Bourgeois, M.; Chedevergne, F.; Bailly, C.; et al. Predictive factors for lumacaftor/ivacaftor clinical response. J. Cyst. Fibros. 2019, 18, 368–374. [Google Scholar] [CrossRef]
- Burgel, P.R.; Munck, A.; Durieu, I.; Chiron, R.; Mely, L.; Prevotat, A.; Murris-Espin, M.; Porzio, M.; Abely, M.; Reix, P.; et al. Real-Life Safety and Effectiveness of Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 188–197. [Google Scholar] [CrossRef]
- Calder, A.D.; Bush, A.; Brody, A.S.; Owens, C.M. Scoring of chest CT in children with cystic fibrosis: State of the art. Pediatr. Radiol. 2014, 44, 1496–1506. [Google Scholar] [CrossRef]
- Loeve, M.; van Hal, P.T.W.; Robinson, P.; de Jong, P.A.; Lequin, M.H.; Hop, W.C.; Williams, T.J.; Nossent, G.D.; Tiddens, H.A. The spectrum of structural abnormalities on CT scans from patients with CF with severe advanced lung disease. Thorax 2009, 64, 876–882. [Google Scholar] [CrossRef] [Green Version]
- Brody, A.S.; Klein, J.S.; Molina, P.L.; Quan, J.; Bean, J.A.; Wilmott, R.W. High-resolution computed tomography in young patients with cystic fibrosis: Distribution of abnormalities and correlation with pulmonary function tests. J. Pediatr. 2004, 145, 32l. [Google Scholar] [CrossRef]
- Chassagnon, G.; Hubert, D.; Fajac, I.; Burgel, P.-R.; Revel, M.-P. Long-term computed tomographic changes in cystic fibrosis patients treated with ivacaftor. Eur. Respir. J. 2016, 48, 249–252. [Google Scholar] [CrossRef] [Green Version]
- Sheikh, S.I.; Long, F.R.; McCoy, K.S.; Johnson, T.; Ryan-Wenger, N.A.; Hayes, D. Computed tomography correlates with improvement with ivacaftor in cystic fibrosis patients with G551D mutation. J. Cyst. Fibros. 2015, 14, 84–89. [Google Scholar] [CrossRef] [Green Version]
- Ratjen, F.; Hug, C.; Marigowda, G.; Tian, S.; Huang, X.; Stanojevic, S.; Milla, C.E.; Robinson, P.D.; Waltz, D.; Davies, J.C. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6–11 years with cystic fibrosis homozygous for F508del-CFTR: A randomised, placebo-controlled phase 3 trial. Lancet Respir. Med. 2017, 5, 557–567. [Google Scholar] [CrossRef]
- McNamara, J.J.; McColley, S.A.; Marigowda, G.; Liu, F.; Tian, S.; Owen, C.A.; Stiles, D.; Li, C.; Waltz, D.; Wang, L.T. Safety, pharmacokinetics, and pharmacodynamics of lumacaftor and ivacaftor combination therapy in children aged 2–5 years with cystic fibrosis homozygous for F508del-CFTR: An open-label phase 3 study. Lancet Respir. Med. 2019, 7, 325–335. [Google Scholar] [CrossRef]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445–Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef]
- Davies, J.C.; Moskowitz, S.M.; Brown, C.; Horsley, A.; Mall, M.A.; McKone, E.F.; Plant, B.J.; Prais, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; et al. VX-659–Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1599–1611. [Google Scholar] [CrossRef]
- Holguin, F. Triple CFTR Modulator Therapy for Cystic Fibrosis. N. Engl. J. Med. 2018, 379, 1671–1672. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, A.M.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Burgel, P.R.; Durieu, I.; Chiron, R.; Ramel, S.; Danner-Boucher, I.; Prevotat, A.; Grenet, D.; Marguet, C.; Reynaud-Gaubert, M.; Macey, J.; et al. Rapid Improvement After Starting Elexacaftor-tezacaftor-ivacaftor in Patients with Cystic Fibrosis and Advanced Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2021. [Google Scholar] [CrossRef]


{kind=link}
| Before Treatment | After/Under Treatment | p-Value | ||
|---|---|---|---|---|
| BMI (kg/m2) | 19.2 ± 2.4 | 20.2 ± 2.8 | <0.001 | |
| FEV1 (%) | 74.8 ± 27.0 | 75.4 ± 25.4 | 0.737 | |
| Sweat chloride test (n = 20) (kg/m2) | 112.7 ± 13.3 | 87.3 ± 17.1 | <0.001 | |
| Brody’s score | ||||
| Total score | 65.5 ± 33.4 | 60.3 ± 29.6 | 0.049 | |
| Bronchiectasis | 23.3 ± 13.7 | 23.3 ± 14.5 | 0.991 | |
| Mucous plugging | 12.3 ± 8.8 | 8.7 ± 7.4 | 0.009 | |
| Peribronchial thickening | 23.8 ± 10.6 | 22.3 ± 9.1 | 0.241 | |
| Parenchyma score | 1.5 ± 1.7 | 1.5 ± 1.7 | 0.768 | |
| Hyperinfilation score | 6.7 ± 5.3 | 6.1 ± 5.1 | 0.373 |
| Bronchiectasis | Mucous Plugging | Peribronchial Thickening | Parenchyma Score | Hyperinfilation Score | Total Score | ||
|---|---|---|---|---|---|---|---|
| Evolution of sweat chloride test (n = 20) | Pearson correlation coefficient (p-value) | 0.12 (0.615) | 0.16 (0.505) | 0.21 (0.371) | 0.25 (0.288) | 0.28 (0.292) * | 0.26 (0.274) |
| Evolution of FEV1 (n = 34) | Pearson correlation coefficient (p-value) | −0.06 (0.723) | −0.48 (0.004) | −0.43 (0.01) | −0.11 (0.547) | −0.26 (0.224) ** | −0.51 (0.002) |
| Before Treatment | After/Under Treatment | p-Value | |
|---|---|---|---|
| BMI (kg/m2) | 19.2 ± 2.4 | 20.2 ± 2.8 | <0.001 a |
| FEV1 (%) | 74.8 ± 27.0 | 75.4 ± 25.4 | 0.737 |
| Sweat chloride test (kg/m2) | 112.6 ± 13.3 | 87.2 ± 17.1 | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnaud, F.; Stremler-Le Bel, N.; Reynaud-Gaubert, M.; Mancini, J.; Gaubert, J.-Y.; Gorincour, G. Computed Tomographic Changes in Patients with Cystic Fibrosis Treated by Combination Therapy with Lumacaftor and Ivacaftor. J. Clin. Med. 2021, 10, 1999. https://doi.org/10.3390/jcm10091999
Arnaud F, Stremler-Le Bel N, Reynaud-Gaubert M, Mancini J, Gaubert J-Y, Gorincour G. Computed Tomographic Changes in Patients with Cystic Fibrosis Treated by Combination Therapy with Lumacaftor and Ivacaftor. Journal of Clinical Medicine. 2021; 10(9):1999. https://doi.org/10.3390/jcm10091999
Chicago/Turabian StyleArnaud, François, Nathalie Stremler-Le Bel, Martine Reynaud-Gaubert, Julien Mancini, Jean-Yves Gaubert, and Guillaume Gorincour. 2021. "Computed Tomographic Changes in Patients with Cystic Fibrosis Treated by Combination Therapy with Lumacaftor and Ivacaftor" Journal of Clinical Medicine 10, no. 9: 1999. https://doi.org/10.3390/jcm10091999
APA StyleArnaud, F., Stremler-Le Bel, N., Reynaud-Gaubert, M., Mancini, J., Gaubert, J. -Y., & Gorincour, G. (2021). Computed Tomographic Changes in Patients with Cystic Fibrosis Treated by Combination Therapy with Lumacaftor and Ivacaftor. Journal of Clinical Medicine, 10(9), 1999. https://doi.org/10.3390/jcm10091999