
Current Options and Future Perspectives in the Treatment of Dyslipidemia

 ,
, 
Abstract
:1. Introduction
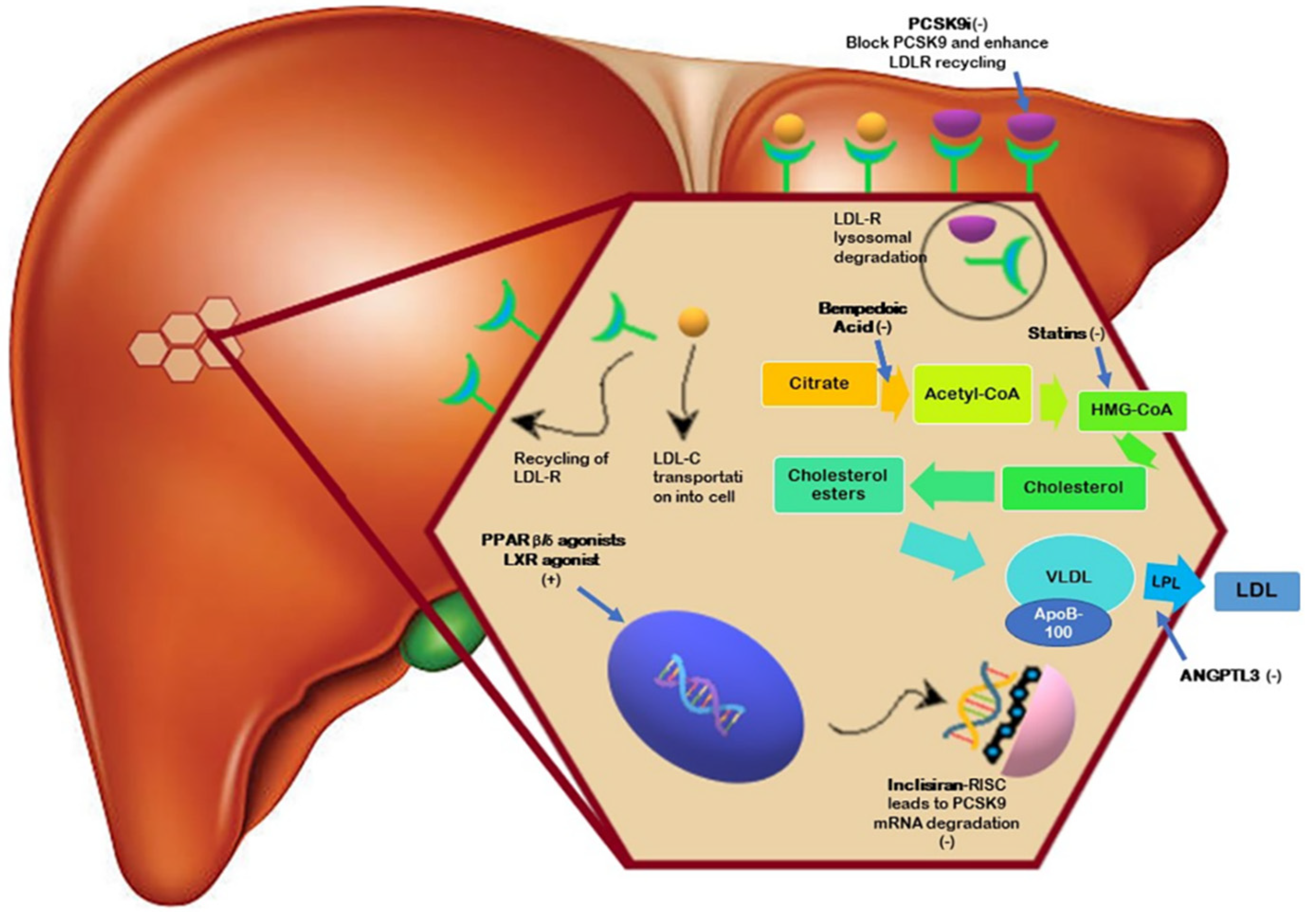
2. PCSK9 Inhibitors
3. Bempedoic Acid
4. Inclisiran
5. LXR Agonists
6. PPARβ/δ Agonists
7. ANGPTL3 Inhibitors
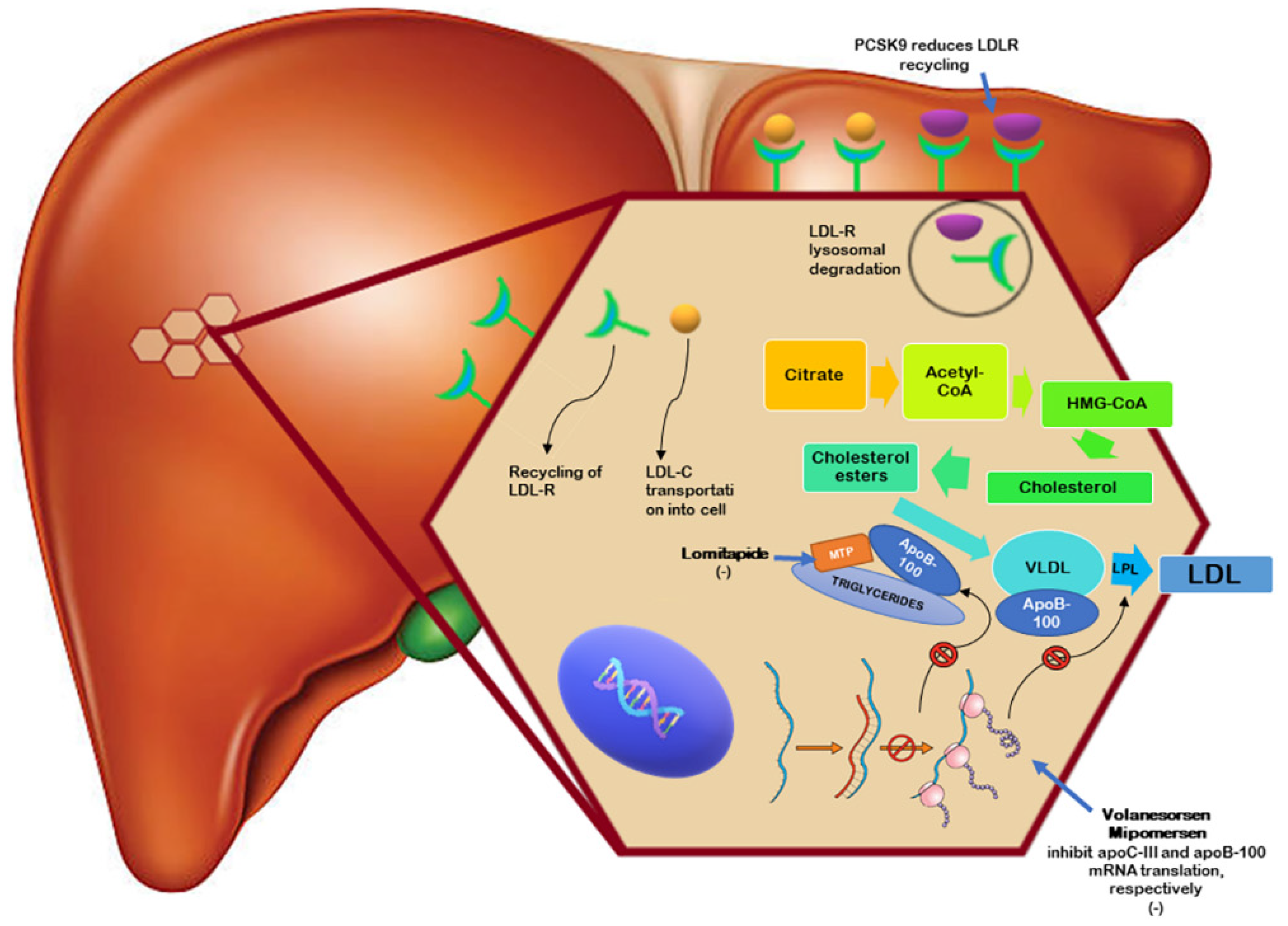
8. Mipomersen
9. Lomitapide
10. Volanesorsen
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABC | Adenosine Triphosphate-Binding Cassette |
| ACSVL1 | Acyl-CoA synthetase 1 |
| ADEs | Adverse Events |
| AHA | American Heart Association |
| AMI | Acute Myocardial infarction |
| ANGPTLs | Angiopoietin-like proteins |
| ApoB | Apolipoprotein B |
| ApoC-III | Apolipoprotein C-III |
| ASCVD | Atherosclerotic Cardiovascular Disease |
| BA | Bempedoic Acid |
| CABG | Coronary Artery Bypass Grafting |
| CCS | Chronic Coronary Syndrome |
| CV | Cardiovascular |
| CVD | Cardiovascular Disease |
| EMA | European Medicines Agency |
| ESC | European Society of Cardiology |
| FDA | Food and Drug Administration |
| FCS | Familial Chylomicronemia Syndrome |
| FH | Familial Hypercholesterolemia |
| HDL-C | High-Density Lipoprotein Cholesterol |
| HMG-CoA | 3-Hydroxy-3-Methylglutaryl-Coenzyme A |
| HeFH | Heterozygous Familial Hypercholesterolemia |
| HoFH | Homozygous Familial Hypercholesterolemia |
| hs-CRP | High-Sensitivity C-Reactive Protein |
| IDL | Intermediate-Density Lipoprotein |
| IVUS | Intravascular Ultrasonography |
| LDL-C | Low-Density Lipoprotein Cholesterol |
| LDL-R | LDL-Receptors |
| Lp(a) | Lipoprotein(a) |
| LPL | Lipoprotein Lipase |
| LXRs | Liver X Receptors |
| MOA | Mechanism of Action |
| MTP | Microsomal Triglyceride Transfer Protein |
| NIRS | Near-Infrared Spectroscopy |
| OCT | Optical Coherence Tomography |
| PPARs | Peroxisome Proliferator-Activated Receptors |
| PCI | Percutaneous Coronary Intervention |
| PCSK9i | Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitors |
| RCT | Randomized Clinical Trial |
| ReCT | Reverse Cholesterol Transport |
| SI | Statin Intolerance |
| siRNA | Small Interfering RNA |
| TG | Triglycerides |
| TRL | Triglycerides Rich Lipoproteins |
| VLDL | Very-Low-Density Lipoproteins |
References
- Available online: https://www.who.int/news/item/09-12-2020-who-reveals-leading-causes-of-death-and-disability-worldwide-2000-2019 (accessed on 7 March 2022).
- Kosmas, C.E.; Pantou, D.; Sourlas, A.; Papakonstantinou, E.J.; Echavarria Uceta, R.; Guzman, E. New and Emerging Lipid-Modifying Drugs to Lower LDL Cholesterol. Drugs Context 2021, 10, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Colin, S.; Chinetti-Gbaguidi, G.; Kuivenhoven, J.A.; Staels, B. Emerging Small Molecule Drugs. Handb. Exp. Pharmacol. 2015, 224, 617–630. [Google Scholar] [PubMed]
- Baigent, C.; Keech, A.; Kearney, P.M.; Blackwell, L.; Buck, G.; Pollicino, C.; Kirby, A.; Sourjina, T.; Peto, R.; Collins, R.; et al. Efficacy and Safety of Cholesterol-Lowering Treatment: Prospective Meta-Analysis of Data from 90,056 Participants in 14 Randomised Trials of Statins. Lancet Lond. Engl. 2005, 366, 1267–1278. [Google Scholar]
- Reiner, Ž. Resistance and Intolerance to Statins. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, A.; Catapano, A.L. Understanding Efficacy and Safety of Lomitapide in Homozygous Familial Hypercholesterolemia. Eur. J. Prev. Cardiol. 2022, 29, 829–831. [Google Scholar] [CrossRef]
- Stoekenbroek, R.M.; Lambert, G.; Cariou, B.; Hovingh, G.K. Inhibiting PCSK9—Biology beyond LDL Control. Nat. Rev. Endocrinol. 2018, 15, 52–62. [Google Scholar] [CrossRef]
- Kosmas, C.E.; Dejesus, E. Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitors: An Emerging Chapter in The Field of Clinical Lipidology. Enliven Clin. Cardiol. Res. 2015, 2, E1. [Google Scholar]
- Leren, T.P. Sorting an LDL Receptor with Bound PCSK9 to Intracellular Degradation. Atherosclerosis 2014, 237, 76–81. [Google Scholar] [CrossRef]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; Mcevoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, E596–E646. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the Management of Dyslipidaemias: Lipid Modification to Reduce Cardiovascular Risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Wiviott, S.D.; Raal, F.J.; Blom, D.J.; Robinson, J.; Ballantyne, C.M.; Somaratne, R.; Legg, J.; Wasserman, S.M.; et al. Efficacy and Safety of Evolocumab in Reducing Lipids and Cardiovascular Events. N. Engl. J. Med. 2015, 372, 1500–1509. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.G.; Farnier, M.; Krempf, M.; Bergeron, J.; Luc, G.; Averna, M.; Stroes, E.S.; Langslet, G.; Raal, F.J.; El Shahawy, M.; et al. Efficacy and Safety of Alirocumab in Reducing Lipids and Cardiovascular Events. N. Engl. J. Med. 2015, 372, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, B.; Hu, M.; Zhang, Y.; Chan, P.; Liu, Z.-M. Alirocumab for the Treatment of Hypercholesterolemia. Expert Opin. Biol. Ther. 2017, 17, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, P.M.; Thompson, P.D.; Cannon, C.P.; Guyton, J.R.; Bergeron, J.; Zieve, F.J.; Bruckert, E.; Jacobson, T.A.; Kopecky, S.L.; Baccara-Dinet, M.T.; et al. Efficacy and Safety of Alirocumab vs Ezetimibe in Statin-Intolerant Patients, with a Statin Rechallenge Arm: The ODYSSEY ALTERNATIVE Randomized Trial. J. Clin. Lipidol. 2015, 9, 758–769. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Hadjiphilippou, S.; Ray, K.K. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. J. R. Coll. Physicians Edinb. 2017, 47, 153–155. [Google Scholar] [CrossRef]
- Giugliano, R.P.; Pedersen, T.R.; Park, J.-G.; De Ferrari, G.M.; Gaciong, Z.A.; Ceska, R.; Toth, K.; Gouni-Berthold, I.; Lopez-Miranda, J.; Schiele, F.; et al. Clinical Efficacy and Safety of Achieving Very Low LDL-Cholesterol Concentrations with the PCSK9 Inhibitor Evolocumab: A Prespecified Secondary Analysis of the FOURIER Trial. Lancet Lond. Engl. 2017, 390, 1962–1971. [Google Scholar] [CrossRef]
- Giugliano, R.P.; Sabatine, M.S.; Ott, B.R. Cognitive Function in a Randomized Trial of Evolocumab. N. Engl. J. Med. 2017, 377, 1997. [Google Scholar] [CrossRef]
- Kosmas, C.E.; Skavdis, A.; Sourlas, A.; Papakonstantinou, E.J.; Peña Genao, E.; Echavarria Uceta, R.; Guzman, E. Safety and Tolerability of PCSK9 Inhibitors: Current Insights. Clin. Pharmacol. Adv. Appl. 2020, 12, 191–202. [Google Scholar] [CrossRef]
- Chaudhary, R.; Garg, J.; Shah, N.; Sumner, A. PCSK9 Inhibitors: A New Era of Lipid Lowering Therapy. World J. Cardiol. 2017, 9, 76–91. [Google Scholar] [CrossRef]
- Sun, J.; Lepor, N.E.; Cantón, G.; Contreras, L.; Hippe, D.S.; Isquith, D.A.; Balu, N.; Kedan, I.; Simonini, A.A.; Yuan, C.; et al. Serial Magnetic Resonance Imaging Detects a Rapid Reduction in Plaque Lipid Content Under PCSK9 Inhibition with Alirocumab. Int. J. Cardiovasc. Imaging 2021, 37, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Lepor, N.E.; Sun, J.; Canton, G.; Contreras, L.; Hippe, D.S.; Isquith, D.A.; Balu, N.; Kedan, I.; Simonini, A.A.; Yuan, C.; et al. Regression in Carotid Plaque Lipid Content and Neovasculature with PCSK9 Inhibition: A Time Course Study. Atherosclerosis 2021, 327, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Aranzulla, T.C.; Piazza, S.; Ricotti, A.; Musumeci, G.; Gaggiano, A. Carotid Plaque Stabilization and Regression with Evolocumab: Rationale and Design of the CARUSO Study. Catheter. Cardiovasc. Interv. Off. J. Soc. Card. Angiogr. Interv. 2021, 98, E115–E121. [Google Scholar] [CrossRef] [PubMed]
- Oyama, K.; Furtado, R.H.M.; Fagundes, A.; Zelniker, T.A.; Tang, M.; Kuder, J.; Murphy, S.A.; Hamer, A.; Wang, H.; Keech, A.C.; et al. Effect of Evolocumab on Complex Coronary Disease Requiring Revascularization. J. Am. Coll. Cardiol. 2021, 77, 259–267. [Google Scholar] [CrossRef]
- Räber, L.; Ueki, Y.; Otsuka, T.; Losdat, S.; Häner, J.D.; Lonborg, J.; Fahrni, G.; Iglesias, J.F.; Van Geuns, R.-J.; Ondracek, A.S.; et al. Effect of Alirocumab Added to High-Intensity Statin Therapy on Coronary Atherosclerosis in Patients with Acute Myocardial Infarction: The Pacman-Ami Randomized Clinical Trial. Ama J. Am. Med Assoc. 2022, 327, 1771. [Google Scholar] [CrossRef]
- Bilen, O.; Ballantyne, C.M. Bempedoic Acid (ETC-1002): An Investigational Inhibitor of ATP Citrate Lyase. Curr. Atheroscler. Rep. 2016, 18, 61. [Google Scholar] [CrossRef]
- Filippov, S.; Pinkosky, S.L.; Lister, R.J.; Pawloski, C.; Hanselman, J.C.; Cramer, C.T.; Srivastava, R.A.K.; Hurley, T.R.; Bradshaw, C.D.; Spahr, M.A.; et al. ETC-1002 Regulates Immune Response, Leukocyte Homing, and Adipose Tissue Inflammation via LKB1-Dependent Activation of Macrophage AMPK. J. Lipid Res. 2013, 54, 2095–2108. [Google Scholar] [CrossRef]
- Sirtori, C.R.; Yamashita, S.; Greco, M.F.; Corsini, A.; Watts, G.F.; Ruscica, M. Recent Advances in Synthetic Pharmacotherapies for Dyslipidaemias. Eur. J. Prev. Cardiol. 2020, 27, 1576–1596. [Google Scholar] [CrossRef]
- Pinkosky, S.L.; Newton, R.S.; Day, E.A.; Ford, R.J.; Lhotak, S.; Austin, R.C.; Birch, C.M.; Smith, B.K.; Filippov, S.; Groot, P.H.E.; et al. Liver-Specific ATP-Citrate Lyase Inhibition by Bempedoic Acid Decreases LDL-C and Attenuates Atherosclerosis. Nat. Commun. 2016, 7, 13457. [Google Scholar] [CrossRef]
- Gutierrez, M.J.; Rosenberg, N.L.; Macdougall, D.E.; Hanselman, J.C.; Margulies, J.R.; Strange, P.; Milad, M.A.; Mcbride, S.J.; Newton, R.S. Efficacy and Safety of ETC-1002, a Novel Investigational Low-Density Lipoprotein-Cholesterol-Lowering Therapy for the Treatment of Patients with Hypercholesterolemia and Type 2 Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 676–683. [Google Scholar] [CrossRef]
- Ballantyne, C.M.; Mckenney, J.M.; Macdougall, D.E.; Margulies, J.R.; Robinson, P.L.; Hanselman, J.C.; Lalwani, N.D. Effect of ETC-1002 on Serum Low-Density Lipoprotein Cholesterol in Hypercholesterolemic Patients Receiving Statin Therapy. Am. J. Cardiol. 2016, 117, 1928–1933. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.D.; Macdougall, D.E.; Newton, R.S.; Margulies, J.R.; Hanselman, J.C.; Orloff, D.G.; Mckenney, J.M.; Ballantyne, C.M. Treatment with ETC-1002 Alone and in Combination with Ezetimibe Lowers LDL Cholesterol in Hypercholesterolemic Patients with or without Statin Intolerance. J. Clin. Lipidol. 2016, 10, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Bays, H.E.; Catapano, A.L.; Lalwani, N.D.; Bloedon, L.T.; Sterling, L.R.; Robinson, P.L.; Ballantyne, C.M.; CLEAR Harmony Trial. Safety and Efficacy of Bempedoic Acid to Reduce LDL Cholesterol. N. Engl. J. Med. 2019, 380, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.C.; Leiter, L.A.; Stroes, E.S.G.; Baum, S.J.; Hanselman, J.C.; Bloedon, L.T.; Lalwani, N.D.; Patel, P.M.; Zhao, X.; Duell, P.B. Effect of Bempedoic Acid vs Placebo Added to Maximally Tolerated Statins on Low-Density Lipoprotein Cholesterol in Patients at High Risk for Cardiovascular Disease: The CLEAR Wisdom Randomized Clinical Trial. JAMA 2019, 322, 1780–1788. [Google Scholar] [CrossRef]
- Jia, X.; Virani, S.S. CLEAR Serenity Trial: More Clarity for the Future of Bempedoic Acid in Patients Unable to Take Statins? J. Am. Heart Assoc. 2019, 8, E012352. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, C.M.; Banach, M.; Mancini, G.B.J.; Lepor, N.E.; Hanselman, J.C.; Zhao, X.; Leiter, L.A. Efficacy and Safety of Bempedoic Acid Added to Ezetimibe in Statin-Intolerant Patients with Hypercholesterolemia: A Randomized, Placebo-Controlled Study. Atherosclerosis 2018, 277, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.G.; Pontremoli, R.; Fogacci, F.; Viazzi, F.; Borghi, C. Effect of Bempedoic Acid on Serum Uric Acid and Related Outcomes: A Systematic Review and Meta-Analysis of the Available Phase 2 and Phase 3 Clinical Studies. Drug Saf. 2020, 43, 727–736. [Google Scholar] [CrossRef]
- Cicero, A.F.G.; Fogacci, F.; Hernandez, A.V.; Banach, M.; Lipid and Blood Pressure Meta-Analysis Collaboration (LBPMC) Group and the International Lipid Expert Panel (ILEP). Efficacy and Safety of Bempedoic Acid for the Treatment of Hypercholesterolemia: A Systematic Review and Meta-Analysis. PLoS Med. 2020, 17, e1003121. [Google Scholar] [CrossRef]
- Nicholls, S.; Lincoff, A.M.; Bays, H.E.; Cho, L.; Grobbee, D.E.; Kastelein, J.J.; Libby, P.; Moriarty, P.M.; Plutzky, J.; Ray, K.K.; et al. Rationale and Design of the CLEAR-Outcomes Trial: Evaluating the Effect of Bempedoic Acid on Cardiovascular Events in Patients with Statin Intolerance. Am. Heart J. 2021, 235, 104–112. [Google Scholar] [CrossRef]
- Khvorova, A. Oligonucleotide Therapeutics—A New Class of Cholesterol-Lowering Drugs. N. Engl. J. Med. 2017, 376, 4–7. [Google Scholar] [CrossRef]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.S.; Collins, M.G.; Stoekenbroek, R.M.; Robson, R.; Wijngaard, P.L.J.; Landmesser, U.; Leiter, L.A.; Kastelein, J.J.P.; Ray, K.K.; Kallend, D. Effects of Renal Impairment on the Pharmacokinetics, Efficacy, and Safety of Inclisiran: An Analysis of the ORION-7 and ORION-1 Studies. Mayo Clin. Proc. 2020, 95, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.J.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Haghikia, A.; Leiter, L.A.; Wright, R.S.; Kallend, D.; Wijngaard, P.; Stoekenbroek, R.; Kastelein, J.J.; Ray, K.K. Effect of Inclisiran, the Small-Interfering RNA against Proprotein Convertase Subtilisin/Kexin Type 9, on Platelets, Immune Cells, and Immunological Biomarkers: A Pre-Specified Analysis from ORION-1. Cardiovasc. Res. 2021, 117, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Dyrbuś, K.; Gąsior, M.; Penson, P.; Ray, K.K.; Banach, M. Inclisiran-New Hope in the Management of Lipid Disorders? J. Clin. Lipidol. 2020, 14, 16–27. [Google Scholar] [CrossRef]
- German, C.A.; Shapiro, M.D. Small Interfering RNA Therapeutic Inclisiran: A New Approach to Targeting PCSK9. Biodrugs Clin. Immunother. Biopharm. Gene Ther. 2020, 34, 1–9. [Google Scholar] [CrossRef]
- Bardolia, C.; Amin, N.S.; Turgeon, J. Emerging Non-Statin Treatment Options for Lowering Low-Density Lipoprotein Cholesterol. Front. Cardiovasc. Med. 2021, 8, 789931. [Google Scholar] [CrossRef]
- Willy, P.J.; Umesono, K.; Ong, E.S.; Evans, R.M.; Heyman, R.A.; Mangelsdorf, D.J. LXR, a Nuclear Receptor that Defines a Distinct Retinoid Response Pathway. Genes Dev. 1995, 9, 1033–1045. [Google Scholar] [CrossRef]
- Miyata, K.S.; Mccaw, S.E.; Patel, H.V.; Rachubinski, R.A.; Capone, J.P. The Orphan Nuclear Hormone Receptor LXR Alpha Interacts with the Peroxisome Proliferator-Activated Receptor and Inhibits Peroxisome Proliferator Signaling. J. Biol. Chem. 1996, 271, 9189–9192. [Google Scholar] [CrossRef]
- Tall, A.R. Cholesterol Efflux Pathways and Other Potential Mechanisms Involved in the Athero-Protective Effect of High Density Lipoproteins. J. Intern. Med. 2008, 263, 256–273. [Google Scholar] [CrossRef]
- Edwards, P.A.; Kennedy, M.A.; Mak, P.A. LXRs; Oxysterol-Activated Nuclear Receptors That Regulate Genes Controlling Lipid Homeostasis. Vascul. Pharmacol. 2002, 38, 249–256. [Google Scholar] [CrossRef]
- Auboeuf, D.; Rieusset, J.; Fajas, L.; Vallier, P.; Frering, V.; Riou, J.P.; Staels, B.; Auwerx, J.; Laville, M.; Vidal, H. Tissue Distribution and Quantification of the Expression of mRNAs of Peroxisome Proliferator-Activated Receptors and Liver X Receptor-Alpha in Humans: No Alteration in Adipose Tissue of Obese and NIDDM Patients. Diabetes 1997, 46, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Heine, G.; Dahten, A.; Hilt, K.; Ernst, D.; Milovanovic, M.; Hartmann, B.; Worm, M. Liver X Receptors Control IgE Expression in B Cells. J. Immunol. Baltim. Md. 2009, 182, 5276–5282. [Google Scholar] [CrossRef] [PubMed]
- Wiebel, F.F.; Steffensen, K.R.; Treuter, E.; Feltkamp, D.; Gustafsson, J.A. Ligand-Independent Coregulator Recruitment by the Triply Activatable OR1/Retinoid X Receptor-Alpha Nuclear Receptor Heterodimer. Mol. Endocrinol. Baltim. Md. 1999, 13, 1105–1118. [Google Scholar]
- Wang, D.Q.-H. Regulation of Intestinal Cholesterol Absorption. Annu. Rev. Physiol. 2007, 69, 221–248. [Google Scholar] [CrossRef]
- Zhang, S.; Li, L.; Wang, J.; Zhang, T.; Ye, T.; Wang, S.; Xing, D.; Chen, W. Recent Advances in the Regulation of ABCA1 and ABCG1 by lncRNAs. Clin. Chim. Acta Int. J. Clin. Chem. 2021, 516, 100–110. [Google Scholar] [CrossRef]
- Repa, J.J.; Berge, K.E.; Pomajzl, C.; Richardson, J.A.; Hobbs, H.; Mangelsdorf, D.J. Regulation of ATP-Binding Cassette Sterol Transporters ABCG5 and ABCG8 by the Liver X Receptors Alpha and Beta. J. Biol. Chem. 2002, 277, 18793–18800. [Google Scholar] [CrossRef]
- Parikh, N.; Frishman, W.H. Liver X Receptors: A Potential Therapeutic Target for Modulating the Atherosclerotic Process. Cardiol. Rev. 2010, 18, 269–274. [Google Scholar] [CrossRef]
- Parikh, M.; Patel, K.; Soni, S.; Gandhi, T. Liver X Receptor: A Cardinal Target for Atherosclerosis and Beyond. J. Atheroscler. Thromb. 2014, 21, 519–531. [Google Scholar] [CrossRef]
- Duval, C.; Touche, V.; Tailleux, A.; Fruchart, J.-C.; Fievet, C.; Clavey, V.; Staels, B.; Lestavel, S. Niemann-Pick C1 Like 1 Gene Expression Is Down-Regulated by LXR Activators in the Intestine. Biochem. Biophys. Res. Commun. 2006, 340, 1259–1263. [Google Scholar] [CrossRef]
- Hong, C.; Tontonoz, P. Liver X Receptors in Lipid Metabolism: Opportunities for Drug Discovery. Nat. Rev. Drug Discov. 2014, 13, 433–444. [Google Scholar] [CrossRef]
- Terasaka, N.; Hiroshima, A.; Koieyama, T.; Ubukata, N.; Morikawa, Y.; Nakai, D.; Inaba, T. T-0901317, a Synthetic Liver X Receptor Ligand, Inhibits Development of Atherosclerosis in LDL Receptor-Deficient Mice. Febs Lett. 2003, 536, 6–11. [Google Scholar] [CrossRef]
- Baranowski, M. Biological Role of Liver X Receptors. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2008, 59, 31–55. [Google Scholar]
- Quinet, E.M.; Basso, M.D.; Halpern, A.R.; Yates, D.W.; Steffan, R.J.; Clerin, V.; Resmini, C.; Keith, J.C.; Berrodin, T.J.; Feingold, I.; et al. LXR Ligand Lowers LDL Cholesterol in Primates, Is Lipid Neutral in Hamster, and Reduces Atherosclerosis in Mouse. J. Lipid Res. 2009, 50, 2358–2370. [Google Scholar] [CrossRef] [PubMed]
- Bradley, M.N.; Hong, C.; Chen, M.; Joseph, S.B.; Wilpitz, D.C.; Wang, X.; Lusis, A.J.; Collins, A.; Hseuh, W.A.; Collins, J.L.; et al. Ligand Activation of LXR Beta Reverses Atherosclerosis and Cellular Cholesterol Overload in Mice Lacking LXR Alpha and Apoe. J. Clin. Invest. 2007, 117, 2337–2346. [Google Scholar] [CrossRef] [PubMed]
- Katz, A.; Udata, C.; Ott, E.; Hickey, L.; Burczynski, M.E.; Burghart, P.; Vesterqvist, O.; Meng, X. Safety, Pharmacokinetics, and Pharmacodynamics of Single Doses of LXR-623, a Novel Liver X-Receptor Agonist, in Healthy Participants. J. Clin. Pharmacol. 2009, 49, 643–649. [Google Scholar] [CrossRef]
- Kirchgessner, T.G.; Martin, R.; Sleph, P.; Grimm, D.; Liu, X.; Lupisella, J.; Smalley, J.; Narayanan, R.; Xie, Y.; Ostrowski, J.; et al. Pharmacological Characterization of a Novel Liver X Receptor Agonist with Partial LXRα Activity and a Favorable Window in Nonhuman Primates. J. Pharmacol. Exp. Ther. 2015, 352, 305–314. [Google Scholar] [CrossRef]
- Kirchgessner, T.G.; Sleph, P.; Ostrowski, J.; Lupisella, J.; Ryan, C.S.; Liu, X.; Fernando, G.; Grimm, D.; Shipkova, P.; Zhang, R.; et al. Beneficial and Adverse Effects of an LXR Agonist on Human Lipid and Lipoprotein Metabolism and Circulating Neutrophils. Cell Metab. 2016, 24, 223–233. [Google Scholar] [CrossRef]
- Li, N.; Wang, X.; Xu, Y.; Lin, Y.; Zhu, N.; Liu, P.; Lu, D.; Si, S. Identification of a Novel Liver X Receptor Agonist that Regulates the Expression of Key Cholesterol Homeostasis Genes with Distinct Pharmacological Characteristics. Mol. Pharmacol. 2017, 91, 264–276. [Google Scholar] [CrossRef]
- Han, L.; Shen, W.-J.; Bittner, S.; Kraemer, F.B.; Azhar, S. PPARs: Regulators of Metabolism and As Therapeutic Targets in Cardiovascular Disease. Part II: PPAR-β/δ and PPAR-γ. Future Cardiol. 2017, 13, 279–296. [Google Scholar] [CrossRef]
- Shende, V.R.; Singh, A.B.; Liu, J. A Novel Peroxisome Proliferator Response Element Modulates Hepatic Low-Density Lipoprotein Receptor Gene Transcription in Response to PPARδ Activation. Biochem. J. 2015, 472, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Zhai, Y.; Wang, J. The Role of PPAR and Its Cross-Talk with Car and LXR in Obesity and Atherosclerosis. Int. J. Mol. Sci. 2018, 19, 1260. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S. Angiopoietin-like 3 in Lipoprotein Metabolism. Nat. Rev. Endocrinol. 2017, 13, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Biterova, E.; Esmaeeli, M.; Alanen, H.I.; Saaranen, M.; Ruddock, L.W. Structures of Angptl3 and Angptl4, Modulators of Triglyceride Levels and Coronary Artery Disease. Sci. Rep. 2018, 8, 6752. [Google Scholar] [CrossRef]
- Lu, X. Structure and Function of Angiopoietin-like Protein 3 (ANGPTL3) in Atherosclerosis. Curr. Med. Chem. 2020, 27, 5159–5174. [Google Scholar] [CrossRef]
- Ono, M.; Shimizugawa, T.; Shimamura, M.; Yoshida, K.; Noji-Sakikawa, C.; Ando, Y.; Koishi, R.; Furukawa, H. Protein Region Important for Regulation of Lipid Metabolism in Angiopoietin-like 3 (ANGPTL3): ANGPTL3 Is Cleaved and Activated In Vivo. J. Biol. Chem. 2003, 278, 41804–41809. [Google Scholar] [CrossRef]
- Dewey, F.E.; Gusarova, V.; Dunbar, R.L.; O’dushlaine, C.; Schurmann, C.; Gottesman, O.; Mccarthy, S.; Van Hout, C.V.; Bruse, S.; Dansky, H.M.; et al. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 211–221. [Google Scholar] [CrossRef]
- Kersten, S. Bypassing the LDL Receptor in Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 383, 775–776. [Google Scholar] [CrossRef]
- Cuchel, M.; Bruckert, E.; Ginsberg, H.N.; Raal, F.J.; Santos, R.D.; Hegele, R.A.; Kuivenhoven, J.A.; Nordestgaard, B.G.; Descamps, O.S.; Steinhagen-Thiessen, E.; et al. Homozygous Familial Hypercholesterolaemia: New Insights and Guidance for Clinicians to Improve Detection and Clinical Management. A Position Paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur. Heart J. 2014, 35, 2146–2157. [Google Scholar] [CrossRef]
- Musunuru, K.; Pirruccello, J.P.; Do, R.; Peloso, G.M.; Guiducci, C.; Sougnez, C.; Garimella, K.V.; Fisher, S.; Abreu, J.; Barry, A.J.; et al. Exome Sequencing, ANGPTL3 Mutations, and Familial Combined Hypolipidemia. N. Engl. J. Med. 2010, 363, 2220–2227. [Google Scholar] [CrossRef]
- Wang, Y.; Gusarova, V.; Banfi, S.; Gromada, J.; Cohen, J.C.; Hobbs, H.H. Inactivation of ANGPTL3 Reduces Hepatic VLDL-Triglyceride Secretion. J. Lipid Res. 2015, 56, 1296–1307. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, D.; Gipe, D.A.; Pordy, R.; Ahmad, Z.; Cuchel, M.; Shah, P.K.; Chyu, K.-Y.; Sasiela, W.J.; Chan, K.-C.; Brisson, D.; et al. ANGPTL3 Inhibition in Homozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2017, 377, 296–297. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Rosenson, R.S.; Reeskamp, L.F.; Hovingh, G.K.; Kastelein, J.J.P.; Rubba, P.; Ali, S.; Banerjee, P.; Chan, K.-C.; Gipe, D.A.; et al. Evinacumab for Homozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 383, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.J.; Lee, R.G.; Brandt, T.A.; Tai, L.-J.; Fu, W.; Peralta, R.; Yu, R.; Hurh, E.; Paz, E.; Mcevoy, B.W.; et al. Cardiovascular and Metabolic Effects of ANGPTL3 Antisense Oligonucleotides. N. Engl. J. Med. 2017, 377, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Nurmohamed, N.S.; Dallinga-Thie, G.M.; Stroes, E.S.G. Targeting apoC-III and ANGPTL3 in the Treatment of Hypertriglyceridemia. Expert Rev. Cardiovasc. Ther. 2020, 18, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Agarwala, A.; Jones, P.; Nambi, V. The Role of Antisense Oligonucleotide Therapy in Patients with Familial Hypercholesterolemia: Risks, Benefits, and Management Recommendations. Curr. Atheroscler. Rep. 2015, 17, 467. [Google Scholar] [CrossRef] [PubMed]
- Ricotta, D.N.; Frishman, W. Mipomersen: A Safe and Effective Antisense Therapy Adjunct to Statins in Patients with Hypercholesterolemia. Cardiol. Rev. 2012, 20, 90–95. [Google Scholar] [CrossRef]
- Wong, E.; Goldberg, T. Mipomersen (Kynamro): A Novel Antisense Oligonucleotide Inhibitor for the Management of Homozygous Familial Hypercholesterolemia. P T Peer-Rev. J. Formul. Manag. 2014, 39, 119–122. [Google Scholar]
- Kastelein, J.J.P.; Wedel, M.K.; Baker, B.F.; Su, J.; Bradley, J.D.; Yu, R.Z.; Chuang, E.; Graham, M.J.; Crooke, R.M. Potent Reduction of Apolipoprotein B and Low-Density Lipoprotein Cholesterol by Short-Term Administration of an Antisense Inhibitor of Apolipoprotein, B. Circulation 2006, 114, 1729–1735. [Google Scholar] [CrossRef]
- Akdim, F.; Stroes, E.S.G.; Sijbrands, E.J.G.; Tribble, D.L.; Trip, M.D.; Jukema, J.W.; Flaim, J.D.; Su, J.; Yu, R.; Baker, B.F.; et al. Efficacy and Safety of Mipomersen, an Antisense Inhibitor of Apolipoprotein B, in Hypercholesterolemic Subjects Receiving Stable Statin Therapy. J. Am. Coll. Cardiol. 2010, 55, 1611–1618. [Google Scholar] [CrossRef]
- Akdim, F.; Visser, M.E.; Tribble, D.L.; Baker, B.F.; Stroes, E.S.G.; Yu, R.; Flaim, J.D.; Su, J.; Stein, E.A.; Kastelein, J.J.P. Effect of Mipomersen, an Apolipoprotein B Synthesis Inhibitor, on Low-Density Lipoprotein Cholesterol in Patients with Familial Hypercholesterolemia. Am. J. Cardiol. 2010, 105, 1413–1419. [Google Scholar] [CrossRef] [PubMed]
- Akdim, F.; Tribble, D.L.; Flaim, J.D.; Yu, R.; Su, J.; Geary, R.S.; Baker, B.F.; Fuhr, R.; Wedel, M.K.; Kastelein, J.J.P. Efficacy of Apolipoprotein B Synthesis Inhibition in Subjects with Mild-To-Moderate Hyperlipidaemia. Eur. Heart J. 2011, 32, 2650–2659. [Google Scholar] [CrossRef] [PubMed]
- Mcgowan, M.P.; Tardif, J.-C.; Ceska, R.; Burgess, L.J.; Soran, H.; Gouni-Berthold, I.; Wagener, G.; Chasan-Taber, S. Randomized, Placebo-Controlled Trial of Mipomersen in Patients with Severe Hypercholesterolemia Receiving Maximally Tolerated Lipid-Lowering Therapy. PLoS ONE 2012, 7, e49006. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.A.; Dufour, R.; Gagne, C.; Gaudet, D.; East, C.; Donovan, J.M.; Chin, W.; Tribble, D.L.; Mcgowan, M. Apolipoprotein B Synthesis Inhibition with Mipomersen in Heterozygous Familial Hypercholesterolemia: Results of a Randomized, Double-Blind, Placebo-Controlled Trial to Assess Efficacy and Safety as Add-On Therapy in Patients with Coronary Artery Disease. Circulation 2012, 126, 2283–2292. [Google Scholar] [CrossRef]
- Thomas, G.S.; Cromwell, W.C.; Ali, S.; Chin, W.; Flaim, J.D.; Davidson, M. Mipomersen, an Apolipoprotein B Synthesis Inhibitor, Reduces Atherogenic Lipoproteins in Patients with Severe Hypercholesterolemia at High Cardiovascular Risk: A Randomized, Double-Blind, Placebo-Controlled Trial. J. Am. Coll. Cardiol. 2013, 62, 2178–2184. [Google Scholar] [CrossRef]
- Khoury, E.; Brisson, D.; Roy, N.; Tremblay, G.; Gaudet, D. Review of the Long-Term Safety of Lomitapide: A Microsomal Triglycerides Transfer Protein Inhibitor for Treating Homozygous Familial Hypercholesterolemia. Expert Opin. Drug Saf. 2019, 18, 403–414. [Google Scholar] [CrossRef]
- Liu, X.; Men, P.; Wang, Y.; Zhai, S.; Zhao, Z.; Liu, G. Efficacy and Safety of Lomitapide in Hypercholesterolemia. Am. J. Cardiovasc. Drugs Drugs Devices Interv. 2017, 17, 299–309. [Google Scholar] [CrossRef]
- Vuorio, A.; Tikkanen, M.J.; Kovanen, P.T. Inhibition of Hepatic Microsomal Triglyceride Transfer Protein—A Novel Therapeutic Option for Treatment of Homozygous Familial Hypercholesterolemia. Vasc. Health Risk Manag. 2014, 10, 263–270. [Google Scholar] [CrossRef]
- Stefanutti, C. Lomitapide-A Microsomal Triglyceride Transfer Protein Inhibitor for Homozygous Familial Hypercholesterolemia. Curr. Atheroscler. Rep. 2020, 22, 38. [Google Scholar] [CrossRef]
- Available online: https://www.ema.europa.eu/en/documents/overview/lojuxta-epar-summary-public_it.pdf (accessed on 25 February 2022).
- Cuchel, M.; Bloedon, L.T.; Szapary, P.O.; Kolansky, D.M.; Wolfe, M.L.; Sarkis, A.; Millar, J.S.; Ikewaki, K.; Siegelman, E.S.; Gregg, R.E.; et al. Inhibition of Microsomal Triglyceride Transfer Protein in Familial Hypercholesterolemia. N. Engl. J. Med. 2007, 356, 148–156. [Google Scholar] [CrossRef]
- Cuchel, M.; Meagher, E.A.; Du Toit Theron, H.; Blom, D.J.; Marais, A.D.; Hegele, R.A.; Averna, M.R.; Sirtori, C.R.; Shah, P.K.; Gaudet, D.; et al. Efficacy and Safety of a Microsomal Triglyceride Transfer Protein Inhibitor in Patients with Homozygous Familial Hypercholesterolaemia: A Single-Arm, Open-Label, Phase 3 Study. Lancet Lond. Engl. 2013, 381, 40–46. [Google Scholar] [CrossRef]
- Blom, D.J.; Averna, M.R.; Meagher, E.A.; Du Toit Theron, H.; Sirtori, C.R.; Hegele, R.A.; Shah, P.K.; Gaudet, D.; Stefanutti, C.; Vigna, G.B.; et al. Long-Term Efficacy and Safety of the Microsomal Triglyceride Transfer Protein Inhibitor Lomitapide in Patients with Homozygous Familial Hypercholesterolemia. Circulation 2017, 136, 332–335. [Google Scholar] [CrossRef] [PubMed]
- Samaha, F.F.; Mckenney, J.; Bloedon, L.T.; Sasiela, W.J.; Rader, D.J. Inhibition of Microsomal Triglyceride Transfer Protein Alone or with Ezetimibe in Patients with Moderate Hypercholesterolemia. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Stefanutti, C.; Blom, D.J.; Averna, M.R.; Meagher, E.A.; Theron, H.D.; Marais, A.D.; Hegele, R.A.; Sirtori, C.R.; Shah, P.K.; Gaudet, D.; et al. The Lipid-Lowering Effects of Lomitapide Are Unaffected by Adjunctive Apheresis in Patients with Homozygous Familial Hypercholesterolaemia—A Post-Hoc Analysis of A Phase 3, Single-Arm, Open-Label Trial. Atherosclerosis 2015, 240, 408–414. [Google Scholar] [CrossRef]
- Blom, D.J.; Fayad, Z.A.; Kastelein, J.J.P.; Larrey, D.; Makris, L.; Schwamlein, C.; Bloeden, L.; Underberg, J.; LOWER Investigators. LOWER, a Registry of Lomitapide-Treated Patients with Homozygous Familial Hypercholesterolemia: Rationale and Design. J. Clin. Lipidol. 2016, 10, 273–282. [Google Scholar] [CrossRef]
- Goulooze, S.C.; Cohen, A.F.; Rissmann, R. Lomitapide. Br. J. Clin. Pharmacol. 2015, 80, 179–181. [Google Scholar] [CrossRef]
- Gaudet, D.; Alexander, V.J.; Baker, B.F.; Brisson, D.; Tremblay, K.; Singleton, W.; Geary, R.S.; Hughes, S.G.; Viney, N.J.; Graham, M.J.; et al. Antisense Inhibition of Apolipoprotein C-III in Patients with Hypertriglyceridemia. N. Engl. J. Med. 2015, 373, 438–447. [Google Scholar] [CrossRef]
- Gouni-Berthold, I.; Alexander, V.J.; Yang, Q.; Hurh, E.; Steinhagen-Thiessen, E.; Moriarty, P.M.; Hughes, S.G.; Gaudet, D.; Hegele, R.A.; O’dea, L.S.L.; et al. Efficacy and Safety of Volanesorsen in Patients with Multifactorial Chylomicronaemia (COMPASS): A Multicentre, Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Diabetes Endocrinol. 2021, 9, 264–275. [Google Scholar] [CrossRef]
- Digenio, A.; Dunbar, R.L.; Alexander, V.J.; Hompesch, M.; Morrow, L.; Lee, R.G.; Graham, M.J.; Hughes, S.G.; Yu, R.; Singleton, W.; et al. Antisense-Mediated Lowering of Plasma Apolipoprotein C-III by Volanesorsen Improves Dyslipidemia and Insulin Sensitivity in Type 2 Diabetes. Diabetes Care 2016, 39, 1408–1415. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drugs | MOA | Efficacy | EMA Approval | FDA Approval |
|---|---|---|---|---|
| PCSK9-i | Monoclonal antibodies that inhibit PCSK9, diminishing the recycling of LDL-R | Medium decrease of LDL-C: 61.9% [12,13] |  |  |
| Bempedoic Acid | The active metabolite inhibits adenosine triphosphate citrate lyase, leading to reduced acetyl CoA levels in the cholesterol synthesis pathway upstream of HMG-CoA reductase | Medium decrease of LDL-C: 27% [31,32] BA plus ezetimibe decrease LDL-C up to 48% [37] |  |  |
| Inclisiran | siRNA targeting 3′ UTR of the PCSK9 mRNA leading to suppression of hepatic PCSK9 production | Medium decrease of LDL-C: 52.3% [45] |  |  |
| LXR agonists | Activation of LXR in macrophages induces cholesterol efflux. LXR reduce cholesterol absorption and promote intestinal excretion, increasing Apo E expression and downregulating Niemann-Pick C1 like 1 | No RCTs available No human |  |  |
| PPARs agonists | Proteins that modulate the transcription of the target genes regulating glucose, TG and lipoprotein metabolism, cell proliferation, inflammation, and vascular tissue function | Seladelpar induce a medium decrease of LDL-C by 18–43% [71,72,73] |  |  |
| ANGPTL3 inhibitors | ANGPTL3 inhibits LPL from combining with the glycosylphosphatidylinositol-anchored HDL binding protein 1: Inhibition of ANGPTL3 results in lower plasma TG, ApoB, LDL-C and HDL-C | Evinacumab induce a medium decrease of TG levels up to 76% and LDL-C levels up to 23% [78] The addition of i.v. evinacumab to the treatment regimen of HoFH patients resulted in a mean reduction of LDL-C levels by 49% ± 23% after 4 weeks [78] |  |  |
| Mipomersen | Antisense oligonucleotide capable of binding ApoB-100 mRNA leading to decrease production of LDL, VLDL and Lp(a) | Medium decrease of LDL-C up to 71% [91,92,93] |  |  |
| Lomitapide | MTP inhibitor leading to reduced lipoprotein production (especially of Apo B) | Used for the treatment of FHMedium decrease of LDL-C levels up to 30% [106] In combination with ezetimibe LDL-C was reduced up to 46% [106] |  |  |
| Volanesorsen | Antisense oligo-nucleotide target to reduce hepatic ApoC-III mRNA | Medium decrease of TG levels up to 71.2% [110] |  |  |
| Drugs | Indication | Contraindications | Side Effects |
|---|---|---|---|
| PCSK9-i | Adults with primary hyperlipidemia (including HeFH) as an adjunct to diet, alone or in combination with other lipid-lowering therapies; In patients with HoFH as an adjunct to diet and other LDL-lowering therapies (e.g., statins, ezetimibe, LDL apheresis) | Patients with a history of a serious hypersensitivity reaction to PCSK9-i | Nasopharyngitis, upper respiratory tract infection, influenza, back pain, and injection site reactions [20,21] |
| Bempedoic Acid | Adults with HeFH or established ASCVD who require additional lowering of LDL-C as an adjunct to diet and maximally tolerated statin therapy | None | Upper respiratory tract infection, muscle spasms, hyperuricemia, back pain, abdominal pain or discomfort, bronchitis, pain in extremity, anemia and elevated liver enzymes [33] |
| Inclisiran | Adults with HeFH or ASCVD, who require additional lowering of LDL-C as an adjunct to diet and maximally tolerated statin therapy | None | Injection site reaction, arthralgia, urinary tract infection, diarrhea, bronchitis, pain in extremity, and dyspnea [45,46,47] |
| LXR agonists | Not yet approved | No human trials available | No human trials available |
| PPARs β/δ agonists | Not yet approved | No human trials available | No human trials available |
| ANGPTL3 inhibitors | Adult and pediatric patients, aged 12 years and older, with HoFH as an adjunct to other LDL-C lowering therapies | History of serious hypersensitivity reactions to ANGPTL3 inhibitors | Nasopharyngitis, influenza-like illness, dizziness, rhinorrhea, and nausea [85,86] |
| Mipomersen | In patients with HoFH as an adjunct to lipid-lowering medications and diet to reduce LDL-C, ApoB, TC, and non HDL-C | Moderate or severe hepatic impairment, or active liver disease, including unexplained persistent elevations of serum transaminases; Known sensitivity to product components | Injection site reactions, flu-like symptoms, nausea, headache, and elevations in serum transaminases, specifically ALT [87,96] |
| Lomitapide | In patients with HoFH to reduce LDL-C, TC, apo B, and non-HDL-C as an adjunct to a low-fat diet and other lipid-lowering treatments, including LDL apheresis | Pregnancy, concomitant use with strong or moderate CYP3A4 inhibitors, moderate or severe hepatic impairment or active liver disease including unexplained persistent abnormal liver function tests | Diarrhea, nausea, vomiting, dyspepsia, and abdominal pain [103,104,108] |
| Volanesorsen | In adult patients with genetically confirmed FCS and at high risk for pancreatitis, in whom response to diet and triglyceride lowering therapy has been inadequate | Hypersensitivity to the drug; Chronic or unexplained thrombocytopenia. Treatment should not be initiated in patients with platelet count < 1.40 × 1011/L | Injection site reactions, serum sickness, and thrombocytopenia [110] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muscoli, S.; Ifrim, M.; Russo, M.; Candido, F.; Sanseviero, A.; Milite, M.; Di Luozzo, M.; Marchei, M.; Sangiorgi, G.M. Current Options and Future Perspectives in the Treatment of Dyslipidemia. J. Clin. Med. 2022, 11, 4716. https://doi.org/10.3390/jcm11164716
Muscoli S, Ifrim M, Russo M, Candido F, Sanseviero A, Milite M, Di Luozzo M, Marchei M, Sangiorgi GM. Current Options and Future Perspectives in the Treatment of Dyslipidemia. Journal of Clinical Medicine. 2022; 11(16):4716. https://doi.org/10.3390/jcm11164716
Chicago/Turabian StyleMuscoli, Saverio, Mihaela Ifrim, Massimo Russo, Francesco Candido, Angela Sanseviero, Marialucia Milite, Marco Di Luozzo, Massimo Marchei, and Giuseppe Massimo Sangiorgi. 2022. "Current Options and Future Perspectives in the Treatment of Dyslipidemia" Journal of Clinical Medicine 11, no. 16: 4716. https://doi.org/10.3390/jcm11164716
APA StyleMuscoli, S., Ifrim, M., Russo, M., Candido, F., Sanseviero, A., Milite, M., Di Luozzo, M., Marchei, M., & Sangiorgi, G. M. (2022). Current Options and Future Perspectives in the Treatment of Dyslipidemia. Journal of Clinical Medicine, 11(16), 4716. https://doi.org/10.3390/jcm11164716