

In Vitro Study of Ineffective Erythropoiesis in Thalassemia: Diverse Intrinsic Pathophysiological Features of Erythroid Cells Derived from Various Thalassemia Syndromes
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participant Samples
2.2. Hematopoietic CD34+ Cell Isolation and In Vitro Erythroid Cell Culture
2.3. Flow Cytometric Analysis
2.4. RNA Isolation and Quantitative RT-PCR Analysis
2.5. Hemoglobin Quantification
2.6. Statistical Analysis
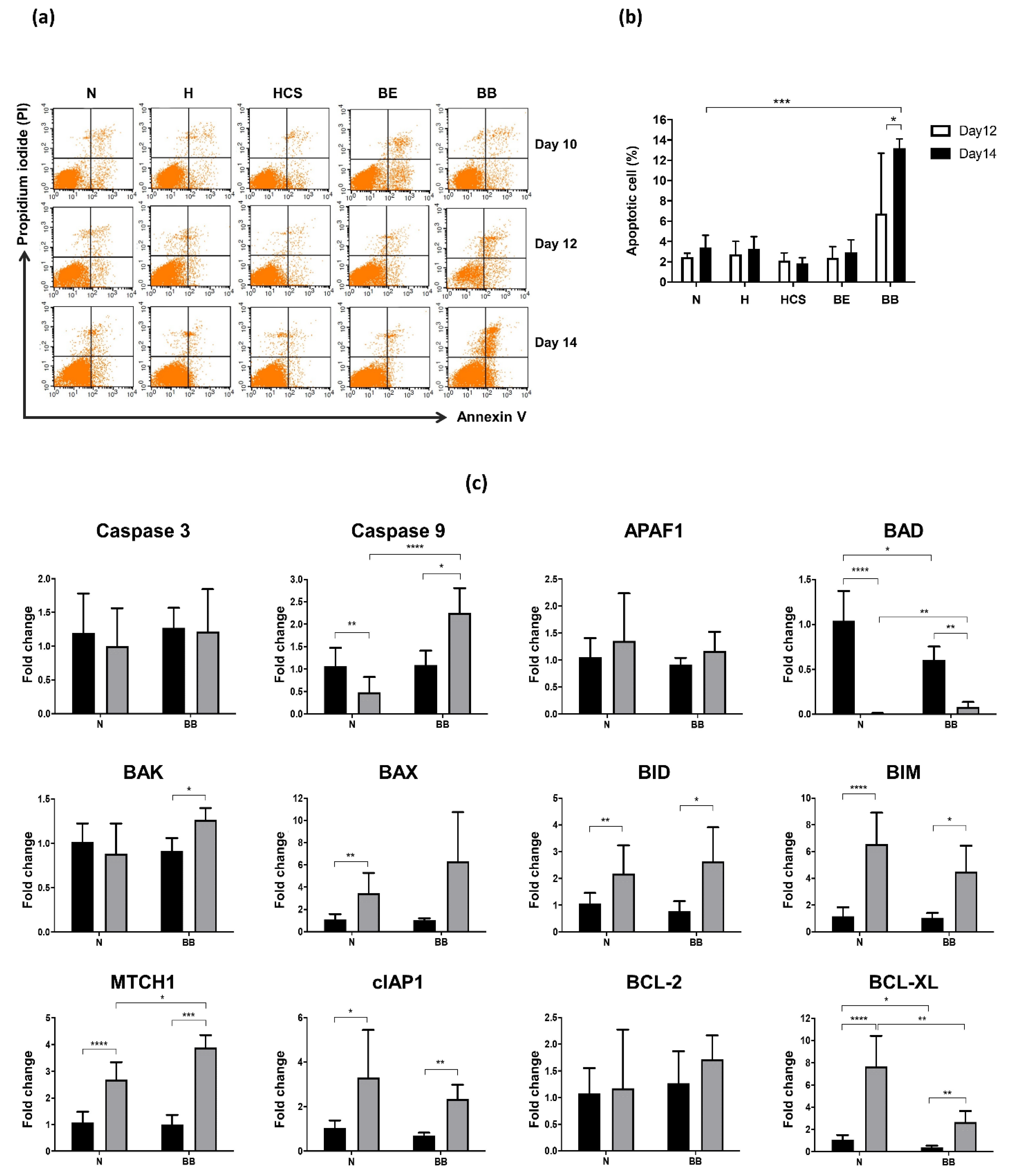
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fucharoen, S.; Weatherall, D.J. Progress toward the control and management of the thalassemias. Hematol. Oncol. Clin. N. Am. 2016, 30, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Weatherall, D.J.; Cappellini, M.D. Thalassaemia. Lancet 2018, 391, 155–167. [Google Scholar] [CrossRef]
- Olivieri, N.F. The beta-thalassemias. N. Engl. J. Med. 1999, 341, 99–109. [Google Scholar] [CrossRef]
- Fucharoen, S.; Weatherall, D.J. The hemoglobin E thalassemias. Cold Spring Harb. Perspect. Med. 2012, 2, a011734. [Google Scholar] [CrossRef]
- Fucharoen, S.; Viprakasit, V. Hb H disease: Clinical course and disease modifiers. Hematol. Am. Soc. Hematol. Educ. Program 2009, 2009, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Musallam, K.M.; Taher, A.T.; Rivella, S. Ineffective erythropoiesis: Anemia and iron overload. Hematol. Oncol. Clin. N. Am. 2018, 32, 213–221. [Google Scholar] [CrossRef]
- Mathias, L.A.; Fisher, T.C.; Zeng, L.; Meiselman, H.J.; Weinberg, K.I.; Hiti, A.L.; Malik, P. Ineffective erythropoiesis in beta-thalassemia major is due to apoptosis at the polychromatophilic normoblast stage. Exp. Hematol. 2000, 28, 1343–1353. [Google Scholar] [CrossRef]
- Kittikalayawong, Y.; Sila-asna, M.; Bunyaratvej, A. Enhanced maturation and proliferation of beta-thalassemia/Hb E erythroid precursor cells in culture. Southeast Asian J. Trop. Med. Public Health 2005, 36, 1298–1303. [Google Scholar]
- Wannatung, T.; Lithanatudom, P.; Leecharoenkiat, A.; Svasti, S.; Fucharoen, S.; Smith, D.R. Increased erythropoiesis of beta-thalassaemia/Hb E proerythroblasts is mediated by high basal levels of ERK1/2 activation. Br. J. Haematol. 2009, 146, 557–568. [Google Scholar] [CrossRef]
- Suriyun, T.; Kaewsakulthong, W.; Khamphikham, P.; Chumchuen, S.; Hongeng, S.; Fucharoen, S.; Sripichai, O. Association of the degree of erythroid expansion and maturation arrest with the clinical severity of β0-thalassemia/hemoglobin E patients. Acta Haematol. 2021, 144, 660–671. [Google Scholar] [CrossRef]
- Suriyun, T.; Winichagoon, P.; Fucharoen, S.; Sripichai, O. Impaired terminal erythroid maturation in β0-thalassemia/HbE patients with different clinical severity. J. Clin. Med. 2022, 11, 1755. [Google Scholar] [CrossRef] [PubMed]
- Sriiam, S.; Leecharoenkiat, A.; Lithanatudom, P.; Wannatung, T.; Svasti, S.; Fucharoen, S.; Svasti, J.; Chokchaichamnankit, D.; Srisomsap, C.; Smith, D.R. Proteomic analysis of hemoglobin H-constant spring (Hb H-CS) erythroblasts. Blood Cells Mol. Dis. 2012, 48, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Angelucci, E.; Lucarelli, G.; Aljurf, M.; Snyder, L.M.; Kiefer, C.R.; Ma, L.; Schrier, S.L. Accelerated programmed cell death (apoptosis) in erythroid precursors of patients with severe beta-thalassemia (Cooley’s anemia). Blood 1993, 82, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Centis, F.; Tabellini, L.; Lucarelli, G.; Buffi, O.; Tonucci, P.; Persini, B.; Annibali, M.; Emiliani, R.; Iliescu, A.; Rapa, S.; et al. The importance of erythroid expansion in determining the extent of apoptosis in erythroid precursors in patients with beta-thalassemia major. Blood 2000, 96, 3624–3629. [Google Scholar] [CrossRef] [PubMed]
- Pootrakul, P.; Sirankapracha, P.; Hemsorach, S.; Moungsub, W.; Kumbunlue, R.; Piangitjagum, A.; Wasi, P.; Ma, L.; Schrier, S.L. A correlation of erythrokinetics, ineffective erythropoiesis, and erythroid precursor apoptosis in thai patients with thalassemia. Blood 2000, 96, 2606–2612. [Google Scholar] [CrossRef] [PubMed]
- Forster, L.; Cornwall, S.; Finlayson, J.; Ghassemifar, R. Cell cycle, proliferation and apoptosis in erythroblasts cultured from patients with β-thalassaemia major. Br. J. Haematol. 2016, 175, 539–542. [Google Scholar] [CrossRef]
- Kheansaard, W.; Panichob, P.; Fucharoen, S.; Tanyong, D.I. Cytokine-induced apoptosis of beta-thalassemia/hemoglobin E erythroid progenitor cells via nitric oxide-mediated process in vitro. Acta Haematol. 2011, 126, 224–230. [Google Scholar] [CrossRef]
- Ponnikorn, S.; Panichakul, T.; Sresanga, K.; Wongborisuth, C.; Roytrakul, S.; Hongeng, S.; Tungpradabkul, S. Phosphoproteomic analysis of apoptotic hematopoietic stem cells from hemoglobin E/β-thalassemia. J. Transl. Med. 2011, 9, 96. [Google Scholar] [CrossRef]
- Sripichai, O.; Munkongdee, T.; Kumkhaek, C.; Svasti, S.; Winichagoon, P.; Fucharoen, S. Coinheritance of the different copy numbers of alpha-globin gene modifies severity of beta-thalassemia/Hb E disease. Ann. Hematol. 2008, 87, 375–379. [Google Scholar] [CrossRef]
- Tangprasittipap, A.; Kaewprommal, P.; Sripichai, O.; Sathirapongsasuti, N.; Satirapod, C.; Shaw, P.J.; Piriyapongsa, J.; Hongeng, S. Comparison of gene expression profiles between human erythroid cells derived from fetal liver and adult peripheral blood. PeerJ 2018, 6, e5527. [Google Scholar] [CrossRef]
- Schrier, S.L.; Bunyaratvej, A.; Khuhapinant, A.; Fucharoen, S.; Aljurf, M.; Snyder, L.M.; Keifer, C.R.; Ma, L.; Mohandas, N. The unusual pathobiology of hemoglobin constant spring red blood cells. Blood 1997, 89, 1762–1769. [Google Scholar] [CrossRef] [Green Version]
- Libani, I.V.; Guy, E.C.; Melchiori, L.; Schiro, R.; Ramos, P.; Breda, L.; Scholzen, T.; Chadburn, A.; Liu, Y.; Kernbach, M.; et al. Decreased differentiation of erythroid cells exacerbates ineffective erythropoiesis in beta-thalassemia. Blood 2008, 112, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Mathangasinghe, Y.; Fauvet, B.; Jane, S.M.; Goloubinoff, P.; Nillegoda, N.B. The Hsp70 chaperone system: Distinct roles in erythrocyte formation and maintenance. Haematologica 2021, 106, 1519–1534. [Google Scholar] [CrossRef] [PubMed]
- Ribeil, J.A.; Zermati, Y.; Vandekerckhove, J.; Cathelin, S.; Kersual, J.; Dussiot, M.; Coulon, S.; Moura, I.C.; Zeuner, A.; Kirkegaard-Sørensen, T.; et al. Hsp70 regulates erythropoiesis by preventing caspase-3-mediated cleavage of GATA-1. Nature 2007, 445, 102–105. [Google Scholar] [CrossRef]
- Arlet, J.B.; Ribeil, J.A.; Guillem, F.; Negre, O.; Hazoume, A.; Marcion, G.; Beuzard, Y.; Dussiot, M.; Moura, I.C.; Demarest, S.; et al. HSP70 sequestration by free α-globin promotes ineffective erythropoiesis in β-thalassaemia. Nature 2014, 514, 242–246. [Google Scholar] [CrossRef]
- Guillem, F.; Dussiot, M.; Colin, E.; Suriyun, T.; Arlet, J.B.; Goudin, N.; Marcion, G.; Seigneuric, R.; Causse, S.; Gonin, P.; et al. XPO1 regulates erythroid differentiation and is a new target for the treatment of β-thalassemia. Haematologica 2020, 105, 2240–2249. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Sinha, S.C.; Kroemer, G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy 2008, 4, 600–606. [Google Scholar] [CrossRef]
- Kim, R. Unknotting the roles of Bcl-2 and Bcl-xL in cell death. Biochem. Biophys. Res. Commun. 2005, 333, 336–343. [Google Scholar] [CrossRef]
- Afreen, S.; Bohler, S.; Müller, A.; Demmerath, E.M.; Weiss, J.M.; Jutzi, J.S.; Schachtrup, K.; Kunze, M.; Erlacher, M. BCL-XL expression is essential for human erythropoiesis and engraftment of hematopoietic stem cells. Cell Death Dis. 2020, 11, 8. [Google Scholar] [CrossRef]
- Yang, E.; Zha, J.; Jockel, J.; Boise, L.H.; Thompson, C.B.; Korsmeyer, S.J. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 1995, 80, 285–291. [Google Scholar] [CrossRef]
- Zha, J.; Harada, H.; Osipov, K.; Jockel, J.; Waksman, G.; Korsmeyer, S.J. BH3 domain of BAD is required for heterodimerization with BCL-XL and pro-apoptotic activity. J. Biol. Chem. 1997, 272, 24101–24104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamarca, V.; Marzo, I.; Sanz-Clemente, A.; Carrodeguas, J.A. Exposure of any of two proapoptotic domains of presenilin 1-associated protein/mitochondrial carrier homolog 1 on the surface of mitochondria is sufficient for induction of apoptosis in a Bax/Bak-independent manner. Eur. J. Cell Biol. 2008, 87, 325–334. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
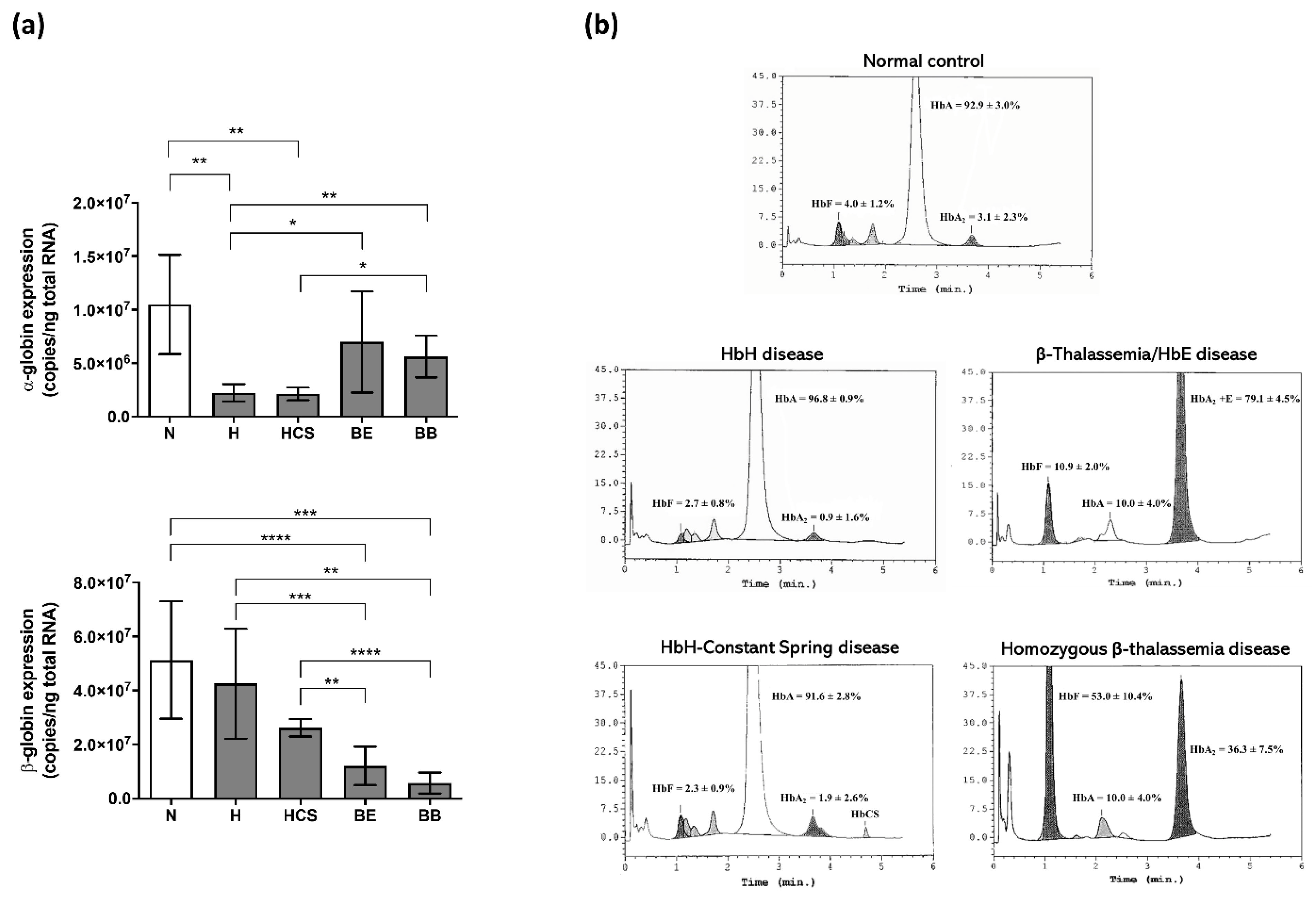
| Study Participants | No. | RBC (×106 cells/uL) | Hb (g/dL) | Hct (%) | MCV (fL) | MCH (pg) | MCHC (g/dL) |
|---|---|---|---|---|---|---|---|
| Healthy normal | 5 | 4.5 ± 0.2 | 12.8 ± 0.8 | 38.5 ± 1.8 | 86.5 ± 3.5 | 28.6 ± 1.3 | 33.1 ± 0.6 |
| Hemoglobin H disease | 5 | 5.6 ± 0.5 | 9.1 ± 1.2 | 31.5 ± 3.3 | 55.8 ± 2.7 | 16.3 ± 1.4 | 29.1 ± 2.1 |
| Hemoglobin H-Constant Spring disease | 5 | 4.0 ± 0.6 | 7.8 ± 0.5 | 30.9 ± 4.7 | 77.4 ± 4.3 | 19.8 ± 2.6 | 25.6 ± 2.7 |
| β0-Thalassemia/hemoglobin E disease | 5 * | 3.9 ± 0.3 | 8.3 ± 1.1 | 26.9 ± 2.6 | 68.2 ± 4.1 | 21.2 ± 2.4 | 31.0 ± 2.1 |
| Homozygous β0-thalassemia disease | 4 * | 2.7 ± 0.8 | 7.0 ± 2.0 | 21.0 ± 5.6 | 77.8 ± 3.2 | 26.0 ± 0.9 | 33.5 ± 1.9 |
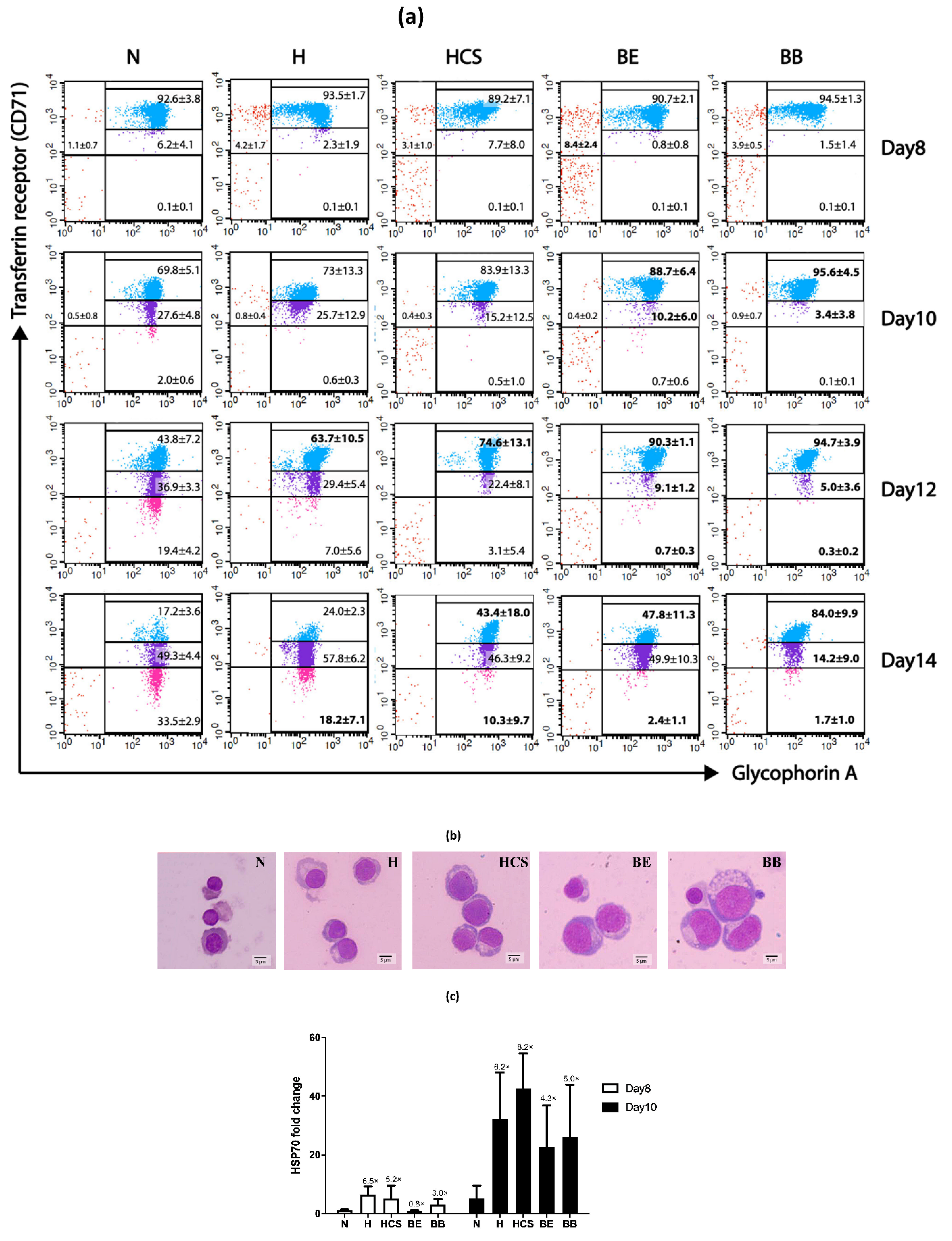
| Day of Culture | Erythroid Populations | Normal | HbH Disease | HbH-CS Disease | β0-Thalassemia/ HbE Disease | Homozygous β0-Thalassemia |
|---|---|---|---|---|---|---|
| Day 8 | CD71 high/GPA negative | 1.1 ± 0.7 | 4.2 ± 1.7 | 3.1 ± 1.0 | 8.4 ± 2.4 * | 3.9 ± 0.5 |
| CD71 high/GPA positive | 92.6 ± 3.8 | 93.5 ± 1.7 | 89.2 ± 7.1 | 90.7 ± 2.1 | 94.5 ± 1.3 | |
| CD71 moderate/GPA positive | 6.2 ± 4.1 | 2.3 ± 1.9 | 7.7 ± 8.0 | 0.8 ± 0.8 | 1.5 ± 1.4 | |
| CD71 low/GPA positive | 0.1 ± 0.1 | 0.1 ± 0.1 | 0.1 ± 0.1 | 0.1 ± 0.1 | 0.1 ± 0.1 | |
| Day 10 | CD71 high/GPA negative | 0.5 ± 0.8 | 0.8 ± 0.4 | 0.4 ± 0.3 | 0.4 ± 0.2 | 0.9 ± 0.7 |
| CD71 high/GPA positive | 69.8 ± 5.1 | 73.0 ± 13.3 | 83.9 ± 13.3 | 88.7 ± 6.4 * | 95.6 ± 4.5 * | |
| CD71 moderate/GPA positive | 27.6 ± 4.8 | 25.7 ± 12.9 | 15.2 ± 12.5 | 10.2 ± 6.0 * | 3.4 ± 3.8 * | |
| CD71 low/GPA positive | 2.0 ± 0.6 | 0.6 ± 0.3 | 0.5 ± 1.0 | 0.7 ± 0.6 | 0.0 ± 0.1 | |
| Day 12 | CD71 high/GPA negative | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| CD71 high/GPA positive | 43.8 ± 7.2 | 63.7 ± 10.5 * | 74.6 ± 13.1 * | 90.3 ± 1.1 * | 94.7 ± 3.9* | |
| CD71 moderate/GPA positive | 36.9 ± 3.3 | 29.4 ± 5.4 | 22.4 ± 8.1 | 9.1 ± 1.2 * | 5.0 ± 3.6 * | |
| CD71 low/GPA positive | 19.4 ± 4.2 | 7.0 ± 5.6 | 3.1 ± 5.4 | 0.7 ± 0.3 * | 0.3 ± 0.2 * | |
| Day 14 | CD71 high/GPA negative | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| CD71 high/GPA positive | 17.2 ± 3.6 | 24.0 ± 2.3 | 43.4 ± 18.0 * | 47.8 ± 11.3 * | 84.0 ± 9.9 * | |
| CD71 moderate/GPA positive | 49.3 ± 4.4 | 57.8 ± 6.2 | 46.3 ± 9.2 | 49.9 ± 10.3 | 14.2 ± 9.0 * | |
| CD71 low/GPA positive | 33.5 ± 2.9 | 18.2 ± 7.1 * | 10.3 ± 9.7 * | 2.4 ± 1.1 * | 1.7 ± 1.0 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaewsakulthong, W.; Suriyun, T.; Chumchuen, S.; Anurathapan, U.; Hongeng, S.; Fucharoen, S.; Sripichai, O. In Vitro Study of Ineffective Erythropoiesis in Thalassemia: Diverse Intrinsic Pathophysiological Features of Erythroid Cells Derived from Various Thalassemia Syndromes. J. Clin. Med. 2022, 11, 5356. https://doi.org/10.3390/jcm11185356
Kaewsakulthong W, Suriyun T, Chumchuen S, Anurathapan U, Hongeng S, Fucharoen S, Sripichai O. In Vitro Study of Ineffective Erythropoiesis in Thalassemia: Diverse Intrinsic Pathophysiological Features of Erythroid Cells Derived from Various Thalassemia Syndromes. Journal of Clinical Medicine. 2022; 11(18):5356. https://doi.org/10.3390/jcm11185356
Chicago/Turabian StyleKaewsakulthong, Woratree, Thunwarat Suriyun, Sukanya Chumchuen, Usanarat Anurathapan, Suradej Hongeng, Suthat Fucharoen, and Orapan Sripichai. 2022. "In Vitro Study of Ineffective Erythropoiesis in Thalassemia: Diverse Intrinsic Pathophysiological Features of Erythroid Cells Derived from Various Thalassemia Syndromes" Journal of Clinical Medicine 11, no. 18: 5356. https://doi.org/10.3390/jcm11185356
APA StyleKaewsakulthong, W., Suriyun, T., Chumchuen, S., Anurathapan, U., Hongeng, S., Fucharoen, S., & Sripichai, O. (2022). In Vitro Study of Ineffective Erythropoiesis in Thalassemia: Diverse Intrinsic Pathophysiological Features of Erythroid Cells Derived from Various Thalassemia Syndromes. Journal of Clinical Medicine, 11(18), 5356. https://doi.org/10.3390/jcm11185356