

Lipoprotein(a): Evidence for Role as a Causal Risk Factor in Cardiovascular Disease and Emerging Therapies


Abstract
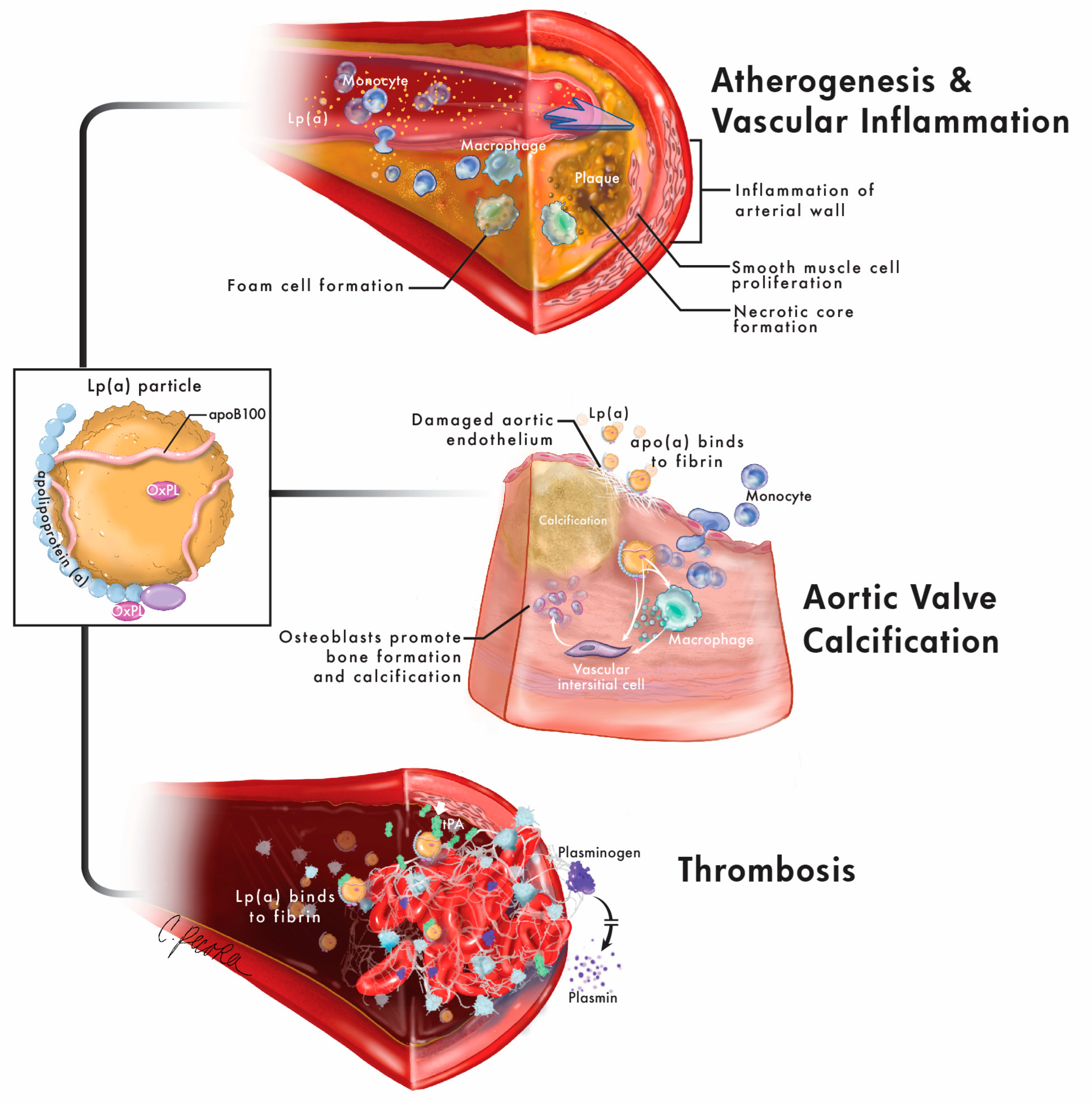
:1. Introduction
2. Lipoprotein(a) and Risk for Atherosclerotic Cardiovascular Disease
2.1. Epidemiologic Studies of Lp(a) and Risk for ASCVD
2.2. Genetic Studies of Lp(a) and Risk for ASCVD
3. Lipoprotein(a) and Risk for Aortic Stenosis/Calcific Aortic Valve Disease
3.1. Epidemiologic, Imaging, and Mechanistic Studies of Lp(a) and Calcific Aortic Valve Disease
3.2. Genetic Studies of Lp(a) and Calcific Aortic Valve Disease
4. Current Therapies and Lipoprotein(a)
5. Emerging Therapies to Lower Lipoprotein(a)
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Schmidt, K.; Noureen, A.; Kronenberg, F.; Utermann, G. Structure, function, and genetics of lipoprotein (a). J. Lipid Res. 2016, 57, 1339–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, R.; Peden, J.F.; Hopewell, J.C.; Kyriakou, T.; Goel, A.; Heath, S.; Parish, S.; Barlera, S.; Franzosi, M.G.; Rust, S.; et al. Genetic Variants Associated with Lp(a) Lipoprotein Level and Coronary Disease. N. Engl. J. Med. 2009, 361, 2518–2528. [Google Scholar] [CrossRef] [Green Version]
- The CARDIoGRAMplusC4D Consortium; Deloukas, P.; Kanoni, S.; Willenborg, C.; Farrall, M.; Assimes, T.L.; Thompson, J.R.; Ingelsson, E.; Saleheen, D.; Erdmann, J.; et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat. Genet. 2013, 45, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Varvel, S.; McConnell, J.P.; Tsimikas, S. Prevalence of Elevated Lp(a) Mass Levels and Patient Thresholds in 532 359 Patients in the United States. Arter. Thromb. Vasc. Biol. 2016, 36, 2239–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-R.; Prasad, A.; Choi, Y.-S.; Xing, C.; Clopton, P.; Witztum, J.L.; Tsimikas, S. LPA Gene, Ethnicity, and Cardiovascular Events. Circulation 2017, 135, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Thanassoulis, G.; Campbell, C.Y.; Owens, D.S.; Smith, J.G.; Smith, A.V.; Peloso, G.M.; Kerr, K.F.; Pechlivanis, S.; Budoff, M.J.; Harris, T.B.; et al. Genetic Associations with Valvular Calcification and Aortic Stenosis. N. Engl. J. Med. 2013, 368, 503–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langsted, A.; Nordestgaard, B.G.; Kamstrup, P.R. Elevated Lipoprotein(a) and Risk of Ischemic Stroke. J. Am. Coll. Cardiol. 2019, 74, 54–66. [Google Scholar] [CrossRef]
- Colantonio, L.D.; Bittner, V.; Safford, M.M.; Marcovina, S.; Brown, T.M.; Jackson, E.A.; Li, M.; López, J.A.G.; Monda, K.L.; Plante, T.B.; et al. Lipoprotein(a) and the risk for coronary heart disease and ischemic stroke events among black and white adults with cardiovascular disease. J. Am. Heart Assoc. 2022, 11, e025397. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, P.R.; Nordestgaard, B.G. Elevated Lipoprotein(a) Levels, LPA Risk Genotypes, and Increased Risk of Heart Failure in the General Population. JACC: Heart Fail. 2016, 4, 78–87. [Google Scholar] [CrossRef]
- Mohammadi-Shemirani, P.; Chong, M.; Narula, S.; Perrot, N.; Conen, D.; Roberts, J.D.; Thériault, S.; Bossé, Y.; Lanktree, M.B.; Pigeyre, M.; et al. Elevated Lipoprotein(a) and Risk of Atrial Fibrillation. J. Am. Coll. Cardiol. 2022, 79, 1579–1590. [Google Scholar] [CrossRef] [PubMed]
- Masson, W.; Lobo, M.; Barbagelata, L.; Molinero, G.; Bluro, I.; Nogueira, J.P. Elevated lipoprotein (a) levels and risk of peripheral artery disease outcomes: A systematic review. Vasc. Med. 2022, 1358863X221091320. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S. A Test in Context: Lipoprotein(a): Diagnosis, prognosis, controversies, and emerging therapies. J. Am. Coll. Cardiol. 2017, 69, 692–711. [Google Scholar] [CrossRef]
- Taleb, A.; Witztum, J.L.; Tsimikas, S. Oxidized phospholipids on apoB-100-containing lipoproteins: A biomarker predicting cardiovascular disease and cardiovascular events. Biomarkers Med. 2011, 5, 673–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsimikas, S.; Brilakis, E.S.; Miller, E.R.; McConnell, J.P.; Lennon, R.J.; Kornman, K.S.; Witztum, J.L.; Berger, P.B. Oxidized Phospholipids, Lp(a) Lipoprotein, and Coronary Artery Disease. N. Engl. J. Med. 2005, 353, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Schnitzler, J.G.; Hoogeveen, R.M.; Ali, L.; Prange, K.H.; Waissi, F.; Van Weeghel, M.; Bachmann, J.C.; Versloot, M.; Borrelli, M.J.; Yeang, C.; et al. Atherogenic Lipoprotein(a) Increases Vascular Glycolysis, Thereby Facilitating Inflammation and Leukocyte Extravasation. Circ. Res. 2020, 126, 1346–1359. [Google Scholar] [CrossRef]
- Van Der Valk, F.M.; Bekkering, S.; Kroon, J.; Yeang, C.; Van den Bossche, J.; Van Buul, J.D.; Ravandi, A.; Nederveen, A.J.; Verberne, H.J.; Scipione, C.; et al. Oxidized Phospholipids on Lipoprotein(a) Elicit Arterial Wall Inflammation and an Inflammatory Monocyte Response in Humans. Circulation 2016, 134, 611–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boffa, M.B.; Koschinsky, M.L. Lipoprotein (a): Truly a direct prothrombotic factor in cardiovascular disease? J. Lipid Res. 2016, 57, 745–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeang, C.; Wilkinson, M.J.; Tsimikas, S. Lipoprotein(a) and oxidized phospholipids in calcific aortic valve stenosis. Curr. Opin. Cardiol. 2016, 31, 440–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: A report of the American college of cardiology/American heart association task force on clinical practice guidelines. J. Am. Coll. Cardiol. 2019, 74, e177–e232. [Google Scholar] [CrossRef]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; De Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/AdA/AGS/AphA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2019, 73, e285–e350. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, G.J.; Thanassoulis, G.; Anderson, T.J.; Barry, A.R.; Couture, P.; Dayan, N.; Francis, G.A.; Genest, J.; Grégoire, J.; Grover, S.A.; et al. 2021 canadian cardiovascular society guidelines for the management of dyslipidemia for the prevention of cardiovascular disease in adults. Can. J. Cardiol. 2021, 37, 1129–1150. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Fazio, S.; Ferdinand, K.C.; Ginsberg, H.N.; Koschinsky, M.L.; Marcovina, S.M.; Moriarty, P.M.; Rader, D.J.; Remaley, A.T.; Reyes-Soffer, G.; et al. NHLBI Working Group Recommendations to Reduce Lipoprotein(a)-Mediated Risk of Cardiovascular Disease and Aortic Stenosis. J. Am. Coll. Cardiol. 2018, 71, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Soffer, G.; Ginsberg, H.N.; Berglund, L.; Duell, P.B.; Heffron, S.P.; Kamstrup, P.R.; Lloyd-Jones, D.M.; Marcovina, S.M.; Yeang, C.; Koschinsky, M.L. Lipoprotein(a): A Genetically Determined, Causal, and Prevalent Risk Factor for Atherosclerotic Cardiovascular Disease: A Scientific Statement From the American Heart Association. Arter. Thromb. Vasc. Biol. 2022, 42, e48–e60. [Google Scholar] [CrossRef] [PubMed]
- Yeang, C.; Witztum, J.L.; Tsimikas, S. Novel method for quantification of lipoprotein(a)-cholesterol: Implications for improving accuracy of LDL-C measurements. J. Lipid Res. 2021, 62, 100053. [Google Scholar] [CrossRef] [PubMed]
- Emerging Risk Factors Collaboration; Sebhat Erqou, S.; Kaptoge, S.; Perry, P.L.; Di Angelantonio, E.; Thompson, A.; White, I.R.; Marcovina, S.M.; Collins, R.; Thompson, S.G.; et al. Lipoprotein(a) Concentration and the Risk of Coronary Heart Disease, Stroke, and Nonvascular Mortality. JAMA 2009, 302, 412–423. [Google Scholar]
- O’Donoghue, M.L.; Morrow, D.A.; Tsimikas, S.; Sloan, S.; Ren, A.F.; Hoffman, E.B.; Desai, N.R.; Solomon, S.D.; Domanski, M.; Arai, K.; et al. Lipoprotein(a) for Risk Assessment in Patients With Established Coronary Artery Disease. J. Am. Coll. Cardiol. 2014, 63, 520–527. [Google Scholar] [CrossRef] [Green Version]
- Willeit, P.; Ridker, P.M.; Nestel, P.J.; Simes, J.; Tonkin, A.M.; Pedersen, T.R.; Schwartz, G.G.; Olsson, A.G.; Colhoun, H.M.; Kronenberg, F.; et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: Individual patient-data meta-analysis of statin outcome trials. Lancet 2018, 392, 1311–1320. [Google Scholar] [CrossRef] [Green Version]
- Bennet, A.; Di Angelantonio, E.; Erqou, S.; Eiriksdottir, G.; Sigurdsson, G.; Woodward, M.; Rumley, A.; Lowe, G.D.O.; Danesh, J.; Gudnason, V. Lipoprotein(a) Levels and Risk of Future Coronary Heart DiseaseLarge-Scale Prospective Data. Arch. Intern. Med. 2008, 168, 598–608. [Google Scholar] [CrossRef] [Green Version]
- Kamstrup, P.R.; Benn, M.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Extreme Lipoprotein(a) Levels and Risk of Myocardial Infarction in the General Population. Circulation 2008, 117, 176–184. [Google Scholar] [CrossRef] [Green Version]
- Virani, S.; Brautbar, A.; Davis, B.C.; Nambi, V.; Hoogeveen, R.; Sharrett, A.R.; Coresh, J.; Mosley, T.H.; Morrisett, J.D.; Catellier, D.J.; et al. Associations Between Lipoprotein(a) Levels and Cardiovascular Outcomes in Black and White Subjects. Circulation 2012, 125, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Paré, G.; Çaku, A.; McQueen, M.; Anand, S.S.; Enas, E.; Clarke, R.; Boffa, M.B.; Koschinsky, M.; Wang, X.; Yusuf, S.; et al. Lipoprotein(a) Levels and the Risk of Myocardial Infarction Among 7 Ethnic Groups. Circulation 2019, 139, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.-L.; Cao, Y.-X.; Zhang, H.-W.; Sun, D.; Hua, Q.; Li, Y.-F.; Guo, Y.-L.; Wu, N.-Q.; Zhu, C.-G.; Gao, Y.; et al. Lipoprotein(a) and Cardiovascular Outcomes in Patients With Coronary Artery Disease and Prediabetes or Diabetes. Diabetes Care 2019, 42, 1312–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.P.; Wang, M.; Pirruccello, J.P.; Ellinor, P.T.; Ng, K.; Kathiresan, S.; Khera, A.V. Lp(a) (Lipoprotein[a]) Concentrations and Incident Atherosclerotic Cardiovascular Disease: New insights from a large national biobank. Arter. Thromb. Vasc. Biol. 2020, 41, 465–474. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Tybjaerg-Hansen, A.; Steffensen, R.; Nordestgaard, B.G. Genetically Elevated Lipoprotein(a) and Increased Risk of Myocardial Infarction. JAMA 2009, 301, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, P.R.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Extreme Lipoprotein(a) Levels and Improved Cardiovascular Risk Prediction. J. Am. Coll. Cardiol. 2013, 61, 1146–1156. [Google Scholar] [CrossRef] [Green Version]
- Kyriakou, T.; Seedorf, U.; Goel, A.; Hopewell, J.C.; Clarke, R.; Watkins, H.; Farrall, M. A Common LPA Null Allele Associates With Lower Lipoprotein(a) Levels and Coronary Artery Disease Risk. Arter. Thromb. Vasc. Biol. 2014, 34, 2095–2099. [Google Scholar] [CrossRef] [Green Version]
- Lim, T.T.; Würtz, P.; Havulinna, A.S.; Palta, P.; Tukiainen, T.; Rehnström, K.; Esko, T.; Mägi, R.; Inouye, M.; Lappalainen, T.; et al. Distribution and Medical Impact of Loss-of-Function Variants in the Finnish Founder Population. PloS Genet. 2014, 10, e1004494. [Google Scholar] [CrossRef]
- Saleheen, D.; Haycock, P.C.; Zhao, W.; Rasheed, A.; Taleb, A.; Imran, A.; Abbas, S.; Majeed, F.; Akhtar, S.; Qamar, N.; et al. Apolipoprotein(a) isoform size, lipoprotein(a) concentration, and coronary artery disease: A mendelian randomisation analysis. Lancet Diabetes Endocrinol. 2017, 5, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.; Virani, S.S.; Ayers, C.R.; Sun, W.; Hoogeveen, R.C.; Rohatgi, A.; Berry, J.D.; Joshi, P.H.; Ballantyne, C.M.; Khera, A. Lipoprotein(a) and Family History Predict Cardiovascular Disease Risk. J. Am. Coll. Cardiol. 2020, 76, 781–793. [Google Scholar] [CrossRef]
- Trinder, M.; Zekavat, S.M.; Uddin, M.; Pampana, A.; Natarajan, P. Apolipoprotein B is an insufficient explanation for the risk of coronary disease associated with lipoprotein(a). Cardiovasc. Res. 2021, 117, 1245–1247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Speiser, J.L.; Ye, F.; Tsai, M.Y.; Cainzos-Achirica, M.; Nasir, K.; Herrington, D.M.; Shapiro, M.D. High-Sensitivity C-Reactive Protein Modifies the Cardiovascular Risk of Lipoprotein(a). J. Am. Coll. Cardiol. 2021, 78, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, T.; Kuroda, T.; Yamasawa, M.; Nishinaga, M.; Mitsuhashi, T.; Seino, Y.; Nagoh, N.; Kayaba, K.; Yamada, S.; Matsuo, H.; et al. Correlation between lipoprotein(a) and aortic valve sclerosis assessed by echocardiography (the JMS Cardiac Echo and Cohort Study). Am. J. Cardiol. 1995, 76, 928–932. [Google Scholar] [CrossRef]
- Stewart, B.; Siscovick, D.; Lind, B.K.; Gardin, J.M.; Gottdiener, J.S.; Smith, V.E.; Kitzman, D.W.; Otto, C.M. Clinical Factors Associated With Calcific Aortic Valve Disease. J. Am. Coll. Cardiol. 1997, 29, 630–634. [Google Scholar] [CrossRef] [Green Version]
- Torzewski, M.; Ravandi, A.; Yeang, C.; Edel, A.; Bhindi, R.; Kath, S.; Twardowski, L.; Schmid, J.; Yang, X.; Franke, U.F.; et al. Lipoprotein(a)-Associated Molecules Are Prominent Components in Plasma and Valve Leaflets in Calcific Aortic Valve Stenosis. JACC: Basic Transl. Sci. 2017, 2, 229–240. [Google Scholar] [CrossRef]
- Bozbas, H.; Yildirir, A.; Atar, I.; Pirat, B.; Eroglu, S.; Aydinalp, A.; Ozin, B.; Muderrisoglu, H. Effects of serum levels of novel atherosclerotic risk factors on aortic valve calcification. J. Heart Valve Dis. 2007, 16, 387–393. [Google Scholar]
- Vongpromek, R.; Bos, S.; Kate, G.-J.R.T.; Yahya, R.; Verhoeven, A.J.M.; de Feyter, P.J.; Kronenberg, F.; van Lennep, J.E.R.; Sijbrands, E.J.G.; Mulder, M.T. Lipoprotein(a) levels are associated with aortic valve calcification in asymptomatic patients with familial hypercholesterolaemia. J. Intern. Med. 2014, 278, 166–173. [Google Scholar] [CrossRef]
- Bouchareb, R.; Mahmut, A.; Nsaibia, M.J.; Boulanger, M.-C.; Dahou, A.; Lépine, J.-L.; Laflamme, M.-H.; Hadji, F.; Couture, C.; Trahan, S.; et al. Autotaxin Derived From Lipoprotein(a) and Valve Interstitial Cells Promotes Inflammation and Mineralization of the Aortic Valve. Circulation 2015, 132, 677–690. [Google Scholar] [CrossRef]
- Després, A.-A.; Perrot, N.; Poulin, A.; Tastet, L.; Shen, M.; Chen, H.Y.; Bourgeois, R.; Trottier, M.; Tessier, M.; Guimond, J.; et al. Lipoprotein(a), Oxidized Phospholipids, and Aortic Valve Microcalcification Assessed by 18F-Sodium Fluoride Positron Emission Tomography and Computed Tomography. CJC Open 2019, 1, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Zheng, K.H.; Tsimikas, S.; Pawade, T.; Kroon, J.; Jenkins, W.S.; Doris, M.K.; White, A.C.; Timmers, N.K.; Hjortnaes, J.; Rogers, M.A.; et al. Lipoprotein(a) and Oxidized Phospholipids Promote Valve Calcification in Patients With Aortic Stenosis. J. Am. Coll. Cardiol. 2019, 73, 2150–2162. [Google Scholar] [CrossRef]
- Glader, C.; Birgander, L.; Söderberg, S.; Ildgruben, H.; Saikku, P.; Waldenström, A.; Dahlén, G. Lipoprotein(a), Chlamydia pneumoniae, leptin and tissue plasminogen activator as risk markers for valvular aortic stenosis. Eur. Heart J. 2003, 24, 198–208. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Elevated Lipoprotein(a) and Risk of Aortic Valve Stenosis in the General Population. J. Am. Coll. Cardiol. 2014, 63, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Arsenault, B.J.; Boekholdt, M.; Dubé, M.-P.; Rhéaume, E.; Wareham, N.J.; Khaw, K.-T.; Sandhu, M.S.; Tardif, J.-C. Lipoprotein(a) Levels, Genotype, and Incident Aortic Valve Stenosis. Circ. Cardiovasc. Genet. 2014, 7, 304–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langsted, A.; Varbo, A.; Kamstrup, P.R.; Nordestgaard, B.G. Elevated Lipoprotein(a) Does Not Cause Low-Grade Inflammation Despite Causal Association With Aortic Valve Stenosis and Myocardial Infarction: A Study of 100 578 Individuals from the General Population. J. Clin. Endocrinol. Metab. 2015, 100, 2690–2699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamstrup, P.R.; Hung, M.-Y.; Witztum, J.L.; Tsimikas, S.; Nordestgaard, B.G. Oxidized Phospholipids and Risk of Calcific Aortic Valve Disease. Arter. Thromb. Vasc. Biol. 2017, 37, 1570–1578. [Google Scholar] [CrossRef] [Green Version]
- Que, X.; Hung, M.-Y.; Yeang, C.; Gonen, A.; Prohaska, T.A.; Sun, X.; Diehl, C.; Määttä, A.; Gaddis, D.E.; Bowden, K.; et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature 2018, 558, 301–306. [Google Scholar] [CrossRef]
- Capoulade, R.; Chan, K.L.; Yeang, C.; Mathieu, P.; Bossé, Y.; Dumesnil, J.G.; Tam, J.W.; Teo, K.K.; Mahmut, A.; Yang, X.; et al. Oxidized Phospholipids, Lipoprotein(a), and Progression of Calcific Aortic Valve Stenosis. J. Am. Coll. Cardiol. 2015, 66, 1236–1246. [Google Scholar] [CrossRef] [Green Version]
- Capoulade, R.; Yeang, C.; Chan, K.L.; Pibarot, P.; Tsimikas, S. Association of Mild to Moderate Aortic Valve Stenosis Progression With Higher Lipoprotein(a) and Oxidized Phospholipid Levels. JAMA Cardiol. 2018, 3, 1212–1217. [Google Scholar] [CrossRef]
- Tsimikas, S.; Gordts, P.L.S.M.; Nora, C.; Yeang, C.; Witztum, J.L. Statin therapy increases lipoprotein(a) levels. Eur. Heart J. 2020, 41, 2275–2284. [Google Scholar] [CrossRef]
- Chan, K.L.; Teo, K.; Dumesnil, J.G.; Ni, A.; Tam, J.; ASTRONOMER Investigators. Effect of lipid lowering with rosuvastatin on progression of aortic stenosis: Results of the aortic stenosis progression observation: Measuring effects of rosuvastatin (ASTRONOMER) trial. Circulation 2010, 121, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Samaha, F.F.; McKenney, J.; Bloedon, L.T.; Sasiela, W.J.; Rader, D.J. Inhibition of microsomal triglyceride transfer protein alone or with ezetimibe in patients with moderate hypercholesterolemia. Nat. Clin. Pr. Cardiovasc. Med. 2008, 5, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Nozue, T.; Michishita, I.; Mizuguchi, I. Effects of Ezetimibe on Remnant-Like Particle Cholesterol, Lipoprotein (a), and Oxidized Low-Density Lipoprotein in Patients with Dyslipidemia. J. Atheroscler. Thromb. 2010, 17, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Boffa, M.B.; Koschinsky, M.L. Therapeutic Lowering of Lipoprotein(a). Circ. Genom. Precis. Med. 2018, 11, e002052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albers, J.J.; Sle’, A.; O’Brien, K.D.; Robinson, J.G.; Kashyap, M.L.; Kwiterovich, P.O.; Xu, P.; Marcovina, S.M. Relationship of Apolipoproteins A-1 and B, and Lipoprotein(a) to Cardiovascular Outcomes. J. Am. Coll. Cardiol. 2013, 62, 1575–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The HPS2-THRIVE Collaborative Group Effects of Extended-Release Niacin with Laropiprant in High-Risk Patients. N. Engl. J. Med. 2014, 371, 203–212. [CrossRef] [PubMed] [Green Version]
- Santos, R.D.; Raal, F.J.; Catapano, A.L.; Witztum, J.L.; Steinhagen-Thiessen, E.; Tsimikas, S. Mipomersen, an Antisense Oligonucleotide to Apolipoprotein B-100, Reduces Lipoprotein(a) in Various Populations With Hypercholesterolemia. Arter. Thromb. Vasc. Biol. 2015, 35, 689–699. [Google Scholar] [CrossRef] [Green Version]
- O’Donoghue, M.L.; Fazio, S.; Giugliano, R.P.; Stroes, E.S.; Kanevsky, E.; Gouni-Berthold, I.; Im, K.; Pineda, A.L.; Wasserman, S.M.; Češka, R.; et al. Lipoprotein(a), PCSK9 Inhibition, and Cardiovascular Risk. Circulation 2019, 139, 1483–1492. [Google Scholar] [CrossRef]
- Bittner, V.A.; Szarek, M.; Aylward, P.E.; Bhatt, D.L.; Diaz, R.; Edelberg, J.M.; Fras, Z.; Goodman, S.G.; Halvorsen, S.; Hanotin, C.; et al. Effect of Alirocumab on Lipoprotein(a) and Cardiovascular Risk After Acute Coronary Syndrome. J. Am. Coll. Cardiol. 2020, 75, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Bhatia, H.S.; Yeang, C.; Baruch, A.; Yang, X.; Stroes, E.S.; Tsimikas, S. PCSK9 Inhibition and Oxidized Phospholipids. J. Am. Coll. Cardiol. 2021, 78, 1288–1289. [Google Scholar] [CrossRef]
- Waldmann, E.; Parhofer, K.G. Lipoprotein apheresis to treat elevated lipoprotein (a). J. Lipid Res. 2016, 57, 1751–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriarty, P.M.; Gray, J.V.; Gorby, L.K. Lipoprotein apheresis for lipoprotein(a) and cardiovascular disease. J. Clin. Lipidol. 2019, 13, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Roeseler, E.; Julius, U.; Heigl, F.; Spitthoever, R.; Heutling, D.; Breitenberger, P.; Leebmann, J.; Lehmacher, W.; Kamstrup, P.R.; Nordestgaard, B.G.; et al. Lipoprotein Apheresis for Lipoprotein(a)-Associated Cardiovascular Disease. Arter. Thromb. Vasc. Biol. 2016, 36, 2019–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohenstein, B.; Julius, U.; Lansberg, P.; Jaeger, B.; Mellwig, K.-P.; Weiss, N.; Graehlert, X.; Roeder, I.; Ramlow, W. Rationale and design of MultiSELECt: A European Multi center S tudy on the E ffect of L ipoprotein(a) E limination by lipoprotein apheresis on C ardiovascular ou t comes. Atheroscler. Suppl. 2017, 30, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Chasman, D.I.; Shiffman, D.; Zee, R.Y.; Louie, J.Z.; Luke, M.M.; Rowland, C.M.; Catanese, J.J.; Buring, J.E.; Devlin, J.J.; Ridker, P.M. Polymorphism in the apolipoprotein(a) gene, plasma lipoprotein(a), cardiovascular disease, and low-dose aspirin therapy. Atherosclerosis 2009, 203, 371–376. [Google Scholar] [CrossRef] [Green Version]
- Lacaze, P.; Bakshi, A.; Riaz, M.; Polekhina, G.; Owen, A.; Bhatia, H.S.; Natarajan, P.; Wolfe, R.; Beilin, L.; Nicholls, S.J.; et al. Aspirin for primary prevention of cardiovascular events in relation to lipoprotein(a) genotypes. J. Am. Coll. Cardiol. 2022, 80, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Cui, K.; Yin, D.; Zhu, C.; Song, W.; Wang, H.; Jia, L.; Zhang, R.; Wang, H.; Cai, Z.; Feng, L.; et al. How do lipoprotein(a) concentrations affect clinical outcomes for patients with stable coronary artery disease who underwent different dual antiplatelet therapy after percutaneous coronary intervention? J. Am. Heart Assoc. 2022, 11, e023578. [Google Scholar] [CrossRef] [PubMed]
- Danik, J.S.; Rifai, N.; Buring, J.E.; Ridker, P.M. Lipoprotein(a), Hormone Replacement Therapy, and Risk of Future Cardiovascular Events. J. Am. Coll. Cardiol. 2008, 52, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Honigberg, M.C.; Trinder, M.; Natarajan, P. Lipoprotein(a), Menopausal Hormone Therapy, and Risk of Coronary Heart Disease in Postmenopausal Individuals. JAMA Cardiol. 2022, 7, 565. [Google Scholar] [CrossRef]
- Serban, M.-C.; Sahebkar, A.; Mikhailidis, D.P.; Toth, P.P.; Jones, S.R.; Muntner, P.; Blaha, M.J.; Andrica, F.; Martin, S.S.; Borza, C.; et al. Impact of L-carnitine on plasma lipoprotein(a) concentrations: A systematic review and meta-analysis of randomized controlled trials. Sci. Rep. 2016, 6, 19188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witkowski, M.; Weeks, T.L.; Hazen, S.L. Gut Microbiota and Cardiovascular Disease. Circ. Res. 2020, 127, 553–570. [Google Scholar] [CrossRef]
- Tsimikas, S.; Viney, N.J.; Hughes, S.G.; Singleton, W.; Graham, M.J.; Baker, B.F.; Burkey, J.L.; Yang, Q.; Marcovina, S.M.; Geary, R.S.; et al. Antisense therapy targeting apolipoprotein(a): A randomised, double-blind, placebo-controlled phase 1 studyy. Lancet 2015, 386, 1472–1483. [Google Scholar] [CrossRef]
- Viney, N.J.; van Capelleveen, J.C.; Geary, R.S.; Xia, S.; A Tami, J.; Yu, R.Z.; Marcovina, S.M.; Hughes, S.G.; Graham, M.J.; Crooke, R.M.; et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): Two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016, 388, 2239–2253. [Google Scholar] [CrossRef]
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.-C.; Baum, S.J.; Steinhagen-Thiessen, E.; Shapiro, M.D.; Stroes, E.S.; Moriarty, P.M.; Nordestgaard, B.G.; et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N. Engl. J. Med. 2020, 382, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Koren, M.J.; Moriarty, P.M.; Baum, S.J.; Neutel, J.; Hernandez-Illas, M.; Weintraub, H.S.; Florio, M.; Kassahun, H.; Melquist, S.; Varrieur, T.; et al. Preclinical development and phase 1 trial of a novel siRNA targeting lipoprotein(a). Nat. Med. 2022, 28, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Wolski, K.; Balog, C.; Swerdlow, D.I.; Scrimgeour, A.C.; Rambaran, C.; Wilson, R.J.; Boyce, M.; Ray, K.K.; Cho, L.; et al. Single Ascending Dose Study of a Short Interfering RNA Targeting Lipoprotein(a) Production in Individuals With Elevated Plasma Lipoprotein(a) Levels. JAMA 2022, 327, 1679. [Google Scholar] [CrossRef] [PubMed]
- Silence Therapeutics Announces Detailed Results from sln360 Phase 1 Study in High Lipoprotein(a) Presented in Late Breaker at the American College of Cardiology (acc.22) Annual Meeting. 2022. Available online: https://silence-therapeutics.com/investors/press-releases/press-releases-details/2022/Silence-Therapeutics-Announces-Detailed-Results-From-SLN360-Phase-1-Study-in-High-Lipoproteina-Presented-in-Late-Breaker-at-the-American-College-of-Cardiology-ACC.22-Annual-Meeting/default.aspx (accessed on 11 August 2022).
- Szarek, M.; A Bittner, V.; Aylward, P.; Baccara-Dinet, M.; Bhatt, D.L.; Diaz, R.; Fras, Z.; Goodman, S.G.; Halvorsen, S.; A Harrington, R.; et al. Lipoprotein(a) lowering by alirocumab reduces the total burden of cardiovascular events independent of low-density lipoprotein cholesterol lowering: ODYSSEY OUTCOMES trial. Eur. Heart J. 2020, 41, 4245–4255. [Google Scholar] [CrossRef]
- Warden, B.; Minnier, J.; Watts, G.; Fazio, S.; Shapiro, M.D. Impact of PCSK9 inhibitors on plasma lipoprotein(a) concentrations with or without a background of niacin therapy. J. Clin. Lipidol. 2019, 13, 580–585. [Google Scholar] [CrossRef]
- A Stiekema, L.C.; Stroes, E.S.G.; Verweij, S.L.; Kassahun, H.; Chen, L.; Wasserman, S.M.; Sabatine, M.S.; Mani, V.; A Fayad, Z. Persistent arterial wall inflammation in patients with elevated lipoprotein(a) despite strong low-density lipoprotein cholesterol reduction by proprotein convertase subtilisin/kexin type 9 antibody treatment. Eur. Heart J. 2019, 40, 2775–2781. [Google Scholar] [CrossRef] [Green Version]
- Merki, E.; Graham, M.J.; Mullick, A.; Miller, E.R.; Crooke, R.M.; Pitas, R.E.; Witztum, J.L.; Tsimikas, S. Antisense Oligonucleotide Directed to Human Apolipoprotein B-100 Reduces Lipoprotein(a) Levels and Oxidized Phospholipids on Human Apolipoprotein B-100 Particles in Lipoprotein(a) Transgenic Mice. Circulation 2008, 118, 743–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsimikas, S.; Moriarty, P.M.; Stroes, E.S. Emerging RNA Therapeutics to Lower Blood Levels of Lp(a). J. Am. Coll. Cardiol. 2021, 77, 1576–1589. [Google Scholar] [CrossRef] [PubMed]
- Enkhmaa, B.; Berglund, L. Non-genetic influences on lipoprotein(a) concentrations. Atherosclerosis 2022, 349, 53–62. [Google Scholar] [CrossRef]
- Haskell, W.L.; Alderman, E.L.; Fair, J.M.; Maron, D.J.; Mackey, S.F.; Superko, H.R.; Williams, P.T.; Johnstone, I.M.; A Champagne, M.; Krauss, R.M. Effects of intensive multiple risk factor reduction on coronary atherosclerosis and clinical cardiac events in men and women with coronary artery disease. The Stanford Coronary Risk Intervention Project (SCRIP). Circulation 1994, 89, 975–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langsted, A.; Kamstrup, P.R.; Nordestgaard, B.G. Lipoprotein(a): Fasting and nonfasting levels, inflammation, and cardiovascular risk. Atherosclerosis 2014, 234, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Ebbeling, C.B.; Knapp, A.; Johnson, A.; Wong, J.M.W.; Greco, K.F.; Ma, C.; Mora, S.; Ludwig, D.S. Effects of a low-carbohydrate diet on insulin-resistant dyslipoproteinemia—a randomized controlled feeding trial. Am. J. Clin. Nutr. 2021, 115, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Bordi, P.L.; Fleming, J.A.; Hill, A.M.; Kris-Etherton, P. Effect of a Moderate Fat Diet With and Without Avocados on Lipoprotein Particle Number, Size and Subclasses in Overweight and Obese Adults: A Randomized, Controlled Trial. J. Am. Heart Assoc. 2015, 4, e001355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faghihnia, N.; Tsimikas, S.; Miller, E.R.; Witztum, J.L.; Krauss, R.M. Changes in lipoprotein(a), oxidized phospholipids, and LDL subclasses with a low-fat high-carbohydrate diet. J. Lipid Res. 2010, 51, 3324–3330. [Google Scholar] [CrossRef] [Green Version]
- Haring, B.; von Ballmoos, M.C.W.; Appel, L.J.; Sacks, F.M. Healthy Dietary Interventions and Lipoprotein (a) Plasma Levels: Results from the Omni Heart Trial. PloS ONE 2014, 9, e114859. [Google Scholar] [CrossRef] [Green Version]
- Ginsberg, H.N.; Kris-Etherton, P.; Dennis, B.; Elmer, P.J.; Ershow, A.; Lefevre, M.; Pearson, T.; Roheim, P.; Ramakrishnan, R.; Reed, R.; et al. Effects of Reducing Dietary Saturated Fatty Acids on Plasma Lipids and Lipoproteins in Healthy Subjects. Arter. Thromb. Vasc. Biol. 1998, 18, 441–449. [Google Scholar] [CrossRef] [Green Version]
- Berglund, L.; Lefevre, M.; Ginsberg, H.N.; Kris-Etherton, P.M.; Elmer, P.J.; Stewart, P.W.; Ershow, A.; A Pearson, T.; Dennis, B.H.; Roheim, P.S.; et al. Comparison of monounsaturated fat with carbohydrates as a replacement for saturated fat in subjects with a high metabolic risk profile: Studies in the fasting and postprandial states. Am. J. Clin. Nutr. 2007, 86, 1611–1620. [Google Scholar] [CrossRef]
- Jones, J.L.; Comperatore, M.; Barona, J.; Calle, M.C.; Andersen, C.; McIntosh, M.; Najm, W.; Lerman, R.H.; Fernandez, M.L. A Mediterranean-style, low–glycemic-load diet decreases atherogenic lipoproteins and reduces lipoprotein (a) and oxidized low-density lipoprotein in women with metabolic syndrome. Metabolism 2012, 61, 366–372. [Google Scholar] [CrossRef]
- Vu, K.N.; Ballantyne, C.M.; Hoogeveen, R.C.; Nambi, V.; Volcik, K.A.; Boerwinkle, E.; Morrison, A.C. Causal Role of Alcohol Consumption in an Improved Lipid Profile: The Atherosclerosis Risk in Communities (ARIC) Study. PLoS ONE 2016, 11, e0148765. [Google Scholar] [CrossRef]
- Ellis, K.L.; de Isla, L.P.; Alonso, R.; Fuentes, F.; Watts, G.; Mata, P. Value of Measuring Lipoprotein(a) During Cascade Testing for Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 2019, 73, 1029–1039. [Google Scholar] [CrossRef]
- Chakraborty, A.; Chan, D.C.; Ellis, K.L.; Pang, J.; Barnett, W.; Woodward, A.M.; Vorster, M.; Norman, R.; Moses, E.K.; Watts, G.F. Cascade testing for elevated lipoprotein(a) in relatives of probands with high lipoprotein(a). Am. J. Prev. Cardiol. 2022, 10, 100343. [Google Scholar] [CrossRef]
- Piercy, K.L.; Troiano, R.P.; Ballard, R.M.; Carlson, S.A.; Fulton, J.E.; Galuska, D.A.; George, S.M.; Olson, R.D. The Physical Activity Guidelines for Americans. JAMA 2018, 320, 2020–2028. [Google Scholar] [CrossRef]
- Kayikcioglu, M. LDL Apheresis and Lp (a) Apheresis: A Clinician’s Perspective. Curr. Atheroscler. Rep. 2021, 23, 15. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Meta-Analyses | ||
|---|---|---|
| Author | Year | Key Findings |
| The Emerging Risk Factors Collaboration [26] | 2009 | Lp(a) associated with CHD (RR per SD 1.13, 95% CI 1.09–1.18) Ischemic stroke (RR per SD 1.10, 95% CI 1.02–1.18) |
| O’donoghue, et al. [27] | 2014 | Lp(a) associated with MACE in population with CAD: Highest quintile: OR 1.40, 95% CI 1.15–1.71 |
| Willeit, et al. [28] | 2018 | Lp(a) associated linearly with CVD at baseline and on-statin therapy in statin outcomes trials On statins: Lp(a) 15–30 mg/dL: HR 0.95 (95% CI 0.82–1.11) Lp(a) 30–50 mg/dL: HR 1.08 (95% CI 0.95–1.23) Lp(a) ≥50 mg/dL: HR 1.42 (95% CI 1.16–1.74) |
| Epidemiologic Studies | ||
| Author | Year | Key Findings |
| Bennet, et al. [29] | 2008 | Top tertile of Lp(a) associated with CHD with OR 1.60 (95% CI 1.38–1.85) |
| Kamstrup, et al. [30] | 2008 | Lp(a) associated with risk for MI in men and women: Women > 95th percentile: HR 3.6 (95% CI 1.7–7.7) Men > 95th percentile: HR 3.7 (1.7–8.0) |
| Virani, et al. [31] | 2012 | Highest quintile of Lp(a) associated with incident CVD risk in: Black individuals (HR 1.35, 95% CI 1.06–1.74) White individuals (HR 1.27, 95% CI 1.10–1.47) |
| Paré, et al. [32] | 2019 | Lp(a) > 50 mg/dL associated with increased risk of MI (OR 1.48, 95% CI 1.32–1.67) overall and in all ethnic groups studied except African and Arab individuals |
| Jin, et al. [33] | 2019 | Lp(a) ≥ 50 mg/dL associated with increased risk of CVD in: Pre-diabetes (HR 2.67, 95% CI 1.38–5.42) Diabetes (HR 3.47, 95% CI 1.80–6.69) |
| Patel, et al. [34] | 2021 | Lp(a) associated with increased ASCVD risk with HR 1.11 (95% CI 1.10–1.12) per 50 nmol/L increment Consistent results seen in White, South Asian, and Black individuals |
| Genetic Studies | ||
| Author | Year | Key Findings |
| Clarke, et al. [2] | 2009 | LPA locus had strongest association with coronary disease in large study of candidate SNPs LPA SNP rs10455872 OR 1.70 (95% CI 1.49–1.95) for coronary disease LPA SNP rs3798220 OR 1.92 (95% CI 1.48–2.49) for coronary disease |
| Kamstrup, et al. [35] | 2009 | Higher levels of Lp(a) and lower number of kringle IV repeats associated with greater MI risk: >95th percentile of Lp(a): HR 2.6 (95% CI 1.6–4.1) 1st quartile of KIV-2 repeats: HR 1.5 (95% CI 1.2–1.9) |
| CARDIoGRAMplusC4D Consortium [3] | 2013 | LPA SNP rs3798220 associated with CAD: OR 1.28 (p < 0.001) |
| Kamstrup, et al. [36] | 2013 | Addition of Lp(a) levels, KIV-2 repeats and carrier status for LPA SNP rs10455872 to traditional risk factors all improved risk prediction for MI and CHD |
| Kyriakou, et al. [37] | 2014 | A null allele (LPA SNP rs41272114) was associated with decreased Lp(a) levels and decreased CAD risk |
| Lim, et al. [38] | 2014 | Splice variants of Lp(a) associated with reduced Lp(a) levels and protection against CVD (OR 0.84, p < 0.001) |
| Lee, et al. [5] | 2016 | Lp(a) levels and SNPs vary by ethnicity. The addition of SNPs to Lp(a) levels did not appear to be clinically meaningful. |
| Salaheen, et al. [39] | 2017 | OR per 1-SD increment of Lp(a) for MI 1.10 (95% CI 1.05–1.14) |
| Author | Year | Key Findings |
|---|---|---|
| Lp(a) and AV Sclerosis | ||
| Gotoh, et al. [43] | 1995 | Greater prevalence of aortic valve sclerosis in individuals with Lp(a) ≥ 30 mg/dL (36.1%) compared with <30 mg/dL (12.7%, p < 0.001) |
| Stewart, et al. [44] | 1997 | Lp(a) associated with increased risk for aortic valve stenosis or sclerosis (OR 1.23, 95% CI 1.14, 1.32) |
| Torzewski, et al. [45] | 2017 | Lp(a) and associated molecules including OxPL detected in AV leaflets of individuals with calcific AS |
| Lp(a) and AV Calcification | ||
| Bozbas, et al. [46] | 2007 | Lp(a) independently associated with AVC (OR 1.01, 95% CI 1.00–1.03) |
| Vongpromek, et al. [47] | 2015 | OR per 10 mg/dL increase in Lp(a) 1.11 (95% CI 1.01–1.20) for AVC by CT |
| Bouchareb, et al. [48] | 2015 | Lp(a) transports autotaxin to the AV which contributes to inflammation and calcification of the valve |
| Despres, et al. [49] | 2019 | In individuals without clinical AS, elevated Lp(a) associated with AV microcalcification by PET/CT |
| Zheng, et al. [50] | 2019 | Higher Lp(a) and OxPL levels associated with greater aortic valve calcification activity by PET/CT Lp(a) induces osteogenic differentiation of vascular cells, mediated by OxPL |
| Lp(a) and AS | ||
| Glader, et al. [51] | 2003 | Lp(a) ≥ 48 mg/dL associated with increased risk for AS (OR 3.4, 95% CI 1.1–11.2) |
| Kamstrup, et al. [52] | 2014 | Lp(a) associated with AS in a graded fashion: >95th percentile of Lp(a) (>90 mg/dL): OR 2.9 (95% CI 1.8–4.9) |
| Arsenault, et al. [53] | 2014 | Top tertile of Lp(a) associated with increased risk for AS: HR 1.57, 95% CI 1.02–2.42 |
| Langsted, et al. [54] | 2015 | Each 1-SD increase in Lp(a) associated with HR 1.23 (95% CI 1.06–1.41) for AS |
| OxPL-apoB and AS | ||
| Kamstrup, et al. [55] | 2017 | Dose-dependent association between OxPL-apoB and CAVD For >95th percentile of levels, OR 3.4 (95% CI 2.1–5.5) |
| Que, et al. [56] | 2018 | Inactivation of OxPL reduces development of AV calcification and AV gradient in mice |
| Lp(a), OxPL-apoB and AS Progression | ||
| Capoulade, et al. [57] | 2015 | Top tertile of Lp(a) (OR 2.6, 95% CI 1.4–5.0) and top tertile of OxPL-apoB (OR 2.4, 95% CI 1.2–4.6) associated with rapid AS progression |
| Capoulade, et al. [58] | 2018 | Lp(a) (OR 1.10, 95% CI 1.03–1.19 per 10 mg/dL increase) and OxPL-apoB (OR 1.06, 95% CI 1.01–1.12 per 1 nM increase) levels linearly associated with faster AS progression, especially in younger participants. |
| Zheng, et al. [50] | 2019 | Higher Lp(a) and OxPL levels associated with faster progression of AV calcium score by CT and hemodynamic progression by echocardiography |
| Genetic Associations | ||
| Thanassoulis, et al. [6] | 2013 | rs10455872 associated with AVC in GWAS (OR per allele 2.05, p < 0.001) LPA genotype associated with incident AS (HR per allele 1.68, 95% CI 1.32–2.15) and AV replacement (HR 1.54, 95% CI 1.05–2.27) |
| Kamstrup, et al. [52] | 2014 | Genotypes corresponding with Lp(a) levels associated with increased risk of AS (HR 1.6, 95% CI 1.2–2.1 per 10-fold Lp(a) increase) |
| Arsenault, et al. [53] | 2014 | Carriers of rs10455872 SNP have increased risk of AS: Heterozygous: HR 1.78, 95% CI 1.11–2.87 Homozygous: HR 4.83, 95% CI 1.77–13.20 |
| Langsted, et al. [54] | 2015 | Causal risk ratio for AS based on LPA SNPs (rs3798220, rs10455872): 1.38 (95% CI 1.23–1.55) Causal risk ratio for AS based on LPA KIV-2 genotype: 1.21 (95% CI 1.06–1.40) |
| Current Therapies | ||||
|---|---|---|---|---|
| Drug | Target | Mechanism | Effect on Lp(a) | CVD Outcomes |
| Lipid Lowering Therapy | ||||
| Statins | HMG-CoA reductase | Inhibit cholesterol production | Do not lower Lp(a) levels, may increase Lp(a) [59] | Reduced ASCVD risk, but Lp(a) associated risk persists in statin treated individuals [28] |
| Ezetimibe | Nieman Pick C1-like1 protein | Reduces absorption of cholesterol in the small intestine | Limited data (possible 3–29% decrease in Lp(a)) [61,62] | No known effect on Lp(a)-associated risk |
| Niacin | Multifactorial | Downregulates LPA gene promotor and reduces apoB and triglycerides, increases HDL [63] | AIM-HIGH: 21% reduction in Lp(a), low absolute reduction [64] HPS2-THRIVE: low absolute reduction [65] | AIM-HIGH trial: no effect on CVD events [64]. HPS2-THRIVE: no overall effect of niacin on major vascular events [65] |
| Mipomersen | apoB | Anti-sense inhibitor of apoB synthesis | Reduces Lp(a) by median 26% [66] | Unclear effect on CV outcomes. Risk of liver toxicity |
| Lomitapide | Microsomal triglyceride transfer protein (MTP) | Inhibition of MTP inhibits transfer of lipids onto apoB | Reduces Lp(a) by 17% [61] | Unclear effect on CV outcomes. Risk of liver toxicity |
| PCSK9i (alirocumab, evolocumab, inclisiran) | PCSK9 | Inhibit degradation of LDL-receptor | Reduce Lp(a) by 19–27% [67,68,69] | Limited data, however, reduction in Lp(a) associated with a reduction in CVD events (15% per 25 nmol/L in FOURIER, 0.6% per 1 mg/dL in ODYSSEY OUTCOMES) [67,68], but may not address inflammatory risk associated with OxPL [70] |
| Lipoprotein apheresis | apoB-containing lipoproteins | Removal of apoB-containing lipoproteins from plasma | Immediate reduction in Lp(a) levels up to 75%, with 30–35% time-averaged reduction when performed every 1–2 weeks [71] | Reduction in Lp(a) and LDL-C translates into significant reduction in MACE events in observational studies [72,73] MultiSELECt is an ongoing multicenter prospective study [74] |
| Anti-platelet therapy | ||||
| Aspirin | COX (cyclooxygenase) [75] | Reduces platelet aggregation through irreversible inhibition of thromboxane A2 | -- | In White women carriers of LPA rs3798220 SNP, aspirin associated with significant reduction in CVD risk (HR 0.44, 95% CI 0.20–0.94) in the Women’s Health Study [75]. Similar results in the ASPREE trial with the same SNP or high genetic risk score [76]. |
| Dual anti-platelet therapy (DAPT) | Multifactorial | Multifactorial | -- | In CAD patients with Lp(a) >30 mg/dL who underwent PCI, DAPT >1 year resulted in a significant reduction in CVD events (HR 0.54, 95% CI 0.31–0.92) compared with DAPT ≤1 year [77] |
| Other | ||||
| Hormone replacement therapy (estrogen) | -- | Possibly through increased Lp(a) uptake by LDL receptor or decreased Lp(a) production [78] | Reduction in Lp(a) of 7.9 nmol/L [79] | No impact on CHD events |
| L-carnitine | -- | Possibly related to fatty acid oxidation | Reduction in Lp(a) of 8.8 mg/dL [80] | Unclear effect on CV outcomes L-carnitine associated with increased CVD risk [81] |
| Emerging Therapies | ||||
| Drug | Target | Mechanism | Effect on Lp(a) | Current stage in development |
| Pelacarsen | apo(a) mRNA | Antisense oligonucleotide (ASO), binds apo(a) mRNA, targets for degradation | Phase I: Dose-dependent, up to −77.8% [82,83]. Ligand-conjugated form: dose-dependent, up to −92% [83] Phase II: Ligand-conjugated form: dose-dependent, up to −80% [84] | Phase III/cardiovascular outcomes trial underway (80 mg monthly subcutaneous injection vs. placebo) (NCT04023552). |
| Olpasiran | apo(a) mRNA | Small interfering RNA (siRNA), binds apo(a) mRNA, targets for degradation | Phase I: Maximum mean percent change in Lp(a) from baseline: −71% to −97% [85] | Phase II underway (NCT04270760) |
| SLN360 | apo(a) mRNA | siRNA, binds apo(a) mRNA and targets for degradation | Phase I: Maximal median percent reduction in Lp(a), dose-dependent, up to −98% [86] | Phase II planned for 2022 [87] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhatia, H.S.; Wilkinson, M.J. Lipoprotein(a): Evidence for Role as a Causal Risk Factor in Cardiovascular Disease and Emerging Therapies. J. Clin. Med. 2022, 11, 6040. https://doi.org/10.3390/jcm11206040
Bhatia HS, Wilkinson MJ. Lipoprotein(a): Evidence for Role as a Causal Risk Factor in Cardiovascular Disease and Emerging Therapies. Journal of Clinical Medicine. 2022; 11(20):6040. https://doi.org/10.3390/jcm11206040
Chicago/Turabian StyleBhatia, Harpreet S., and Michael J. Wilkinson. 2022. "Lipoprotein(a): Evidence for Role as a Causal Risk Factor in Cardiovascular Disease and Emerging Therapies" Journal of Clinical Medicine 11, no. 20: 6040. https://doi.org/10.3390/jcm11206040
APA StyleBhatia, H. S., & Wilkinson, M. J. (2022). Lipoprotein(a): Evidence for Role as a Causal Risk Factor in Cardiovascular Disease and Emerging Therapies. Journal of Clinical Medicine, 11(20), 6040. https://doi.org/10.3390/jcm11206040