
Post-Transplant Lymphoproliferative Disease (PTLD) after Allogeneic Hematopoietic Stem Cell Transplantation: Biology and Treatment Options
 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
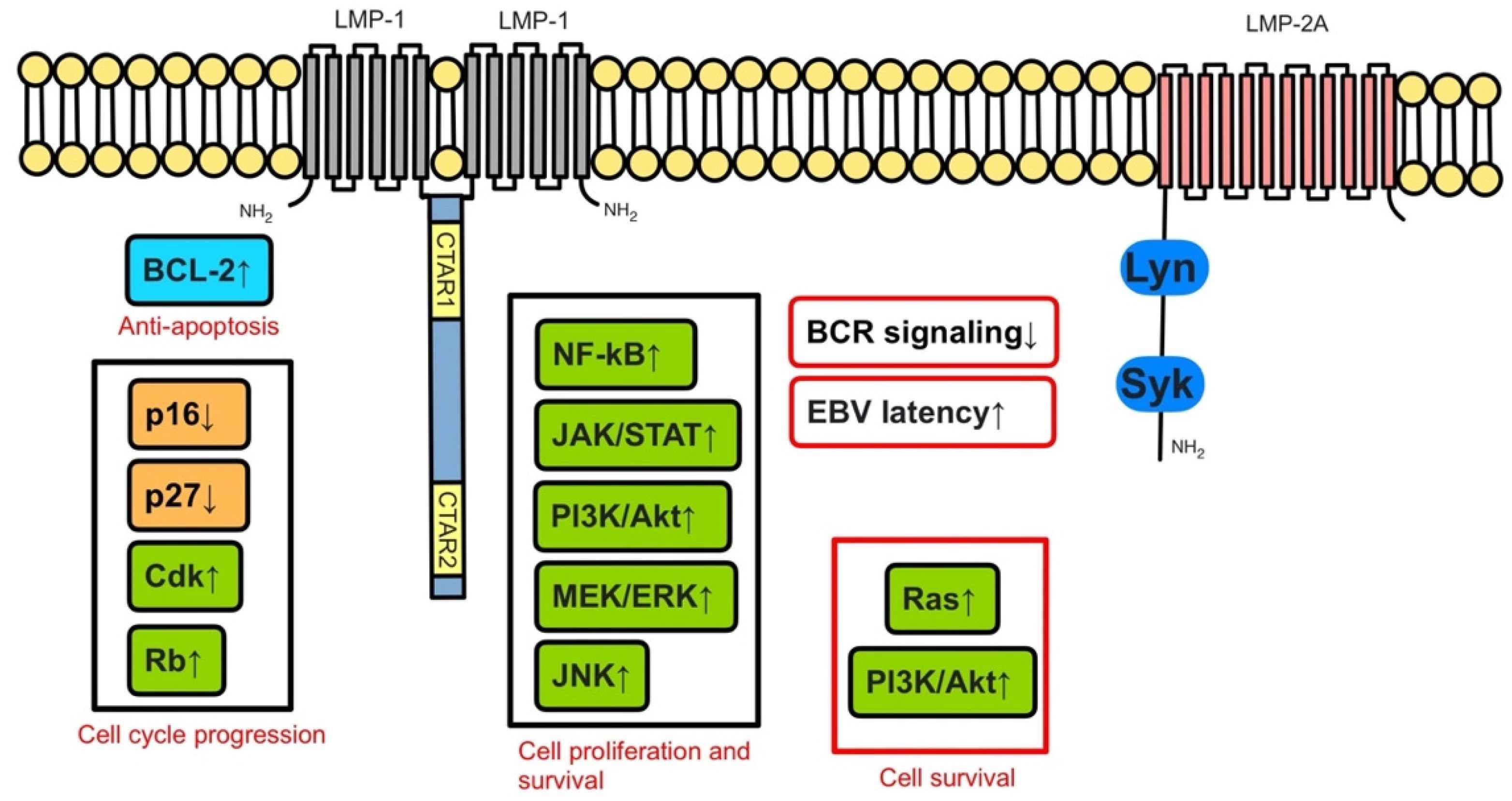
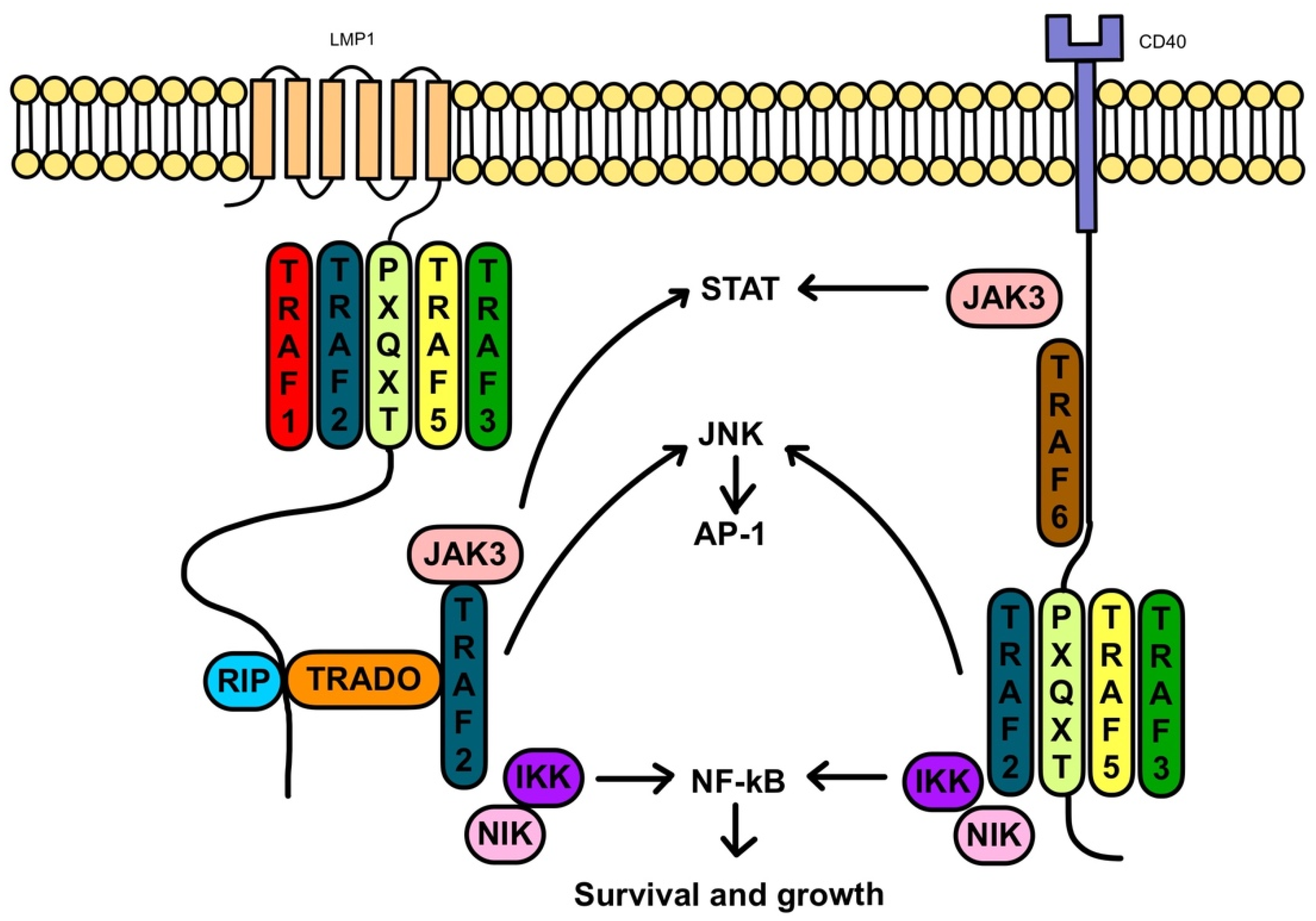
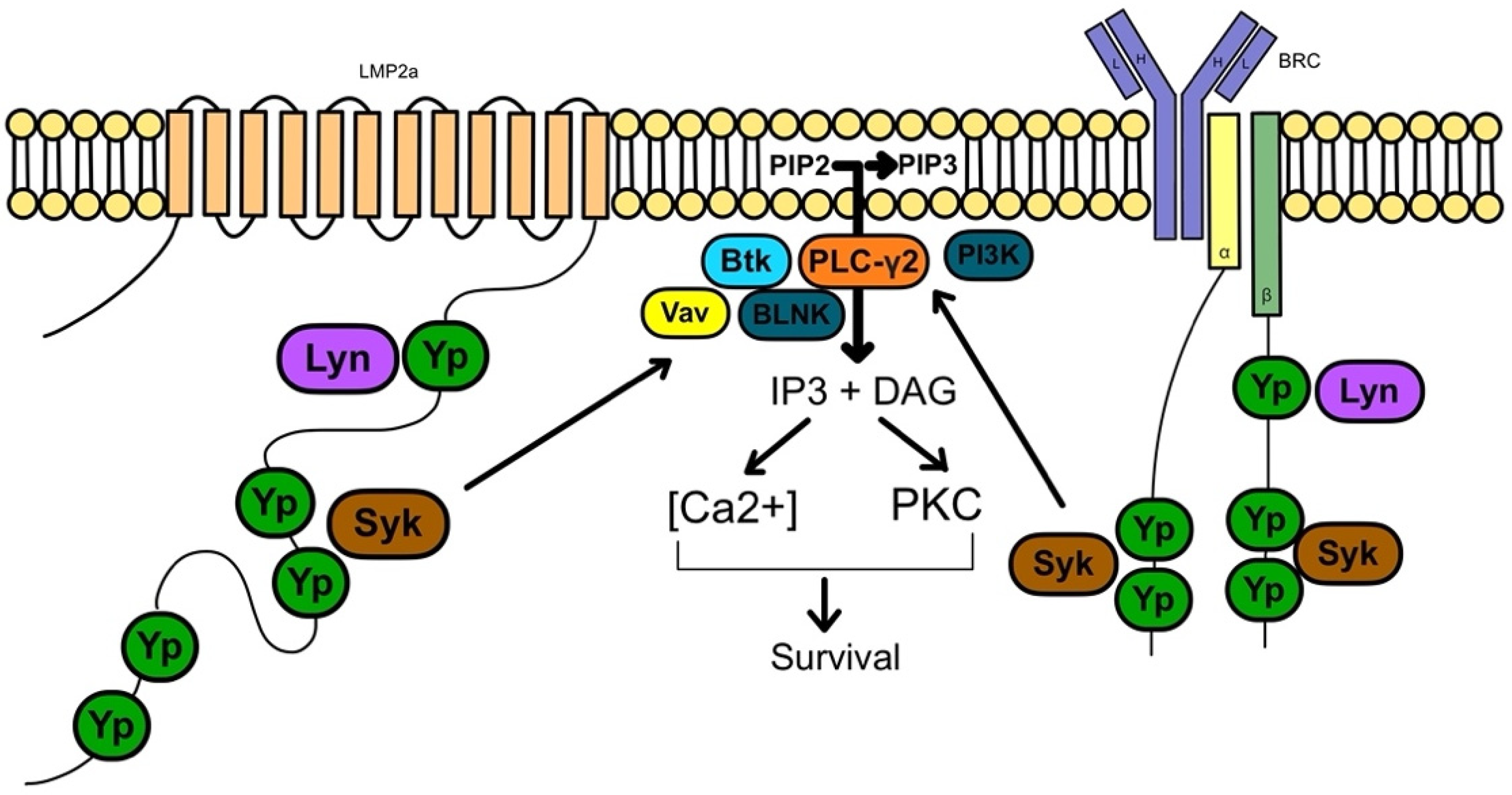
2. Pathophysiology of PTLD
2.1. EBV-Positive PTLD
2.2. EBV-Negative PTLD
3. Risk Factors for PTLD
4. Management
4.1. Reduction of Immunosuppression (RIS)
4.2. Rituximab
4.2.1. Prophylaxis
4.2.2. Preemptive Therapy
4.2.3. Treatment
4.3. Chemotherapy
4.4. Adoptive Immunotherapy
4.4.1. EBV-Specific Cytotoxic T-Lymphocytes for PTLD
4.4.2. Chimeric Antigen Receptor T-Cell Therapy for PTLD
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singavi, A.K.; Harrington, A.M.; Fenske, T.S. Post-transplant lymphoproliferative disorders. Cancer Treat. Res. 2015, 165, 305–327. [Google Scholar] [CrossRef] [PubMed]
- Al-Mansour, Z.; Nelson, B.P.; Evens, A.M. Post-transplant lymphoproliferative disease (PTLD): Risk factors, diagnosis, and current treatment strategies. Curr. Hematol. Malig. Rep. 2013, 8, 173–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, A.L.; Marcus, R.; Bradley, J.A. Post-transplant lymphoproliferative disorders (PTLD) after solid organ transplantation. Crit. Rev. Oncol. Hematol. 2005, 56, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.; Michaels, M.G. Epstein-Barr virus infection and posttransplant lymphoproliferative disorder. Am. J. Transplant. 2013, 13 (Suppl. S3), 41–54. [Google Scholar] [CrossRef]
- Heil, D.S.; Luskin, M.R.; Stadtmauer, E.A.; Schuster, S.J.; Tsai, D.E.; Reshef, R. EBV-negative post-transplant lymphoproliferative disorder: Clinical characteristics, response to therapy, and survival. J. Clin. Oncol. 2013, 31, 8578. [Google Scholar] [CrossRef]
- Roncella, S.; Cutrona, G.; Truini, M.; Airoldi, I.; Pezzolo, A.; Valetto, A.; Di Martino, D.; Dadati, P.; De Rossi, A.; Ulivi, M.; et al. Late Epstein-Barr virus infection of a hepatosplenic gamma delta T-cell lymphoma arising in a kidney transplant recipient. Haematologica 2000, 85, 256–262. [Google Scholar]
- De-The, G.; Day, N.E.; Geser, A.; Lavoue, M.F.; Ho, J.H.; Simons, M.J.; Sohier, R.; Tukei, P.; Vonka, V.; Zavadova, H. Sero-epidemiology of the Epstein-Barr virus: Preliminary analysis of an international study—A review. IARC Sci. Publ. 1975, 11, 3–16. [Google Scholar]
- Dunmire, S.K.; Verghese, P.S.; Balfour, H.H., Jr. Primary Epstein-Barr virus infection. J. Clin. Virol. 2018, 102, 84–92. [Google Scholar] [CrossRef]
- Ruf, S.; Moser, O.; Wössmann, W.; Kreyenberg, H.; Wagner, H.J. Examining the origin of posttransplant lymphoproliferative disorder in a patient after a second allogeneic hematopoeitic stem cell transplantation for relapsed BCR-ABL positive acute lymphoblastic leukemia. J. Pediatr. Hematol. Oncol. 2011, 33, 50–54. [Google Scholar] [CrossRef]
- Hislop, A.D.; Taylor, G.S.; Sauce, D.; Rickinson, A.B. Cellular responses to viral infection in humans: Lessons from Epstein-Barr virus. Annu. Rev. Immunol. 2007, 25, 587–617. [Google Scholar] [CrossRef]
- Hochberg, D.; Souza, T.; Catalina, M.; Sullivan, J.L.; Luzuriaga, K.; Thorley-Lawson, D.A. Acute infection with Epstein-Barr virus targets and overwhelms the peripheral memory B-cell compartment with resting, latently infected cells. J. Virol. 2004, 78, 5194–5204. [Google Scholar] [CrossRef] [PubMed]
- Cesaro, S.; Pegoraro, A.; Tridello, G.; Calore, E.; Pillon, M.; Varotto, S.; Abate, D.; Barzon, L.; Mengoli, C.; Carli, M.; et al. A prospective study on modulation of immunosuppression for Epstein-Barr virus reactivation in pediatric patients who underwent unrelated hematopoietic stem-cell transplantation. Transplantation 2010, 89, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Murray, P.G. Epstein-Barr virus and oncogenesis: From latent genes to tumours. Oncogene 2003, 22, 5108–5121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorley-Lawson, D.A. Epstein-Barr virus: Exploiting the immune system. Nat. Rev. Immunol. 2001, 1, 75–82. [Google Scholar] [CrossRef]
- Ok, C.Y.; Li, L.; Young, K.H. EBV-driven B-cell lymphoproliferative disorders: From biology, classification and differential diagnosis to clinical management. Exp. Mol. Med. 2015, 47, e132. [Google Scholar] [CrossRef] [Green Version]
- Choi, I.K.; Wang, Z.; Ke, Q.; Hong, M.; Qian, Y.; Zhao, X.; Liu, Y.; Kim, H.J.; Ritz, J.; Cantor, H.; et al. Signaling by the Epstein-Barr virus LMP1 protein induces potent cytotoxic CD4(+) and CD8(+) T cell responses. Proc. Natl. Acad. Sci. USA 2018, 115, E686–E695. [Google Scholar] [CrossRef] [Green Version]
- Minamitani, T.; Ma, Y.; Zhou, H.; Kida, H.; Tsai, C.Y.; Obana, M.; Okuzaki, D.; Fujio, Y.; Kumanogoh, A.; Zhao, B.; et al. Mouse model of Epstein-Barr virus LMP1- and LMP2A-driven germinal center B-cell lymphoproliferative disease. Proc. Natl. Acad. Sci. USA 2017, 114, 4751–4756. [Google Scholar] [CrossRef] [Green Version]
- Leblond, V.; Davi, F.; Charlotte, F.; Dorent, R.; Bitker, M.O.; Sutton, L.; Gandjbakhch, I.; Binet, J.L.; Raphael, M. Posttransplant lymphoproliferative disorders not associated with Epstein-Barr virus: A distinct entity? J. Clin. Oncol. 1998, 16, 2052–2059. [Google Scholar] [CrossRef]
- Courville, E.L.; Yohe, S.; Chou, D.; Nardi, V.; Lazaryan, A.; Thakral, B.; Nelson, A.C.; Ferry, J.A.; Sohani, A.R. EBV-negative monomorphic B-cell post-transplant lymphoproliferative disorders are pathologically distinct from EBV-positive cases and frequently contain TP53 mutations. Mod. Pathol. 2016, 29, 1200–1211. [Google Scholar] [CrossRef]
- Craig, F.E.; Johnson, L.R.; Harvey, S.A.; Nalesnik, M.A.; Luo, J.H.; Bhattacharya, S.D.; Swerdlow, S.H. Gene expression profiling of Epstein-Barr virus-positive and -negative monomorphic B-cell posttransplant lymphoproliferative disorders. Diagn. Mol. Pathol. 2007, 16, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, J.F.; Morscio, J.; Dierickx, D.; Vandenberghe, P.; Gheysens, O.; Verhoef, G.; Zamani, M.; Tousseyn, T.; Wlodarska, I. EBV-Positive and EBV-Negative Posttransplant Diffuse Large B Cell Lymphomas Have Distinct Genomic and Transcriptomic Features. Am. J. Transplant. 2016, 16, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, A.; Capello, D.; Scandurra, M.; Greiner, T.C.; Chan, W.C.; Bhagat, G.; Rossi, D.; Morra, E.; Paulli, M.; Rambaldi, A.; et al. Single nucleotide polymorphism-arrays provide new insights in the pathogenesis of post-transplant diffuse large B-cell lymphoma. Br. J. Haematol. 2010, 149, 569–577. [Google Scholar] [CrossRef] [PubMed]
- DeStefano, C.B.; Desai, S.H.; Shenoy, A.G.; Catlett, J.P. Management of post-transplant lymphoproliferative disorders. Br. J. Haematol. 2018, 182, 330–343. [Google Scholar] [CrossRef] [Green Version]
- McDonald, R.A.; Smith, J.M.; Ho, M.; Lindblad, R.; Ikle, D.; Grimm, P.; Wyatt, R.; Arar, M.; Liereman, D.; Bridges, N.; et al. Incidence of PTLD in pediatric renal transplant recipients receiving basiliximab, calcineurin inhibitor, sirolimus and steroids. Am. J. Transplant. 2008, 8, 984–989. [Google Scholar] [CrossRef]
- Curtis, R.E.; Travis, L.B.; Rowlings, P.A.; Socie, G.; Kingma, D.W.; Banks, P.M.; Jaffe, E.S.; Sale, G.E.; Horowitz, M.M.; Witherspoon, R.P.; et al. Risk of lymphoproliferative disorders after bone marrow transplantation: A multi-institutional study. Blood 1999, 94, 2208–2216. [Google Scholar]
- Abbas, F.; El Kossi, M.; Shaheen, I.S.; Sharma, A.; Halawa, A. Post-transplantation lymphoproliferative disorders: Current concepts and future therapeutic approaches. World J. Transplant. 2020, 10, 29–46. [Google Scholar] [CrossRef]
- Uhlin, M.; Wikell, H.; Sundin, M.; Blennow, O.; Maeurer, M.; Ringden, O.; Winiarski, J.; Ljungman, P.; Remberger, M.; Mattsson, J. Risk factors for Epstein-Barr virus-related post-transplant lymphoproliferative disease after allogeneic hematopoietic stem cell transplantation. Haematologica 2014, 99, 346–352. [Google Scholar] [CrossRef] [Green Version]
- Morton, M.; Coupes, B.; Roberts, S.A.; Johnson, S.L.; Klapper, P.E.; Vallely, P.J.; Picton, M.L. Epstein-Barr virus infection in adult renal transplant recipients. Am. J. Transplant. 2014, 14, 1619–1629. [Google Scholar] [CrossRef]
- Tucci, A.; Rizza, S.; Cocchis, D.; Martini, S.; Romagnoli, R.; Marzano, A. Early and Late Hepatitis B Reactivation After IFN- or DAA-based Therapy of Recurrent Hepatitis C in Anti-HBc-positive Liver Transplant Recipients. Transplantation 2018, 102, e354–e355. [Google Scholar] [CrossRef]
- Zallio, F.; Primon, V.; Tamiazzo, S.; Pini, M.; Baraldi, A.; Corsetti, M.T.; Gotta, F.; Bertassello, C.; Salvi, F.; Rocchetti, A.; et al. Epstein-Barr virus reactivation in allogeneic stem cell transplantation is highly related to cytomegalovirus reactivation. Clin. Transplant. 2013, 27, E491–E497. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Zhu, Q.; Qiu, H.; Chen, F.; Xue, S.; Ma, X.; Sun, A.; Wu, D. Clinical risks analysis of EBV infection in patients with allogeneic hematopoietic stem cell transplantation. Zhonghua Xue Ye Xue Za Zhi 2016, 37, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Landgren, O.; Gilbert, E.S.; Rizzo, J.D.; Socié, G.; Banks, P.M.; Sobocinski, K.A.; Horowitz, M.M.; Jaffe, E.S.; Kingma, D.W.; Travis, L.B.; et al. Risk factors for lymphoproliferative disorders after allogeneic hematopoietic cell transplantation. Blood 2009, 113, 4992–5001. [Google Scholar] [CrossRef] [PubMed]
- Ballen, K.K.; Cutler, C.; Yeap, B.Y.; McAfee, S.L.; Dey, B.R.; Attar, E.C.; Chen, Y.B.; Haspel, R.L.; Liney, D.; Koreth, J.; et al. Donor-derived second hematologic malignancies after cord blood transplantation. Biol. Blood Marrow Transplant. 2010, 16, 1025–1031. [Google Scholar] [CrossRef] [Green Version]
- Strati, P.; Gharaibeh, K.A.; Leung, N.; Cosio, F.G.; Call, T.G.; Shanafelt, T.D. Solid organ transplant in individuals with monoclonal B-cell lymphocytosis and chronic lymphocytic leukaemia. Br. J. Haematol. 2016, 174, 162–165. [Google Scholar] [CrossRef] [Green Version]
- Tiede, C.; Maecker-Kolhoff, B.; Klein, C.; Kreipe, H.; Hussein, K. Risk factors and prognosis in T-cell posttransplantation lymphoproliferative diseases: Reevaluation of 163 cases. Transplantation 2013, 95, 479–488. [Google Scholar] [CrossRef]
- Dierickx, D.; Habermann, T.M. Post-Transplantation Lymphoproliferative Disorders in Adults. N. Engl. J. Med. 2018, 378, 549–562. [Google Scholar] [CrossRef]
- Al Hamed, R.; Bazarbachi, A.H.; Mohty, M. Epstein-Barr virus-related post-transplant lymphoproliferative disease (EBV-PTLD) in the setting of allogeneic stem cell transplantation: A comprehensive review from pathogenesis to forthcoming treatment modalities. Bone Marrow Transplant. 2020, 55, 25–39. [Google Scholar] [CrossRef]
- Trappe, R.U.; Dierickx, D.; Zimmermann, H.; Morschhauser, F.; Mollee, P.; Zaucha, J.M.; Dreyling, M.H.; Duhrsen, U.; Reinke, P.; Verhoef, G.; et al. Response to Rituximab Induction Is a Predictive Marker in B-Cell Post-Transplant Lymphoproliferative Disorder and Allows Successful Stratification Into Rituximab or R-CHOP Consolidation in an International, Prospective, Multicenter Phase II Trial. J. Clin. Oncol. 2017, 35, 536–543. [Google Scholar] [CrossRef] [Green Version]
- Burns, D.M.; Rana, S.; Martin, E.; Nagra, S.; Ward, J.; Osman, H.; Bell, A.I.; Moss, P.; Russell, N.H.; Craddock, C.F.; et al. Greatly reduced risk of EBV reactivation in rituximab-experienced recipients of alemtuzumab-conditioned allogeneic HSCT. Bone Marrow Transplant. 2016, 51, 825–832. [Google Scholar] [CrossRef] [Green Version]
- Van Besien, K.; Bachier-Rodriguez, L.; Satlin, M.; Brown, M.A.; Gergis, U.; Guarneri, D.; Hsu, J.; Phillips, A.A.; Mayer, S.A.; Singh, A.D.; et al. Prophylactic rituximab prevents EBV PTLD in haplo-cord transplant recipients at high risk. Leuk. Lymphoma 2019, 60, 1693–1696. [Google Scholar] [CrossRef] [PubMed]
- McIver, Z.; Stephens, N.; Grim, A.; Barrett, A.J. Rituximab administration within 6 months of T cell-depleted allogeneic SCT is associated with prolonged life-threatening cytopenias. Biol. Blood Marrow Transplant. 2010, 16, 1549–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petropoulou, A.D.; Robin, M.; Rocha, V.; Ribaud, P.; Xhaard, A.; Abboud, I.; Peffault de Latour, R.; Socié, G.; Peraldi, M.-N. Nephrotic Syndrome Associated With Graft Rejection After Unrelated Double Cord Blood Transplantation. Transplantation 2010, 90, 801–802. [Google Scholar] [CrossRef] [PubMed]
- Allen, U.D.; Preiksaitis, J.K. Post-transplant lymphoproliferative disorders, Epstein-Barr virus infection, and disease in solid organ transplantation: Guidelines from the American Society of Transplantation Infectious Diseases Community of Practice. Clin. Transplant. 2019, 33, e13652. [Google Scholar] [CrossRef] [PubMed]
- Tomblyn, M.; Chiller, T.; Einsele, H.; Gress, R.; Sepkowitz, K.; Storek, J.; Wingard, J.R.; Young, J.A.; Boeckh, M.J. Guidelines for preventing infectious complications among hematopoietic cell transplantation recipients: A global perspective. Biol. Blood Marrow Transplant. 2009, 15, 1143–1238. [Google Scholar] [CrossRef] [Green Version]
- Styczynski, J.; van der Velden, W.; Fox, C.P.; Engelhard, D.; de la Camara, R.; Cordonnier, C.; Ljungman, P. Management of Epstein-Barr Virus infections and post-transplant lymphoproliferative disorders in patients after allogeneic hematopoietic stem cell transplantation: Sixth European Conference on Infections in Leukemia (ECIL-6) guidelines. Haematologica 2016, 101, 803–811. [Google Scholar] [CrossRef]
- Van Esser, J.W.; Niesters, H.G.; van der Holt, B.; Meijer, E.; Osterhaus, A.D.; Gratama, J.W.; Verdonck, L.F.; Löwenberg, B.; Cornelissen, J.J. Prevention of Epstein-Barr virus-lymphoproliferative disease by molecular monitoring and preemptive rituximab in high-risk patients after allogeneic stem cell transplantation. Blood 2002, 99, 4364–4369. [Google Scholar] [CrossRef]
- Styczynski, J.; Reusser, P.; Einsele, H.; de la Camara, R.; Cordonnier, C.; Ward, K.N.; Ljungman, P.; Engelhard, D. Management of HSV, VZV and EBV infections in patients with hematological malignancies and after SCT: Guidelines from the Second European Conference on Infections in Leukemia. Bone Marrow Transplant. 2009, 43, 757–770. [Google Scholar] [CrossRef] [Green Version]
- Kinch, A.; Sundström, C.; Baecklund, E.; Backlin, C.; Molin, D.; Enblad, G. Expression of PD-1, PD-L1, and PD-L2 in posttransplant lymphoproliferative disorder after solid organ transplantation. Leuk. Lymphoma 2019, 60, 376–384. [Google Scholar] [CrossRef] [Green Version]
- Delapierre, B.; Reman, O.; Dina, J.; Breuil, C.; Bellal, M.; Johnson-Ansah, H.; Gac, A.C.; Damaj, G.; Chantepie, S. Low dose Rituximab for pre-emptive treatment of Epstein Barr virus reactivation after allogenic hematopoietic stem cell transplantation. Curr. Res. Transl. Med. 2019, 67, 145–148. [Google Scholar] [CrossRef]
- Styczynski, J.; Gil, L.; Tridello, G.; Ljungman, P.; Donnelly, J.P.; van der Velden, W.; Omar, H.; Martino, R.; Halkes, C.; Faraci, M.; et al. Response to rituximab-based therapy and risk factor analysis in Epstein Barr Virus-related lymphoproliferative disorder after hematopoietic stem cell transplant in children and adults: A study from the Infectious Diseases Working Party of the European Group for Blood and Marrow Transplantation. Clin. Infect. Dis. 2013, 57, 794–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.-P.; Zhang, C.-L.; Mo, X.-D.; Zhang, X.-H.; Chen, H.; Han, W.; Chen, Y.-H.; Wang, Y.; Yan, C.-H.; Wang, J.-Z.; et al. Epstein-Barr Virus–Related Post-Transplantation Lymphoproliferative Disorder after Unmanipulated Human Leukocyte Antigen Haploidentical Hematopoietic Stem Cell Transplantation: Incidence, Risk Factors, Treatment, and Clinical Outcomes. Biol. Blood Marrow Transplant. 2015, 21, 2185–2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, C.P.; Burns, D.; Parker, A.N.; Peggs, K.S.; Harvey, C.M.; Natarajan, S.; Marks, D.I.; Jackson, B.; Chakupurakal, G.; Dennis, M.; et al. EBV-associated post-transplant lymphoproliferative disorder following in vivo T-cell-depleted allogeneic transplantation: Clinical features, viral load correlates and prognostic factors in the rituximab era. Bone Marrow Transplant. 2014, 49, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Doubrovina, E.; Oflaz-Sozmen, B.; Prockop, S.E.; Kernan, N.A.; Abramson, S.; Teruya-Feldstein, J.; Hedvat, C.; Chou, J.F.; Heller, G.; Barker, J.N.; et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood 2012, 119, 2644–2656. [Google Scholar] [CrossRef]
- Savoldo, B.; Goss, J.A.; Hammer, M.M.; Zhang, L.; Lopez, T.; Gee, A.P.; Lin, Y.F.; Quiros-Tejeira, R.E.; Reinke, P.; Schubert, S.; et al. Treatment of solid organ transplant recipients with autologous Epstein Barr virus-specific cytotoxic T lymphocytes (CTLs). Blood 2006, 108, 2942–2949. [Google Scholar] [CrossRef]
- Heslop, H.E.; Slobod, K.S.; Pule, M.A.; Hale, G.A.; Rousseau, A.; Smith, C.A.; Bollard, C.M.; Liu, H.; Wu, M.F.; Rochester, R.J.; et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood 2010, 115, 925–935. [Google Scholar] [CrossRef] [Green Version]
- Lindsay, J.; Othman, J.; Heldman, M.R.; Slavin, M.A. Epstein-Barr virus posttransplant lymphoproliferative disorder: Update on management and outcomes. Curr. Opin. Infect. Dis. 2021, 34, 635–645. [Google Scholar] [CrossRef]
- Rooney, C.M.; Smith, C.A.; Ng, C.Y.; Loftin, S.K.; Sixbey, J.W.; Gan, Y.; Srivastava, D.K.; Bowman, L.C.; Krance, R.A.; Brenner, M.K.; et al. Infusion of cytotoxic T cells for the prevention and treatment of Epstein-Barr virus-induced lymphoma in allogeneic transplant recipients. Blood 1998, 92, 1549–1555. [Google Scholar] [CrossRef]
- Bollard, C.M.; Cooper, L.J.; Heslop, H.E. Immunotherapy targeting EBV-expressing lymphoproliferative diseases. Best Pract. Res. Clin. Haematol. 2008, 21, 405–420. [Google Scholar] [CrossRef] [Green Version]
- Moosmann, A.; Bigalke, I.; Tischer, J.; Schirrmann, L.; Kasten, J.; Tippmer, S.; Leeping, M.; Prevalsek, D.; Jaeger, G.; Ledderose, G.; et al. Effective and long-term control of EBV PTLD after transfer of peptide-selected T cells. Blood 2010, 115, 2960–2970. [Google Scholar] [CrossRef] [Green Version]
- Prockop, S.; Doubrovina, E.; Suser, S.; Heller, G.; Barker, J.; Dahi, P.; Perales, M.A.; Papadopoulos, E.; Sauter, C.; Castro-Malaspina, H.; et al. Off-the-shelf EBV-specific T cell immunotherapy for rituximab-refractory EBV-associated lymphoma following transplantation. J. Clin. Investig. 2020, 130, 733–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prockop, S.; Mahadeo, K.M.; Beitinjaneh, A.; Choquet, S.; Stiff, P.; Reshef, R.; Satyanarayana, G.; Dahiya, S.; Parmar, H.; Ye, W.; et al. Multicenter, Open-Label, Phase 3 Study of Tabelecleucel for Solid Organ or Allogeneic Hematopoietic Cell Transplant Recipients with Epstein-Barr Virus-Driven Post Transplant Lymphoproliferative Disease after Failure of Rituximab or Rituximab and Chemotherapy (ALLELE). Blood 2021, 138, 301. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Locke, F.L.; Miklos, D.B.; Jacobson, C.A.; Perales, M.A.; Kersten, M.J.; Oluwole, O.O.; Ghobadi, A.; Rapoport, A.P.; McGuirk, J.; Pagel, J.M.; et al. Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. N. Engl. J. Med. 2022, 386, 640–654. [Google Scholar] [CrossRef]
- Krishnamoorthy, S.; Ghobadi, A.; Santos, R.D.; Schilling, J.D.; Malone, A.F.; Murad, H.; Bartlett, N.L.; Alhamad, T. CAR-T therapy in solid organ transplant recipients with treatment refractory posttransplant lymphoproliferative disorder. Am. J. Transplant. 2021, 21, 809–814. [Google Scholar] [CrossRef]
- Mamlouk, O.; Nair, R.; Iyer, S.P.; Edwards, A.; Neelapu, S.S.; Steiner, R.E.; Adkins, S.A.; Hawkins, M.; Saini, N.; Devashish, K.; et al. Safety of CAR T-cell therapy in kidney transplant recipients. Blood 2021, 137, 2558–2562. [Google Scholar] [CrossRef]
- Luttwak, E.; Hagin, D.; Perry, C.; Wolach, O.; Itchaki, G.; Amit, O.; Bar-On, Y.; Freund, T.; Kay, S.; Eshel, R.; et al. Anti-CD19 CAR-T therapy for EBV-negative posttransplantation lymphoproliferative disease-a single center case series. Bone Marrow Transplant. 2021, 56, 1031–1037. [Google Scholar] [CrossRef]
- Feng, G.; Li, Q.; Zhu, H.; Jiang, Y.; Yuan, J.; Fu, Y.; Deng, Q. Safety and Efficacy of Anti-CD19-Chimeric Antigen Receptor T Cell Combined With Programmed Cell Death 1 Inhibitor Therapy in a Patient With Refractory Post-Transplant Lymphoproliferative Disease: Case Report and Literature Review. Front. Oncol. 2021, 11, 726134. [Google Scholar] [CrossRef]
- Tanaka, K.; Albin, M.J.; Yuan, X.; Yamaura, K.; Habicht, A.; Murayama, T.; Grimm, M.; Waaga, A.M.; Ueno, T.; Padera, R.F.; et al. PDL1 is required for peripheral transplantation tolerance and protection from chronic allograft rejection. J. Immunol. 2007, 179, 5204–5210. [Google Scholar] [CrossRef] [Green Version]
- Yan, N.; Wang, N.; Zhang, P.; Wang, G.; Mao, X.; Peng, D.; Kuang, D.; Chen, L.; Zhu, L.; Zhou, J.; et al. Case Report: Successful Chimeric Antigen Receptor T Cell Therapy in Haploidentical-Allogeneic Stem Cell Transplant Patients With Post-Transplant Lymphoproliferative Disorder. Front. Oncol. 2021, 11, 709370. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Barrett, D.M.; Kenderian, S.S.; Shestova, O.; Hofmann, T.J.; Perazzelli, J.; Klichinsky, M.; Aikawa, V.; Nazimuddin, F.; Kozlowski, M.; et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Investig. 2016, 126, 3814–3826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhou, Y.; Li, W.; Tang, Q.; Chen, R.; Zhu, J.; Feng, Z. T cells expressing a LMP1-specific chimeric antigen receptor mediate antitumor effects against LMP1-positive nasopharyngeal carcinoma cells in vitro and in vivo. J. Biomed. Res. 2014, 28, 468–475. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clerico, M.; Dogliotti, I.; Aroldi, A.; Consoli, C.; Giaccone, L.; Bruno, B.; Cavallo, F. Post-Transplant Lymphoproliferative Disease (PTLD) after Allogeneic Hematopoietic Stem Cell Transplantation: Biology and Treatment Options. J. Clin. Med. 2022, 11, 7542. https://doi.org/10.3390/jcm11247542
Clerico M, Dogliotti I, Aroldi A, Consoli C, Giaccone L, Bruno B, Cavallo F. Post-Transplant Lymphoproliferative Disease (PTLD) after Allogeneic Hematopoietic Stem Cell Transplantation: Biology and Treatment Options. Journal of Clinical Medicine. 2022; 11(24):7542. https://doi.org/10.3390/jcm11247542
Chicago/Turabian StyleClerico, Michele, Irene Dogliotti, Andrea Aroldi, Chiara Consoli, Luisa Giaccone, Benedetto Bruno, and Federica Cavallo. 2022. "Post-Transplant Lymphoproliferative Disease (PTLD) after Allogeneic Hematopoietic Stem Cell Transplantation: Biology and Treatment Options" Journal of Clinical Medicine 11, no. 24: 7542. https://doi.org/10.3390/jcm11247542
APA StyleClerico, M., Dogliotti, I., Aroldi, A., Consoli, C., Giaccone, L., Bruno, B., & Cavallo, F. (2022). Post-Transplant Lymphoproliferative Disease (PTLD) after Allogeneic Hematopoietic Stem Cell Transplantation: Biology and Treatment Options. Journal of Clinical Medicine, 11(24), 7542. https://doi.org/10.3390/jcm11247542