
Cell Death in AMD: The Rationale for Targeting Fas

 ,
, 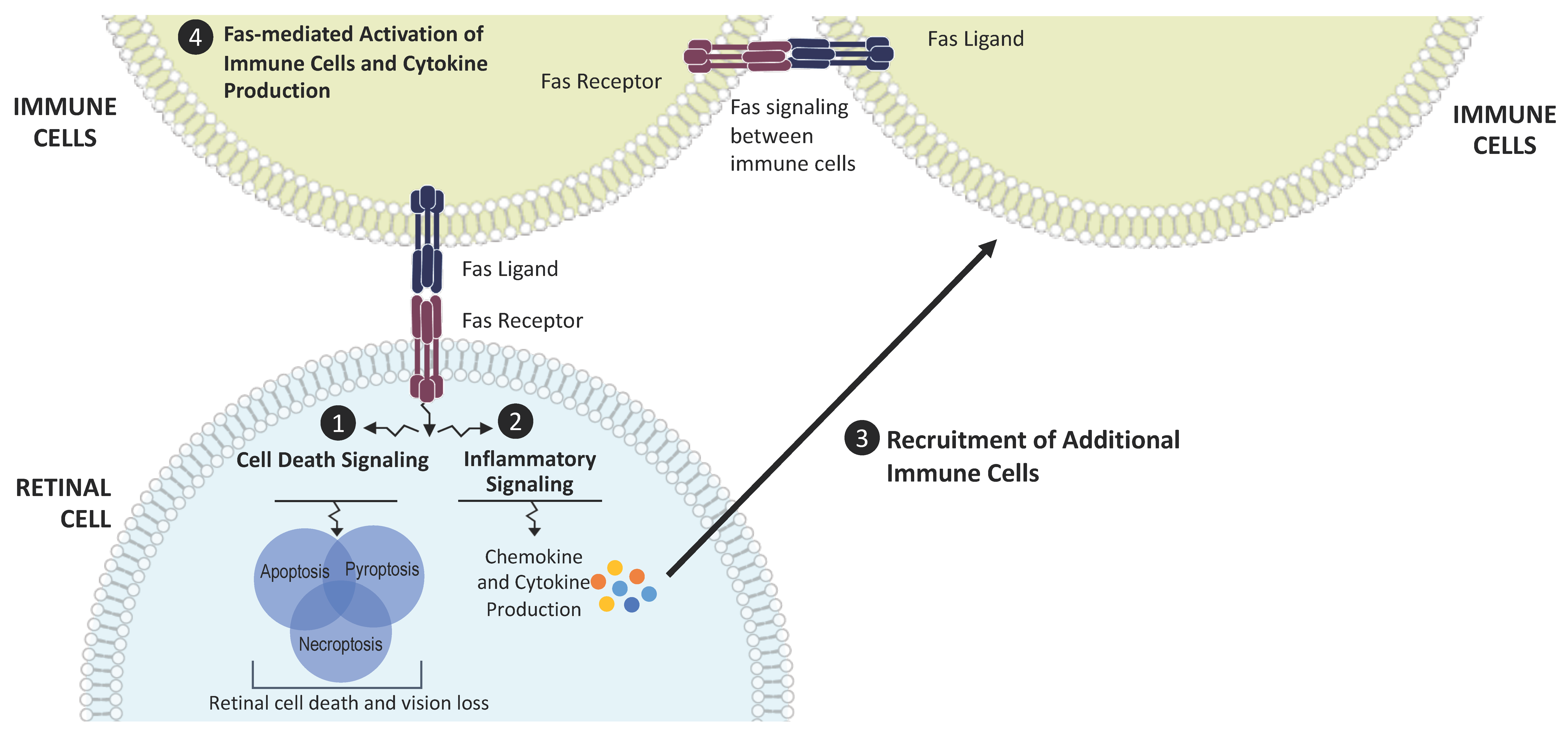{kind=link}
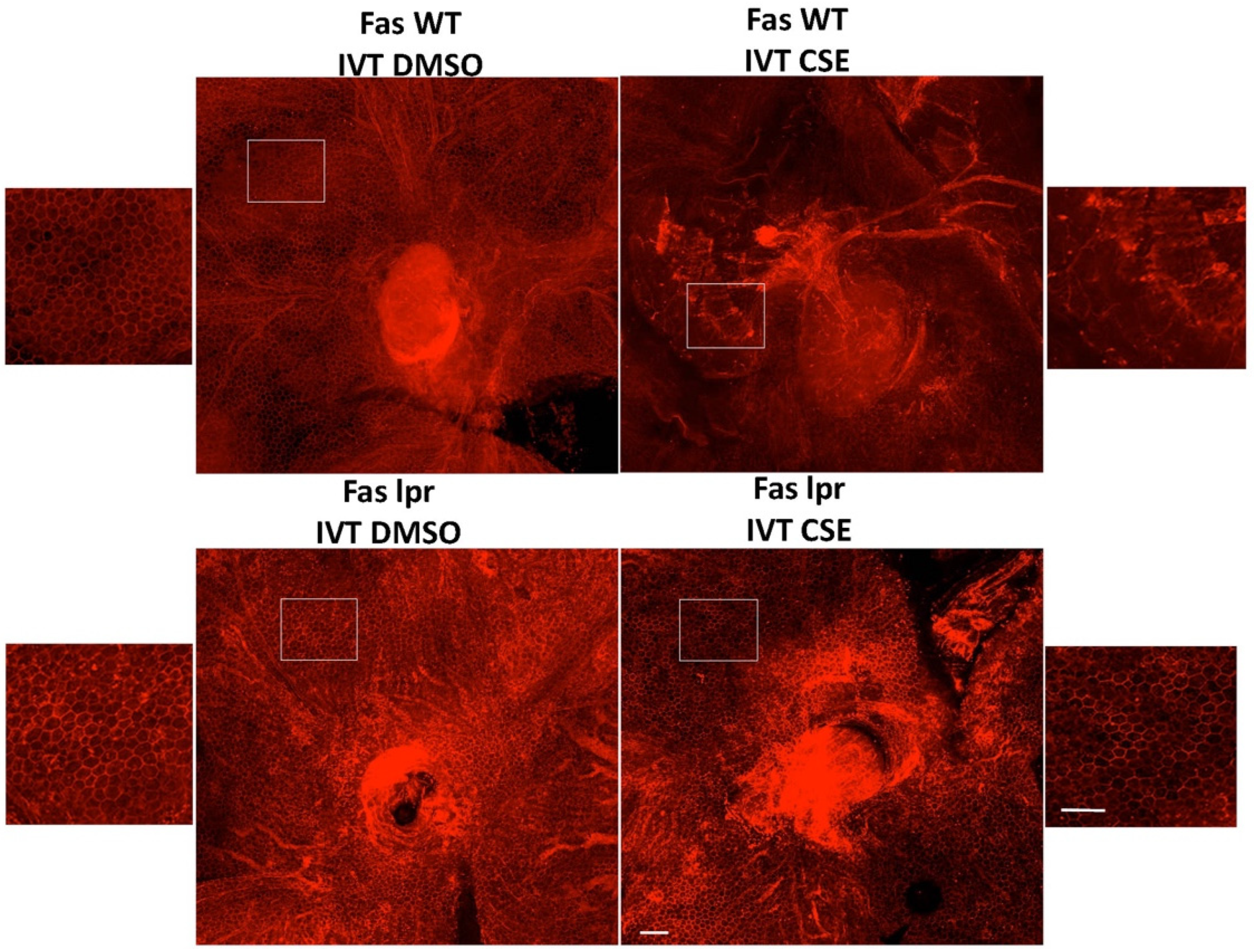{kind=link}
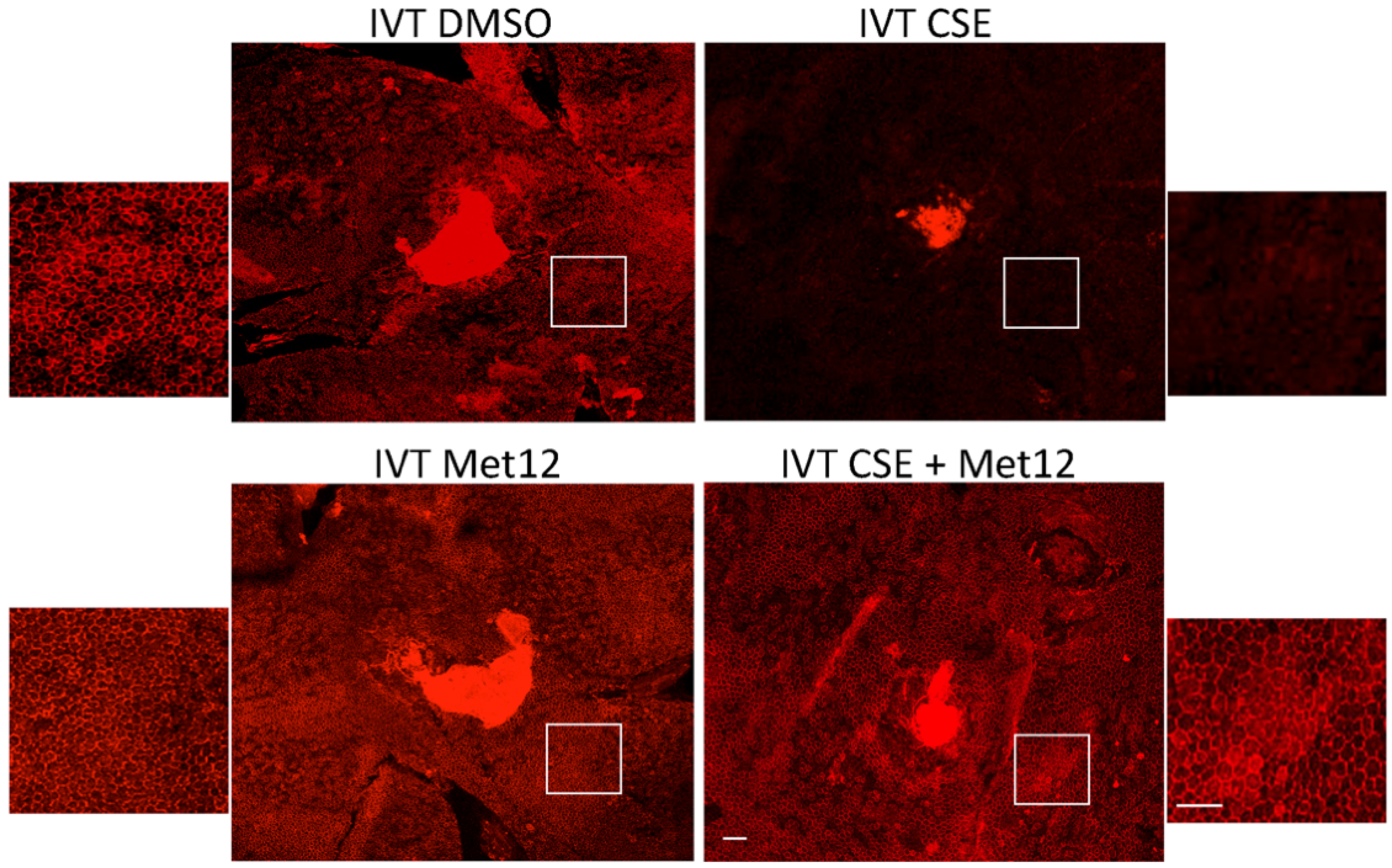{kind=link}
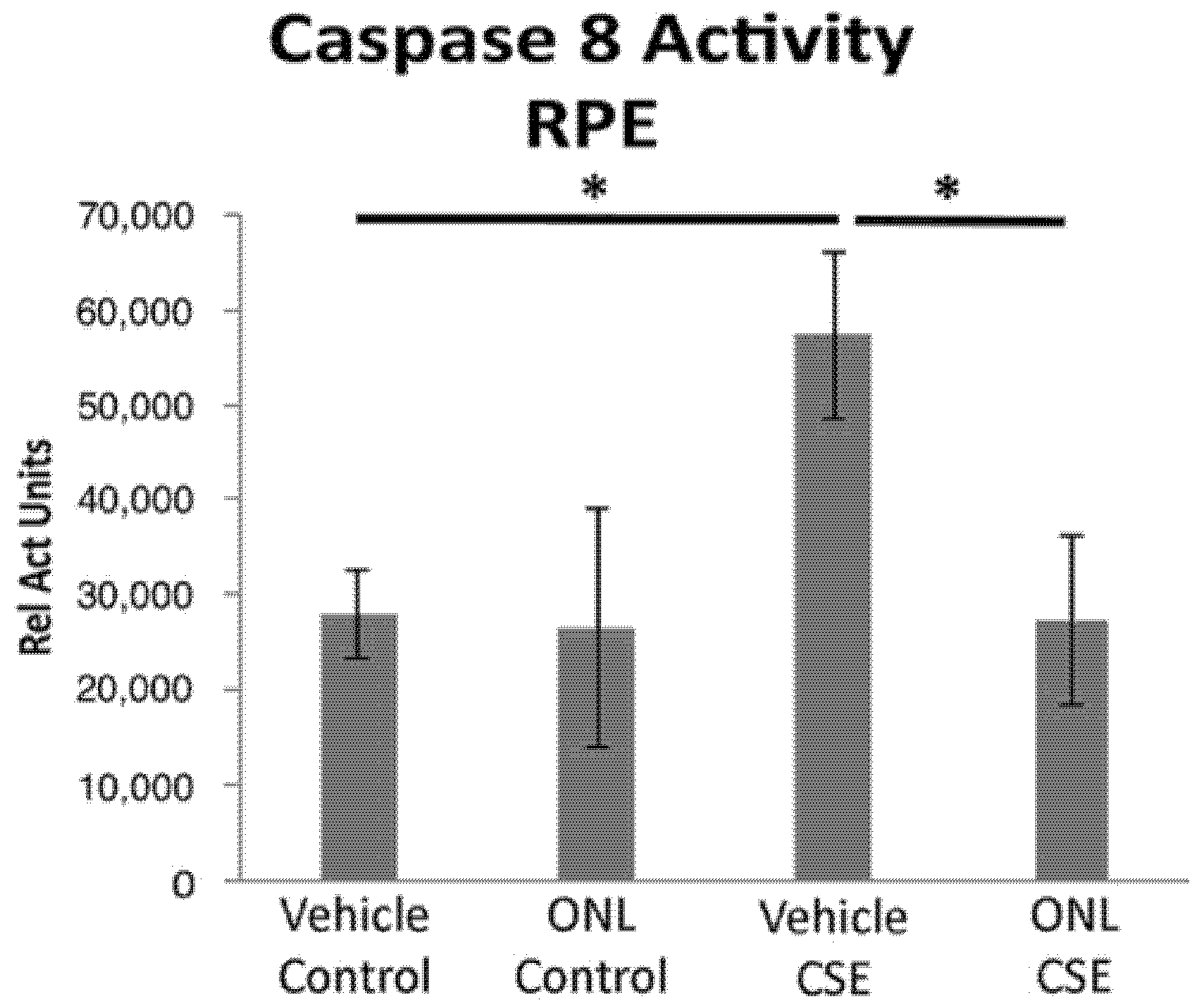{kind=link}
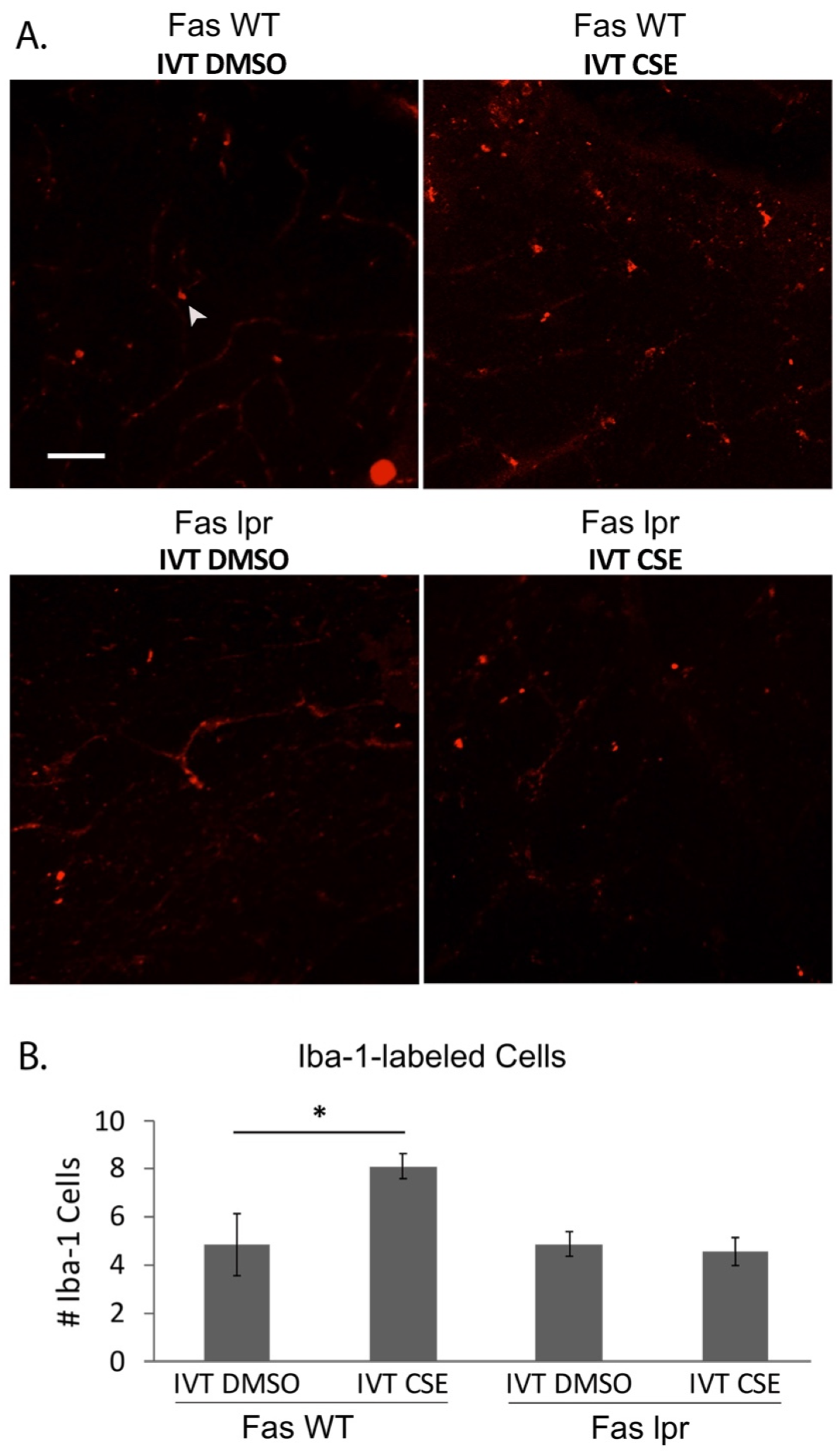{kind=link}
{kind=link}
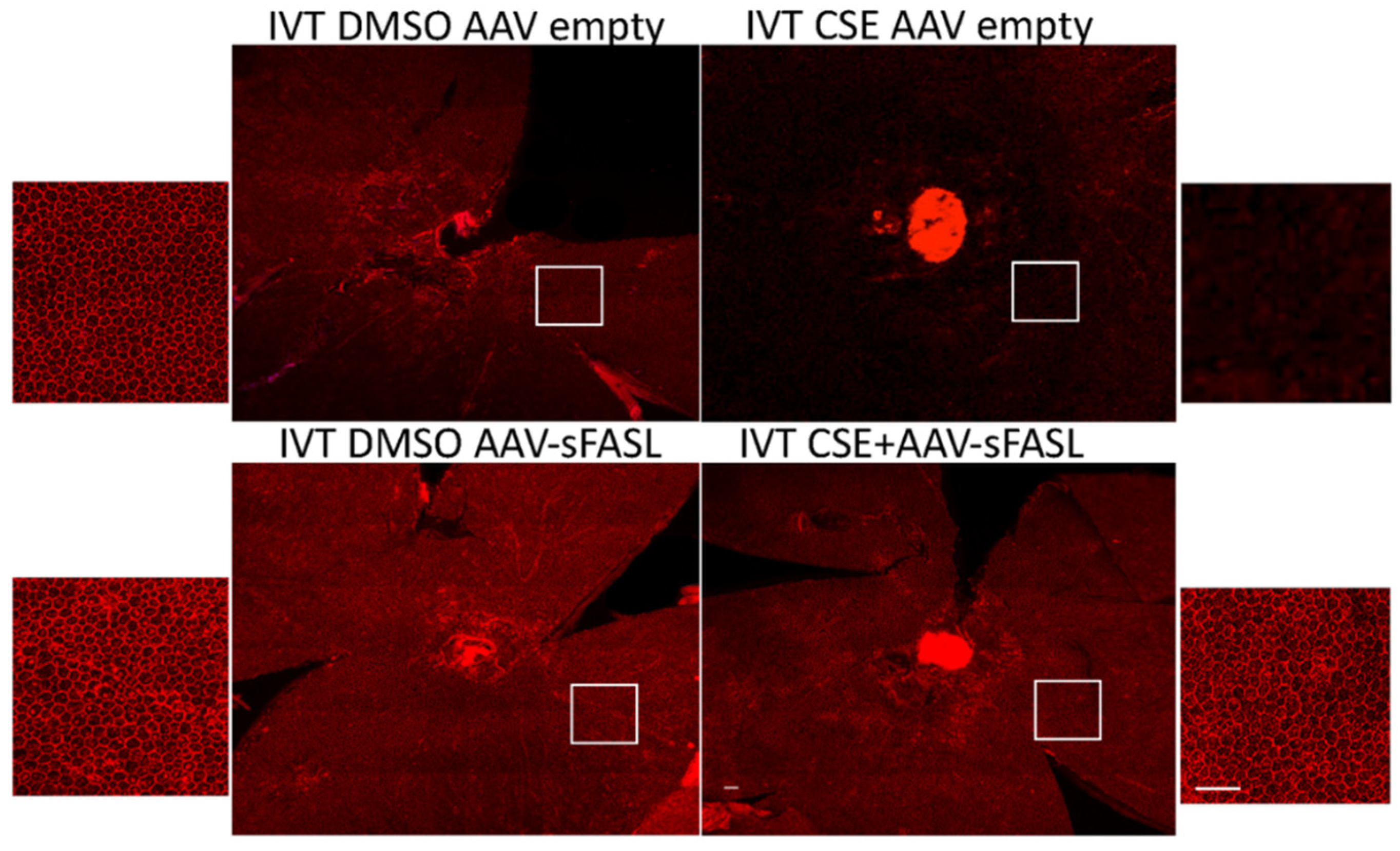{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Overview of Programmed Cell Death
3. Programmed Cell Death in AMD
4. The Rationale for Targeting Fas
5. Fas Inhibition and Retinal Cell Protection
6. Fas Inhibition and Disease-Associated Inflammation
7. Gene Therapy for Inhibiting Fas
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Materials and Methods
References
- Friedman, D.S.; O’Colmain, B.J.; Muñoz, B.; Tomany, S.C.; McCarty, C.; DeJong, P.T.V.M.; Nemesure, B.; Mitchell, P.; Kempen, J.; Congdon, N. Prevalence of Age-Related Macular Degeneration in the United States. Arch. Ophthalmol. 2004, 122, 564–572. [Google Scholar]
- Clemons, T.E.; Milton, R.C.; Klein, R.; Seddon, J.M.; Ferris, F.; Age-Related Eye Disease Study Research Group. Risk Factors for the Incidence of Advanced Age-Related Macular Degeneration in the Age-Related Eye Disease Study (AREDS): AREDS report no. 19. Ophthalmology 2005, 112, 533–539. [Google Scholar] [CrossRef] [Green Version]
- Hyman, L.; Neborsky, R. Risk factors for age-related macular degeneration: An update. Curr. Opin. Ophthalmol. 2002, 13, 171–175. [Google Scholar] [CrossRef]
- Ferris, F.L.; Davis, M.D.; Clemons, T.E.; Lee, L.Y.; Chew, E.Y.; Lindblad, A.S.; Milton, R.C.; Bressler, S.B.; Klein, R. A simplified severity scale for age-related macular degeneration: AREDS report no. 18. Arch. Ophthalmol. 2005, 123, 1570–1574. [Google Scholar]
- Halawa, O.A.; Lin, J.B.; Miller, J.W.; Vavvas, D.G. A Review of Completed and Ongoing Complement Inhibitor Trials for Geographic Atrophy Secondary to Age-Related Macular Degeneration. J. Clin. Med. 2021, 10, 2580. [Google Scholar] [CrossRef]
- Hannah, J.Y.; Wykoff, C.C. Investigational Agents in Development for the Treatment of Geographic Atrophy Secondary to Age-Related Macular Degeneration. BioDrugs 2021, 35, 303–323. [Google Scholar]
- Mahmoudzadeh, R.; Hinkle, J.W.; Hsu, J.; Garg, S.J. Emerging treatments for geographic atrophy in age-related macular degeneration. Curr. Opin. Ophthalmol. 2021, 32, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.D.; Weil, M.; Raff, M.C. Programmed Cell Death in Animal Development. Cell 1997, 88, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Krysko, D.V.; Vanden Berghe, T.; D’Herde, K.; Vandenabeele, P. Apoptosis and necrosis: Detection, discrimination and phagocytosis. Methods 2008, 44, 205–221. [Google Scholar] [CrossRef]
- Vanden Berghe, T.; Grootjans, S.; Goossens, V.; Dondelinger, Y.; Krysko, D.V.; Takahashi, N.; Vandenabeele, P. Determination of apoptotic and necrotic cell death in vitro and in vivo. Methods 2013, 61, 117–129. [Google Scholar] [CrossRef]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef]
- Ketelut-Carneiro, N.; Fitzgerald, K.A. Apoptosis, pyroptosis, and necroptosis—Oh my! The many ways a cell can die. J. Mol. Biol. 2021, 167378. [Google Scholar] [CrossRef]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.-L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Hadian, K.; Stockwell, B.R. SnapShot: Ferroptosis. Cell 2020, 181, 1188. [Google Scholar] [CrossRef]
- Shu, W.; Dunaief, J.L. Potential Treatment of Retinal Diseases with Iron Chelators. Pharmaceuticals 2018, 11, 112. [Google Scholar] [CrossRef] [Green Version]
- Ambati, J.; Fowler, B.J. Mechanisms of Age-Related Macular Degeneration. Neuron 2012, 75, 26–39. [Google Scholar] [CrossRef] [Green Version]
- Kinnunen, K.; Petrovski, G.; Moe, M.C.; Berta, A.; Kaarniranta, K. Molecular mechanisms of retinal pigment epithelium damage and development of age-related macular degeneration. Acta Ophthalmol. 2012, 90, 299–309. [Google Scholar] [CrossRef]
- Hanus, J.; Anderson, C.; Wang, S. RPE necroptosis in response to oxidative stress and in AMD. Ageing Res. Rev. 2015, 24, 286–298. [Google Scholar] [CrossRef] [Green Version]
- Hanus, J.; Anderson, C.; Sarraf, D.; Ma, J.; Wang, S. Retinal pigment epithelial cell necroptosis in response to sodium iodate. Cell Death Discov. 2016, 2, 16054. [Google Scholar] [CrossRef]
- Dunaief, J.L.; Dentchev, T.; Ying, G.-S.; Milam, A.H. The Role of Apoptosis in Age-Related Macular Degeneration. Arch. Ophthalmol. 2002, 120, 1435–1442. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.T.; Gao, J.; Cao, S.; Sandhu, N.; Cui, J.Z.; Chou, C.L.; Fang, E.; Matsubara, J.A. Inflammatory Mediators Induced by Amyloid-Beta in the Retina and RPE In Vivo: Implications for Inflammasome Activation in Age-Related Macular Degeneration. Investig. Opthalmol. Vis. Sci. 2013, 54, 2225–2237. [Google Scholar] [CrossRef] [Green Version]
- Tseng, W.A.; Thein, T.; Kinnunen, K.; Lashkari, K.; Gregory, M.S.; D’Amore, P.A.; Ksander, B.R. NLRP3 Inflammasome Activation in Retinal Pigment Epithelial Cells by Lysosomal Destabilization: Implications for Age-Related Macular Degeneration. Investig. Opthalmol. Vis. Sci. 2013, 54, 110–120. [Google Scholar] [CrossRef]
- Brandstetter, C.; Patt, J.; Holz, F.G.; Krohne, T.U. Inflammasome priming increases retinal pigment epithelial cell susceptibility to lipofuscin phototoxicity by changing the cell death mechanism from apoptosis to pyroptosis. J. Photochem. Photobiol. B Biol. 2016, 161, 177–183. [Google Scholar] [CrossRef]
- Gao, J.; Cui, J.Z.; To, E.; Cao, S.; Matsubara, J.A. Evidence for the activation of pyroptotic and apoptotic pathways in RPE cells associated with NLRP3 inflammasome in the rodent eye. J. Neuroinflamm. 2018, 15, 15. [Google Scholar] [CrossRef]
- Jaffe, G.J.; Westby, K.; Csaky, K.G.; Mones, J.; Pearlman, J.A.; Patel, S.S.; Joondeph, B.C.; Randolph, J.; Masonson, H.; Rezaei, K.A. C5 inhibitor avacincaptad pegol for geographic atrophy due to age-related macular degeneration: A randomized pivotal phase 2/3 trial. Ophthalmology 2021, 128, 576–586. [Google Scholar] [CrossRef]
- Liao, D.S.; Grossi, F.V.; El Mehdi, D.; Gerber, M.R.; Brown, D.M.; Heier, J.S.; Wykoff, C.C.; Singerman, L.J.; Abraham, P.; Grassmann, F.; et al. Complement C3 Inhibitor Pegcetacoplan for Geographic Atrophy Secondary to Age-Related Macular Degeneration: A Randomized Phase 2 Trial. Ophthalmology 2020, 127, 186–195. [Google Scholar] [CrossRef] [Green Version]
- Scholl, H.P.; Boyer, D.; Giani, A.; Chong, V. The Use of Neuroprotective Agents in Treating Geographic Atrophy. Ophthalmic Res. 2021, 64, 888–902. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Notomi, S.; Hisatomi, T.; Nakazawa, T.; Ishibashi, T.; Miller, J.W.; Vavvas, D.G. Photoreceptor cell death and rescue in retinal detachment and degenerations. Prog. Retin. Eye Res. 2013, 37, 114–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huckfeldt, R.; Vavvas, D. Neuroprotection for Retinal Detachment. Int. Ophthalmol. Clin. 2013, 53, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, F.; Beutler, B. The Tumor Necrosis Factor Ligand and Receptor Families. N. Engl. J. Med. 1996, 334, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Wang, R.; Zhang, L.; Yin, D.; Luo, X.; Solomon, J.; Jiang, R.; Markos, K.; Davidson, W.; Scott, D.; et al. Death the Fas way: Regulation and pathophysiology of CD95 and its ligand. Pharmacol. Ther. 2000, 88, 333–347. [Google Scholar] [CrossRef]
- Mouasni, S.; Tourneur, L. FADD at the Crossroads between Cancer and Inflammation. Trends Immunol. 2018, 39, 1036–1053. [Google Scholar] [CrossRef]
- Guégan, J.; Legembre, P. Nonapoptotic functions of Fas/ CD 95 in the immune response. FEBS J. 2018, 285, 809–827. [Google Scholar] [CrossRef]
- Zacks, D.N.; Zheng, Q.-D.; Bakhru, R.; Han, Y.; Miller, J.W. FAS-Mediated Apoptosis and Its Relation to Intrinsic Pathway Activation in an Experimental Model of Retinal Detachment. Investig. Opthalmol. Vis. Sci. 2004, 45, 4563–4569. [Google Scholar] [CrossRef]
- Zacks, D.N.; Boehlke, C.; Richards, A.L.; Zheng, Q.D. Role of the Fas-Signaling Pathway in Photoreceptor Neuroprotection. Arch. Ophthalmol. 2007, 125, 1389–1395. [Google Scholar] [CrossRef] [Green Version]
- Besirli, C.G.; Zheng, Q.-D.; Reed, D.M.; Zacks, D.N. ERK-Mediated Activation of Fas Apoptotic Inhibitory Molecule 2 (Faim2) Prevents Apoptosis of 661W Cells in a Model of Detachment-Induced Photoreceptor Cell Death. PLoS ONE 2012, 7, e46664. [Google Scholar] [CrossRef]
- Pawar, M.; Busov, B.; Chandrasekhar, A.; Yao, J.; Zacks, D.N.; Besirli, C.G. FAS apoptotic inhibitory molecule 2 is a stress-induced intrinsic neuroprotective factor in the retina. Cell Death Differ. 2017, 24, 1799–1810. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Yao, J.; Jia, L.; Lin, C.; Zacks, D.N. Protective Effect of Met12, a Small Peptide Inhibitor of Fas, on the Retinal Pigment Epithelium and Photoreceptor After Sodium Iodate Injury. Investig. Opthalmol. Vis. Sci. 2017, 58, 1801–1810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, A.; Kocab, A.J.; Zacks, D.N.; Marshak-Rothstein, A.; Gregory-Ksander, M. A small peptide antagonist of the Fas receptor inhibits neuroinflammation and prevents axon degeneration and retinal ganglion cell death in an inducible mouse model of glaucoma. J. Neuroinflamm. 2019, 16, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Moriarty-Craige, S.E.; Li, C.; Lynn, M.J.; Cai, J.; Jones, D.P.; Sternberg, P. Associations of Plasma-Soluble Fas Ligand with Aging and Age-Related Macular Degeneration. Investig. Opthalmol. Vis. Sci. 2008, 49, 1345–1349. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kondo, N.; Cano, M.; Ebrahimi, K.; Yoshida, T.; Barnett, B.P.; Biswal, S.; Handa, J.T. Nrf2 signaling modulates cigarette smoke-induced complement activation in retinal pigmented epithelial cells. Free Radic. Biol. Med. 2014, 70, 155–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cano, M.; Wang, L.; Wan, J.; Barnett, B.P.; Ebrahimi, K.; Qian, J.; Handa, J.T. Oxidative stress induces mitochondrial dysfunction and a protective unfolded protein response in RPE cells. Free Radic. Biol. Med. 2014, 69, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Fleckenstein, M.; Keenan, T.D.L.; Guymer, R.H.; Chakravarthy, U.; Schmitz-Valckenberg, S.; Klaver, C.C.; Wong, W.T.; Chew, E.Y. Age-related macular degeneration. Nat. Rev. Dis. Prim. 2021, 7, 31. [Google Scholar] [CrossRef]
- Spindler, J.; Zandi, S.; Pfister, I.B.; Gerhardt, C.; Garweg, J.G. Cytokine profiles in the aqueous humor and serum of patients with dry and treated wet age-related macular degeneration. PLoS ONE 2018, 13, e0203337. [Google Scholar] [CrossRef]
- Nielsen, M.K.; Subhi, Y.; Molbech, C.R.; Falk, M.K.; Nissen, M.H.; Sørensen, T.L. Chemokine Profile and the Alterations in CCR5-CCL5 Axis in Geographic Atrophy Secondary to Age-Related Macular Degeneration. Investig. Opthalmol. Vis. Sci. 2020, 61, 28. [Google Scholar] [CrossRef] [Green Version]
- Camelo, S.; Lavelette, S.; Guillonneau, X.; Raoul, W.; Sennlaub, F. Association of Choroidal Interleukin-17-Producing T Lymphocytes and Macrophages with Geographic Atrophy. Ophthalmologica 2016, 236, 53–58. [Google Scholar] [CrossRef]
- Ambati, J.; Atkinson, J.P.; Gelfand, B.D. Immunology of age-related macular degeneration. Nat. Rev. Immunol. 2013, 13, 438–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozaki, M.; Raisler, B.J.; Sakurai, E.; Sarma, J.V.; Barnum, S.R.; Lambris, J.; Chen, Y.; Zhang, K.; Ambati, B.K.; Baffi, J.Z.; et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc. Natl. Acad. Sci. USA 2006, 103, 2328–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, S.P.; Henry, C.; Kearney, C.J.; Logue, S.; Feoktistova, M.; Tynan, G.A.; Lavelle, E.; Leverkus, M.; Martin, S. Fas/CD95-Induced Chemokines Can Serve as “Find-Me” Signals for Apoptotic Cells. Mol. Cell 2013, 49, 1034–1048. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.; Wang, Y.; Yun, J.; Hajrasouliha, A.R.; Zhao, Y.; Sun, D.; Kaplan, H.J.; Shao, H. HMGB1 release triggered by the interaction of live retinal cells and uveitogenic T cells is Fas/FasL activation-dependent. J. Neuroinflamm. 2015, 12, 179. [Google Scholar] [CrossRef] [Green Version]
- Hohlbaum, A.M.; Moe, S.; Marshak-Rothstein, A. Opposing Effects of Transmembrane and Soluble FAS Ligand Expression on Inflammation and Tumor Cell Survival. J. Exp. Med. 2000, 191, 1209–1220. [Google Scholar] [CrossRef] [Green Version]
- Gregory, M.S.; Hackett, C.G.; Abernathy, E.F.; Lee, K.S.; Saff, R.R.; Hohlbaum, A.M.; Moody, K.-S.L.; Hobson, M.W.; Jones, A.; Kolovou, P.; et al. Opposing Roles for Membrane Bound and Soluble Fas Ligand in Glaucoma-Associated Retinal Ganglion Cell Death. PLoS ONE 2011, 6, e17659. [Google Scholar] [CrossRef]
- Krishnan, A.; Fei, F.; Jones, A.; Busto, P.; Marshak-Rothstein, A.; Ksander, B.R.; Gregory-Ksander, M. Overexpression of Soluble Fas Ligand following Adeno-Associated Virus Gene Therapy Prevents Retinal Ganglion Cell Death in Chronic and Acute Murine Models of Glaucoma. J. Immunol. 2016, 197, 4626–4638. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zacks, D.N.; Kocab, A.J.; Choi, J.J.; Gregory-Ksander, M.S.; Cano, M.; Handa, J.T. Cell Death in AMD: The Rationale for Targeting Fas. J. Clin. Med. 2022, 11, 592. https://doi.org/10.3390/jcm11030592
Zacks DN, Kocab AJ, Choi JJ, Gregory-Ksander MS, Cano M, Handa JT. Cell Death in AMD: The Rationale for Targeting Fas. Journal of Clinical Medicine. 2022; 11(3):592. https://doi.org/10.3390/jcm11030592
Chicago/Turabian StyleZacks, David N., Andrew J. Kocab, Joanne J. Choi, Meredith S. Gregory-Ksander, Marisol Cano, and James T. Handa. 2022. "Cell Death in AMD: The Rationale for Targeting Fas" Journal of Clinical Medicine 11, no. 3: 592. https://doi.org/10.3390/jcm11030592
APA StyleZacks, D. N., Kocab, A. J., Choi, J. J., Gregory-Ksander, M. S., Cano, M., & Handa, J. T. (2022). Cell Death in AMD: The Rationale for Targeting Fas. Journal of Clinical Medicine, 11(3), 592. https://doi.org/10.3390/jcm11030592