
First Stabilize and then Gradually Recruit: A Paradigm Shift in Protective Mechanical Ventilation for Acute Lung Injury

 , , ,
, , ,  and
and
Abstract
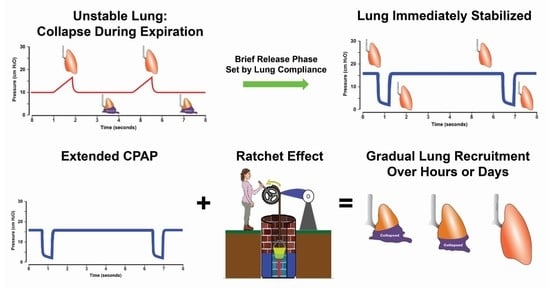
:
1. Introduction
2. Conceptual Approaches to Protective Lung Ventilation
2.1. Protective Lung Approach
2.2. Open Lung Approach
2.3. The Acutely Injured Lung Becomes Time- and Pressure-Dependent
2.4. Stabilize Lung Approach
3. Stabilize the Lung Approach
3.1. Time-Dependent Alveolar Collapse
3.2. TCAV Method Stabilizes and then Opens the Lung
3.3. Basic Scientific Evidence of TCAV Efficacy
3.4. Time-Controlled Alveolar Recruitment
4. How TCAV Works
4.1. Inspiratory and Expiratory Time to First Stabilize and then Gradually Recruit Alveoli
4.2. Inflate-and-Brake Method to Ratchet Open Collapsed Alveoli
5. Physiological Correctness: Scientifically Sound Methods to Set and Adjust the APRV Mode
5.1. Adjusting the CPAP Phase Pressure (PHigh) to Maintain a Low VT [126,127]
5.2. Adjusting TLow to Reduce PaCO2 [128]
5.3. Increasing the CPAP Phase Time (THigh) before the Lung Has Been Adequately Recruited
5.4. Concerns for Safety Using the TCAV Method
- APRV causes barotrauma since ∆P is not controlled. To date, there have been no RCTs showing that APRV causes more barotrauma than other ventilator modes (see Myth #2 [90]).
- APRV can generate large VT as lung compliance improves. There have been no clinical studies using APRV linking an increase in VT to an increase in mortality. Indeed, as lung compliance improves, a larger VT can be used, and ∆P will actually decrease since ∆P = VT/Lung Compliance. It has been shown in multiple studies that a low VT used on a patient with higher lung compliance can actually increase mortality (see Myth #3 [90]).
- APRV will cause hypercapnia. Over the past 25 years, it has been shown that patients on the APRV mode had lower PaCO2 levels than those on conventional mechanical ventilation methods when minute ventilation was matched. This suggests that the volume of CO2 removed with each breath is greater with APRV than with conventional modes (see Myth #5 [90]).
- APRV causes hemodynamic compromise. It is well-known that venous return can be impaired during positive pressure ventilation. There have been no clinical studies indicating that this occurs more with APRV than with any other ventilator mode (see Myth #4 [90]).
- The complexity of the APRV mode limits its widespread use. This suggests that skills necessary to use other modes of ventilation on the ARDS patient are much simpler to grasp, which is not true. Yes, the skill set is different, but it is not inherently more complex and difficult. APRV has been used successfully on tens of thousands of patients for over 30 years and continues to be used to generate a large volume of empirical data (see Myth #1 [90]).
- APRV is an Open Lung Approach (OLA) and, thus, is similar to those OLA using conventional ventilation methods that have not shown reduced mortality. Indeed, the whole point of this proof-of-concept manuscript is that TCAV is not a typical OLA, but rather a novel stabilize and then gradually recruit method, as suggested in the title. We review the current OLA strategies in Section 2.2. Open Lung Approach. Using the TCAV method, the brief expiratory time will very quickly stabilize alveoli, minimizing RACE-induced atelectrauma, a major VILI mechanism. The lung can then be gradually and safely reopened over an extended period of time (hours or days). Lung reopening is accomplished using the inflation and brake ratcheting open of tissue with each breath, combined with an extended CPAP Phase that recruits alveoli with long-opening time constants. Contrast this with a conventional low VT and PEEP method with recruitment maneuvers designed to open the lung in minutes or seconds, which does not afford durable lung recruitment [69]. It is true that the TCAV method will open the lung, but the method and timing are completely different from the methods used with conventional ventilation methods.
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers 2019, 5, 18. [Google Scholar] [CrossRef]
- Slutsky, A.S.; Ranieri, V.M. Ventilator-induced lung injury. N. Engl. J. Med. 2013, 369, 2126–2136. [Google Scholar] [CrossRef] [Green Version]
- Caser, E.B.; Zandonade, E.; Pereira, E.; Gama, A.M.; Barbas, C.S. Impact of distinct definitions of acute lung injury on its incidence and outcomes in Brazilian ICUs: Prospective evaluation of 7133 patients*. Crit. Care Med. 2014, 42, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Laffey, J.G.; Bellani, G.; Pham, T.; Fan, E.; Madotto, F.; Bajwa, E.K.; Brochard, L.; Clarkson, K.; Esteban, A.; Gattinoni, L.; et al. Potentially modifiable factors contributing to outcome from acute respiratory distress syndrome: The LUNG SAFE study. Intensive Care Med. 2016, 42, 1865–1876. [Google Scholar] [CrossRef] [PubMed]
- Fan, E.; Brodie, D.; Slutsky, A.S. Acute Respiratory Distress Syndrome: Advances in Diagnosis and Treatment. JAMA 2018, 319, 698–710. [Google Scholar] [CrossRef]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef]
- Bates, J.H.T.; Smith, B.J. Ventilator-induced lung injury and lung mechanics. Ann. Transl. Med. 2018, 6, 378. [Google Scholar] [CrossRef]
- Cressoni, M.; Chiurazzi, C.; Gotti, M.; Amini, M.; Brioni, M.; Algieri, I.; Cammaroto, A.; Rovati, C.; Massari, D.; di Castiglione, C.B.; et al. Lung inhomogeneities and time course of ventilator-induced mechanical injuries. Anesthesiology 2015, 123, 618–627. [Google Scholar] [CrossRef]
- Cereda, M.; Xin, Y. Alveolar recruitment and lung injury: An issue of timing and location? Crit. Care Med. 2013, 41, 2837–2838. [Google Scholar] [CrossRef]
- Cereda, M.; Xin, Y.; Meeder, N.; Zeng, J.; Jiang, Y.; Hamedani, H.; Profka, H.; Kadlecek, S.; Clapp, J.; Deshpande, C.G.; et al. Visualizing the Propagation of Acute Lung Injury. Anesthesiology 2016, 124, 121–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, K.; Krischer, J.M.; Pfaffenroth, A.; Wilde, S.; Lopez-Rodriguez, E.; Braun, A.; Smith, B.J.; Knudsen, L. Hidden Microatelectases Increase Vulnerability to Ventilation-Induced Lung Injury. Front. Physiol. 2020, 11, 530485. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Pesenti, A. The concept of “baby lung”. Intensive Care Med. 2005, 31, 776–784. [Google Scholar] [CrossRef]
- Marini, J.J.; Gattinoni, L. Time Course of Evolving Ventilator-Induced Lung Injury: The “Shrinking Baby Lung”. Crit. Care Med. 2020, 48, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Writing Group for the Alveolar Recruitment for Acute Respiratory Distress Syndrome Trial (ART) Investigators; Cavalcanti, A.B.; Suzumura, E.A.; Laranjeira, L.N.; Paisani, D.M.; Damiani, L.P.; Guimaraes, H.P.; Romano, E.R.; Regenga, M.M.; Taniguchi, L.N.T.; et al. Effect of Lung Recruitment and Titrated Positive End-Expiratory Pressure (PEEP) vs Low PEEP on Mortality in Patients with Acute Respiratory Distress Syndrome: A Randomized Clinical Trial. JAMA 2017, 318, 1335–1345. [Google Scholar] [CrossRef] [PubMed]
- Brower, R.G.; Lanken, P.N.; MacIntyre, N.; Matthay, M.A.; Morris, A.; Ancukiewicz, M.; Schoenfeld, D.; Thompson, B.T.; The National Heart, Lung, and Blood Institute ARDS Clinical Trials Network. Higher versus lower positive end-expiratory pressures in patients with the acute respiratory distress syndrome. N. Engl. J. Med. 2004, 351, 327–336. [Google Scholar]
- Meade, M.O.; Cook, D.J.; Guyatt, G.H.; Slutsky, A.S.; Arabi, Y.M.; Cooper, D.J.; Davies, A.R.; Hand, L.E.; Zhou, Q.; Thabane, L.; et al. Ventilation strategy using low tidal volumes, recruitment maneuvers, and high positive end-expiratory pressure for acute lung injury and acute respiratory distress syndrome: A randomized controlled trial. JAMA 2008, 299, 637–645. [Google Scholar] [CrossRef] [Green Version]
- Mercat, A.; Richard, J.C.; Vielle, B.; Jaber, S.; Osman, D.; Diehl, J.L.; Lefrant, J.Y.; Prat, G.; Richecoeur, J.; Nieszkowska, A.; et al. Positive end-expiratory pressure setting in adults with acute lung injury and acute respiratory distress syndrome: A randomized controlled trial. JAMA 2008, 299, 646–655. [Google Scholar] [CrossRef] [Green Version]
- Nieman, G.F.; Andrews, P.; Satalin, J.; Wilcox, K.; Kollisch-Singule, M.; Madden, M.; Aiash, H.; Blair, S.J.; Gatto, L.A.; Habashi, N.M. Acute lung injury: How to stabilize a broken lung. Crit. Care 2018, 22, 136. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.C.; Townsley, M.I. Evaluation of lung injury in rats and mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L231–L246. [Google Scholar] [CrossRef]
- Taeusch, H.W.; Bernardino de la Serna, J.; Perez-Gil, J.; Alonso, C.; Zasadzinski, J.A. Inactivation of pulmonary surfactant due to serum-inhibited adsorption and reversal by hydrophilic polymers: Experimental. Biophys. J. 2005, 89, 1769–1779. [Google Scholar] [CrossRef] [Green Version]
- Albert, R.K. The role of ventilation-induced surfactant dysfunction and atelectasis in causing acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2012, 185, 702–708. [Google Scholar] [CrossRef]
- Nieman, G.F.; Bredenberg, C.E. High surface tension pulmonary edema induced by detergent aerosol. J. Appl. Physiol. 1985, 58, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Perlman, C.E.; Lederer, D.J.; Bhattacharya, J. Micromechanics of alveolar edema. Am. J. Respir. Cell. Mol. Biol. 2011, 44, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.L.; Chen, Y.Z.; Hu, Z.Y. A micromechanical model for estimating alveolar wall strain in mechanically ventilated edematous lungs. J. Appl. Physiol. 2014, 117, 586–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makiyama, A.M.; Gibson, L.J.; Harris, R.S.; Venegas, J.G. Stress concentration around an atelectatic region: A finite element model. Respir. Physiol. Neurobiol. 2014, 201, 101–110. [Google Scholar] [CrossRef]
- Retamal, J.; Bergamini, B.C.; Carvalho, A.R.; Bozza, F.A.; Borzone, G.; Borges, J.B.; Larsson, A.; Hedenstierna, G.; Bugedo, G.; Bruhn, A. Non-lobar atelectasis generates inflammation and structural alveolar injury in the surrounding healthy tissue during mechanical ventilation. Crit. Care 2014, 18, 505. [Google Scholar] [CrossRef]
- Acute Respiratory Distress Syndrome Network; Brower, R.G.; Matthay, M.A.; Morris, A.; Schoenfeld, D.; Thompson, B.T.; Wheeler, A. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1301–1308. [Google Scholar] [CrossRef] [Green Version]
- Gattinoni, L.; Pesenti, A.; Avalli, L.; Rossi, F.; Bombino, M. Pressure-volume curve of total respiratory system in acute respiratory failure. Computed tomographic scan study. Am. Rev. Respir. Dis. 1987, 136, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Deans, K.J.; Minneci, P.C.; Cui, X.; Banks, S.M.; Natanson, C.; Eichacker, P.Q. Mechanical ventilation in ARDS: One size does not fit all. Crit. Care Med. 2005, 33, 1141–1143. [Google Scholar] [CrossRef]
- Nieman, G.F.; Gatto, L.A.; Andrews, P.; Satalin, J.; Camporota, L.; Daxon, B.; Blair, S.J.; Al-Khalisy, H.; Madden, M.; Kollisch-Singule, M.; et al. Prevention and treatment of acute lung injury with time-controlled adaptive ventilation: Physiologically informed modification of airway pressure release ventilation. Ann. Intensive Care 2020, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Amato, M.B.; Meade, M.O.; Slutsky, A.S.; Brochard, L.; Costa, E.L.; Schoenfeld, D.A.; Stewart, T.E.; Briel, M.; Talmor, D.; Mercat, A.; et al. Driving pressure and survival in the acute respiratory distress syndrome. N. Engl. J. Med. 2015, 372, 747–755. [Google Scholar] [CrossRef] [Green Version]
- Yehya, N.; Hodgson, C.L.; Amato, M.B.P.; Richard, J.C.; Brochard, L.J.; Mercat, A.; Goligher, E.C. Response to Ventilator Adjustments for Predicting Acute Respiratory Distress Syndrome Mortality. Driving Pressure versus Oxygenation. Ann. Am. Thorac. Soc. 2021, 18, 857–864. [Google Scholar] [CrossRef]
- Costa, E.L.V.; Slutsky, A.; Brochard, L.J.; Brower, R.; Serpa-Neto, A.; Cavalcanti, A.B.; Mercat, A.; Meade, M.; Morais, C.C.A.; Goligher, E.; et al. Ventilatory Variables and Mechanical Power in Patients with Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2021, 204, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, H.; Pettenuzzo, T.; Aoyama, K.; Pinto, R.; Englesakis, M.; Fan, E. Association of Driving Pressure with Mortality Among Ventilated Patients with Acute Respiratory Distress Syndrome: A Systematic Review and Meta-Analysis. Crit. Care Med. 2018, 46, 300–306. [Google Scholar] [CrossRef]
- Chen, Z.; Wei, X.; Liu, G.; Tai, Q.; Zheng, D.; Xie, W.; Chen, L.; Wang, G.; Sun, J.Q.; Wang, S.; et al. Higher vs. Lower DP for Ventilated Patients with Acute Respiratory Distress Syndrome: A Systematic Review and Meta-Analysis. Emerg. Med. Int. 2019, 2019, 4654705. [Google Scholar] [CrossRef]
- Goligher, E.C.; Costa, E.L.V.; Yarnell, C.J.; Brochard, L.J.; Stewart, T.E.; Tomlinson, G.; Brower, R.G.; Slutsky, A.S.; Amato, M.P.B. Effect of Lowering Tidal Volume on Mortality in ARDS Varies with Respiratory System Elastance. Am. J. Respir. Crit. Care Med. 2021, 203, 1378–1385. [Google Scholar] [CrossRef]
- Protti, A.; Andreis, D.T.; Monti, M.; Santini, A.; Sparacino, C.C.; Langer, T.; Votta, E.; Gatti, S.; Lombardi, L.; Leopardi, O.; et al. Lung stress and strain during mechanical ventilation: Any difference between statics and dynamics? Crit. Care Med. 2013, 41, 1046–1055. [Google Scholar] [CrossRef] [Green Version]
- Protti, A.; Andreis, D.T.; Iapichino, G.E.; Monti, M.; Comini, B.; Milesi, M.; Zani, L.; Gatti, S.; Lombardi, L.; Gattinoni, L. High positive end-expiratory pressure: Only a dam against oedema formation? Crit. Care 2013, 17, R131. [Google Scholar] [CrossRef] [Green Version]
- Protti, A.; Maraffi, T.; Milesi, M.; Votta, E.; Santini, A.; Pugni, P.; Andreis, D.T.; Nicosia, F.; Zannin, E.; Gatti, S.; et al. Role of Strain Rate in the Pathogenesis of Ventilator-Induced Lung Edema. Crit. Care Med. 2016, 44, e838–e845. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.V.; Kollisch-Singule, M.; Satalin, J.; Searles, Q.; Dombert, L.; Abdel-Razek, O.; Yepuri, N.; Leonard, A.; Gruessner, A.; Andrews, P.; et al. The role of high airway pressure and dynamic strain on ventilator-induced lung injury in a heterogeneous acute lung injury model. Intensive Care Med. Exp. 2017, 5, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seah, A.S.; Grant, K.A.; Aliyeva, M.; Allen, G.B.; Bates, J.H.T. Quantifying the roles of tidal volume and PEEP in the pathogenesis of ventilator-induced lung injury. Ann. Biomed. Eng. 2011, 39, 1505–1516. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; Patel, B.V.; Takata, M. Ventilation with “clinically relevant” high tidal volumes does not promote stretch-induced injury in the lungs of healthy mice. Crit. Care Med. 2012, 40, 2850–2857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syring, R.S.; Otto, C.M.; Spivack, R.E.; Markstaller, K.; Baumgardner, J.E. Maintenance of end-expiratory recruitment with increased respiratory rate after saline-lavage lung injury. J. Appl. Physiol. 2007, 102, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Hamlington, K.L.; Smith, B.J.; Dunn, C.M.; Charlebois, C.M.; Roy, G.S.; Bates, J.H.T. Linking lung function to structural damage of alveolar epithelium in ventilator-induced lung injury. Respir. Physiol. Neurobiol. 2018, 255, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Otto, C.M.; Markstaller, K.; Kajikawa, O.; Karmrodt, J.; Syring, R.S.; Pfeiffer, B.; Good, V.P.; Frevert, C.W.; Baumgardner, J.E. Spatial and temporal heterogeneity of ventilator-associated lung injury after surfactant depletion. J. Appl. Physiol. 2008, 104, 1485–1494. [Google Scholar] [CrossRef]
- Schiller, H.J.; McCann, U.G., 2nd; Carney, D.E.; Gatto, L.A.; Steinberg, J.M.; Nieman, G.F. Altered alveolar mechanics in the acutely injured lung. Crit. Care Med. 2001, 29, 1049–1055. [Google Scholar] [CrossRef]
- Cereda, M.; Emami, K.; Kadlecek, S.; Xin, Y.; Mongkolwisetwara, P.; Profka, H.; Barulic, A.; Pickup, S.; Mansson, S.; Wollmer, P.; et al. Quantitative imaging of alveolar recruitment with hyperpolarized gas MRI during mechanical ventilation. J. Appl. Physiol. 2011, 110, 499–511. [Google Scholar] [CrossRef] [Green Version]
- Gaver, D.P., 3rd; Nieman, G.F.; Gatto, L.A.; Cereda, M.; Habashi, N.M.; Bates, J.H.T. The POOR get POORer: A hypothesis for the pathogenesis of ventilator-induced lung injury. Am. J. Respir. Crit. Care Med. 2020, 202, 1081–1087. [Google Scholar] [CrossRef]
- Cereda, M.; Xin, Y.; Goffi, A.; Herrmann, J.; Kaczka, D.W.; Kavanagh, B.P.; Perchiazzi, G.; Yoshida, T.; Rizi, R.R. Imaging the Injured Lung: Mechanisms of Action and Clinical Use. Anesthesiology 2019, 131, 716–749. [Google Scholar] [CrossRef]
- Cabrera-Benitez, N.E.; Laffey, J.G.; Parotto, M.; Spieth, P.M.; Villar, J.; Zhang, H.; Slutsky, A.S. Mechanical ventilation-associated lung fibrosis in acute respiratory distress syndrome: A significant contributor to poor outcome. Anesthesiology 2014, 121, 189–198. [Google Scholar] [CrossRef]
- Burkhardt, A. Alveolitis and collapse in the pathogenesis of pulmonary fibrosis. Am. Rev. Respir. Dis. 1989, 140, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, L.; Ruppert, C.; Ochs, M. Tissue remodelling in pulmonary fibrosis. Cell. Tissue Res. 2017, 367, 607–626. [Google Scholar] [CrossRef] [PubMed]
- Lutz, D.; Gazdhar, A.; Lopez-Rodriguez, E.; Ruppert, C.; Mahavadi, P.; Gunther, A.; Klepetko, W.; Bates, J.H.; Smith, B.; Geiser, T.; et al. Alveolar derecruitment and collapse induration as crucial mechanisms in lung injury and fibrosis. Am. J. Respir. Cell. Mol. Biol. 2015, 52, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Duggan, M.; Kavanagh, B.P. Pulmonary atelectasis: A pathogenic perioperative entity. Anesthesiology 2005, 102, 838–854. [Google Scholar] [CrossRef]
- Spinelli, E.; Mauri, T.; Beitler, J.R.; Pesenti, A.; Brodie, D. Respiratory drive in the acute respiratory distress syndrome: Pathophysiology, monitoring, and therapeutic interventions. Intensive Care Med. 2020, 46, 606–618. [Google Scholar] [CrossRef]
- Petersson, J.; Glenny, R.W. Gas exchange and ventilation-perfusion relationships in the lung. Eur. Respir. J. 2014, 44, 1023–1041. [Google Scholar] [CrossRef] [Green Version]
- Andrews, P.L.; Sadowitz, B.; Kollisch-Singule, M.; Satalin, J.; Roy, S.; Snyder, K.; Gatto, L.A.; Nieman, G.F.; Habashi, N.M. Alveolar instability (atelectrauma) is not identified by arterial oxygenation predisposing the development of an occult ventilator-induced lung injury. Intensive Care Med. Exp. 2015, 3, 54. [Google Scholar] [CrossRef] [Green Version]
- Pavone, L.; Albert, S.; DiRocco, J.; Gatto, L.; Nieman, G. Alveolar instability caused by mechanical ventilation initially damages the nondependent normal lung. Crit. Care 2007, 11, R104. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.M.; Viale, J.P.; Hamilton, R.M.; McPeak, H.; Sutton, L.; Hahn, C.E. Within-breath arterial PO2 oscillations in an experimental model of acute respiratory distress syndrome. Br. J. Anaesth. 2000, 85, 456–459. [Google Scholar] [CrossRef] [Green Version]
- Duggan, M.; McCaul, C.L.; McNamara, P.J.; Engelberts, D.; Ackerley, C.; Kavanagh, B.P. Atelectasis causes vascular leak and lethal right ventricular failure in uninjured rat lungs. Am. J. Respir. Crit. Care Med. 2003, 167, 1633–1640. [Google Scholar] [CrossRef]
- Sipmann, F.S.; Santos, A.; Tusman, G. Heart-lung interactions in acute respiratory distress syndrome: Pathophysiology, detection and management strategies. Ann. Transl. Med. 2018, 6, 27. [Google Scholar] [CrossRef]
- Pearse, D.B.; Searcy, R.M.; Mitzner, W.; Permutt, S.; Sylvester, J.T. Effects of tidal volume and respiratory frequency on lung lymph flow. J. Appl. Physiol. 2005, 99, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, A.; Arold, S.P.; Bartolak-Suki, E.; Parameswaran, H.; Suki, B. Jamming dynamics of stretch-induced surfactant release by alveolar type II cells. J. Appl. Physiol. 2012, 112, 824–831. [Google Scholar] [CrossRef] [Green Version]
- Nieman, G.; Kollisch-Singule, M.; Ramcharran, H.; Satalin, J.; Blair, S.; Gatto, L.A.; Andrews, P.; Ghosh, A.; Kaczka, D.W.; Gaver, D.; et al. Unshrinking the baby lung to calm the VILI vortex. Crit. Care 2022, 26, 242. [Google Scholar] [CrossRef]
- Ferguson, N.D.; Cook, D.J.; Guyatt, G.H.; Mehta, S.; Hand, L.; Austin, P.; Zhou, Q.; Matte, A.; Walter, S.D.; Lamontagne, F.; et al. High-frequency oscillation in early acute respiratory distress syndrome. N. Engl. J. Med. 2013, 368, 795–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, D.; Lamb, S.E.; Shah, S.; MacKenzie, I.; Tunnicliffe, W.; Lall, R.; Rowan, K.; Cuthbertson, B.H.; Group, O.S. High-frequency oscillation for acute respiratory distress syndrome. N. Engl. J. Med. 2013, 368, 806–813. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, A.; Drazen, J.M. High-frequency oscillatory ventilation on shaky ground. N. Engl. J. Med. 2013, 368, 863–865. [Google Scholar] [CrossRef] [Green Version]
- van der Zee, P.; Gommers, D. Recruitment Maneuvers and Higher PEEP, the So-Called Open Lung Concept, in Patients with ARDS. Crit. Care 2019, 23, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasso, S.; Fanelli, V.; Cafarelli, A.; Anaclerio, R.; Amabile, M.; Ancona, G.; Fiore, T. Effects of high versus low positive end-expiratory pressures in acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2005, 171, 1002–1008. [Google Scholar] [CrossRef]
- Angriman, F.; Ferreyro, B.L.; Donaldson, L.; Cuthbertson, B.H.; Ferguson, N.D.; Bollen, C.W.; Bachman, T.E.; Lamontagne, F.; Adhikari, N.K.J. The harm of high-frequency oscillatory ventilation (HFOV) in ARDS is not related to a high baseline risk of acute cor pulmonale or short-term changes in hemodynamics. Intensive Care Med. 2020, 46, 132–134. [Google Scholar] [CrossRef]
- Kaczka, D.W. Oscillatory ventilation redux: Alternative perspectives on ventilator-induced lung injury in the acute respiratory distress syndrome. Curr. Opin. Physiol. 2021, 21, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Villar, J.; Blanco, J.; Anon, J.M.; Santos-Bouza, A.; Blanch, L.; Ambros, A.; Gandia, F.; Carriedo, D.; Mosteiro, F.; Basaldua, S.; et al. The ALIEN study: Incidence and outcome of acute respiratory distress syndrome in the era of lung protective ventilation. Intensive Care Med. 2011, 37, 1932–1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Cai, G.; Gong, S.; Dong, L.; Yan, J.; Cai, W. Interaction between low tidal volume ventilation strategy and severity of acute respiratory distress syndrome: A retrospective cohort study. Crit. Care 2019, 23, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parcha, V.; Kalra, R.; Bhatt, S.P.; Berra, L.; Arora, G.; Arora, P. Trends and Geographic Variation in Acute Respiratory Failure and ARDS Mortality in the United States. Chest 2021, 159, 1460–1472. [Google Scholar] [CrossRef] [PubMed]
- Raschke, R.A.; Stoffer, B.; Assar, S.; Fountain, S.; Olsen, K.; Heise, C.W.; Gallo, T.; Padilla-Jones, A.; Gerkin, R.; Parthasarathy, S.; et al. The relationship of tidal volume and driving pressure with mortality in hypoxic patients receiving mechanical ventilation. PLoS ONE 2021, 16, e0255812. [Google Scholar] [CrossRef]
- Sahetya, S.K.; Brower, R.G. Lung Recruitment and Titrated PEEP in Moderate to Severe ARDS: Is the Door Closing on the Open Lung? JAMA 2017, 318, 1327–1329. [Google Scholar] [CrossRef]
- Cereda, M.; Xin, Y.; Emami, K.; Huang, J.; Rajaei, J.; Profka, H.; Han, B.; Mongkolwisetwara, P.; Kadlecek, S.; Kuzma, N.N.; et al. Positive end-expiratory pressure increments during anesthesia in normal lung result in hysteresis and greater numbers of smaller aerated airspaces. Anesthesiology 2013, 119, 1402–1409. [Google Scholar] [CrossRef] [Green Version]
- Dianti, J.; Tisminetzky, M.; Ferreyro, B.L.; Englesakis, M.; Del Sorbo, L.; Sud, S.; Talmor, D.; Ball, L.; Meade, M.; Hodgson, C.; et al. Association of Positive End-Expiratory Pressure and Lung Recruitment Selection Strategies with Mortality in Acute Respiratory Distress Syndrome: A Systematic Review and Network Meta-analysis. Am. J. Respir. Crit. Care Med. 2022, 205, 1300–1310. [Google Scholar] [CrossRef]
- Habashi, N.M. Other approaches to open-lung ventilation: Airway pressure release ventilation. Crit. Care Med. 2005, 33, S228–S240. [Google Scholar] [CrossRef]
- Andrews, P.L.; Shiber, J.R.; Jaruga-Killeen, E.; Roy, S.; Sadowitz, B.; O’Toole, R.V.; Gatto, L.A.; Nieman, G.F.; Scalea, T.; Habashi, N.M. Early application of airway pressure release ventilation may reduce mortality in high-risk trauma patients: A systematic review of observational trauma ARDS literature. J. Trauma. Acute Care Surg. 2013, 75, 635–641. [Google Scholar] [CrossRef] [Green Version]
- Albert, S.P.; DiRocco, J.; Allen, G.B.; Bates, J.H.; Lafollette, R.; Kubiak, B.D.; Fischer, J.; Maroney, S.; Nieman, G.F. The role of time and pressure on alveolar recruitment. J. Appl. Physiol. 2009, 106, 757–765. [Google Scholar] [CrossRef] [Green Version]
- Emr, B.; Gatto, L.A.; Roy, S.; Satalin, J.; Ghosh, A.; Snyder, K.; Andrews, P.; Habashi, N.; Marx, W.; Ge, L.; et al. Airway pressure release ventilation prevents ventilator-induced lung injury in normal lungs. JAMA Surg. 2013, 148, 1005–1012. [Google Scholar] [CrossRef] [Green Version]
- Kollisch-Singule, M.; Emr, B.; Smith, B.; Roy, S.; Jain, S.; Satalin, J.; Snyder, K.; Andrews, P.; Habashi, N.; Bates, J.; et al. Mechanical breath profile of airway pressure release ventilation: The effect on alveolar recruitment and microstrain in acute lung injury. JAMA Surg. 2014, 149, 1138–1145. [Google Scholar] [CrossRef] [Green Version]
- Kollisch-Singule, M.; Emr, B.; Smith, B.; Ruiz, C.; Roy, S.; Meng, Q.; Jain, S.; Satalin, J.; Snyder, K.; Ghosh, A.; et al. Airway pressure release ventilation reduces conducting airway micro-strain in lung injury. J. Am. Coll. Surg. 2014, 219, 968–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollisch-Singule, M.; Jain, S.; Andrews, P.; Smith, B.J.; Hamlington-Smith, K.L.; Roy, S.; DiStefano, D.; Nuss, E.; Satalin, J.; Meng, Q.; et al. Effect of Airway Pressure Release Ventilation on Dynamic Alveolar Heterogeneity. JAMA Surg. 2015, 151, 64–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollisch-Singule, M.; Emr, B.; Jain, S.V.; Andrews, P.; Satalin, J.; Liu, J.; Porcellio, E.; Kenyon, V.; Wang, G.; Marx, W.; et al. The effects of airway pressure release ventilation on respiratory mechanics in extrapulmonary lung injury. Intensive Care Med. Exp. 2015, 3, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, B.J.; Lundblad, L.K.; Kollisch-Singule, M.; Satalin, J.; Nieman, G.; Habashi, N.; Bates, J.H. Predicting the response of the injured lung to the mechanical breath profile. J. Appl. Physiol. 2015, 118, 932–940. [Google Scholar] [CrossRef] [Green Version]
- Kollisch-Singule, M.; Jain, S.V.; Satalin, J.; Andrews, P.; Searles, Q.; Liu, Z.; Zhou, Y.; Wang, G.; Meier, A.H.; Gatto, L.A.; et al. Limiting ventilator-associated lung injury in a preterm porcine neonatal model. J. Pediatr. Surg. 2017, 52, 50–55. [Google Scholar] [CrossRef]
- Jain, S.V.; Kollisch-Singule, M.; Sadowitz, B.; Dombert, L.; Satalin, J.; Andrews, P.; Gatto, L.A.; Nieman, G.F.; Habashi, N.M. The 30-year evolution of airway pressure release ventilation (APRV). Intensive Care Med. Exp. 2016, 4, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, P.; Shiber, J.; Madden, M.; Nieman, G.F.; Camporota, L.; Habashi, N.M. Myths and Misconceptions of Airway Pressure Release Ventilation: Getting Past the Noise and on to the Signal. Front. Physiol. 2022, 13, 928562. [Google Scholar] [CrossRef]
- Boehme, S.; Bentley, A.H.; Hartmann, E.K.; Chang, S.; Erdoes, G.; Prinzing, A.; Hagmann, M.; Baumgardner, J.E.; Ullrich, R.; Markstaller, K.; et al. Influence of inspiration to expiration ratio on cyclic recruitment and derecruitment of atelectasis in a saline lavage model of acute respiratory distress syndrome. Crit. Care Med. 2015, 43, e65–e74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, W.R.; Dominelli, P.B.; Molgat-Seon, Y.; Lipson, R.; Griesdale, D.E.; Sekhon, M.; Ayas, N.; Sheel, A.W. Effect of tidal volume and positive end-expiratory pressure on expiratory time constants in experimental lung injury. Physiol. Rep. 2016, 4, e12737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, P.; Berglund, J.E.; Fernandez Mondejar, E.; Magnusson, A.; Hedenstierna, G. Dynamics of lung collapse and recruitment during prolonged breathing in porcine lung injury. J. Appl. Physiol. 1998, 85, 1533–1543. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.; Gerard, S.E.; Shao, W.; Xin, Y.; Cereda, M.; Reinhardt, J.M.; Christensen, G.E.; Hoffman, E.A.; Kaczka, D.W. Effects of Lung Injury on Regional Aeration and Expiratory Time Constants: Insights From Four-Dimensional Computed Tomography Image Registration. Front. Physiol. 2021, 12, 707119. [Google Scholar] [CrossRef]
- Bates, J.H.; Irvin, C.G. Time dependence of recruitment and derecruitment in the lung: A theoretical model. J. Appl. Physiol. 2002, 93, 705–713. [Google Scholar] [CrossRef] [Green Version]
- Markstaller, K.; Eberle, B.; Kauczor, H.U.; Scholz, A.; Bink, A.; Thelen, M.; Heinrichs, W.; Weiler, N. Temporal dynamics of lung aeration determined by dynamic CT in a porcine model of ARDS. Br. J. Anaesth. 2001, 87, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Lachmann, B. Open up the lung and keep the lung open. Intensive Care Med. 1992, 18, 319–321. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.K.; Emr, B.; Sadowitz, B.; Gatto, L.A.; Ghosh, A.; Satalin, J.M.; Snyder, K.P.; Ge, L.; Wang, G.; Marx, W.; et al. Preemptive application of airway pressure release ventilation prevents development of acute respiratory distress syndrome in a rat traumatic hemorrhagic shock model. Shock 2013, 40, 210–216. [Google Scholar] [CrossRef] [Green Version]
- Nieman, G.F.; Satalin, J.; Kollisch-Singule, M.; Andrews, P.; Aiash, H.; Habashi, N.M.; Gatto, L.A. Physiology in Medicine: Understanding dynamic alveolar physiology to minimize ventilator-induced lung injury. J. Appl. Physiol. 2017, 122, 1516–1522. [Google Scholar] [CrossRef]
- Mead, J.; Takishima, T.; Leith, D. Stress distribution in lungs: A model of pulmonary elasticity. J. Appl. Physiol. 1970, 28, 596–608. [Google Scholar] [CrossRef]
- Suki, B.; Stamenovic, D.; Hubmayr, R. Lung parenchymal mechanics. Compr. Physiol. 2011, 1, 1317–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paré, P.D.; Mitzner, W. Airway-parenchymal interdependence. Compr. Physiol. 2012, 2, 1921–1935. [Google Scholar]
- Ryans, J.M.; Fujioka, H.; Gaver, D.P., 3rd. Micro-scale to Meso-scale Analysis of Parenchymal Tethering: The Effect of Heterogeneous Alveolar Pressures on the Pulmonary Mechanics of Compliant Airways. J. Appl. Physiol. 2019, 126, 1204–1213. [Google Scholar] [CrossRef]
- Fujioka, H.; Halpern, D.; Gaver, D.P., 3rd. A model of surfactant-induced surface tension effects on the parenchymal tethering of pulmonary airways. J. Biomech. 2013, 46, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Kollisch-Singule, M.C.; Jain, S.V.; Andrews, P.L.; Satalin, J.; Gatto, L.A.; Villar, J.; De Backer, D.; Gattinoni, L.; Nieman, G.F.; Habashi, N.M. Looking beyond macroventilatory parameters and rethinking ventilator-induced lung injury. J. Appl. Physiol. (1985) 2018, 124, 1214–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, R.; Widdicombe, J.G. Stress relaxation of the human lung. Clin. Sci. 1960, 20, 19–31. [Google Scholar]
- Suki, B.; Barabasi, A.L.; Lutchen, K.R. Lung tissue viscoelasticity: A mathematical framework and its molecular basis. J. Appl. Physiol. 1994, 76, 2749–2759. [Google Scholar] [CrossRef]
- Coughlin, M.F.; Suki, B.; Stamenovic, D. Dynamic behavior of lung parenchyma in shear. J. Appl. Physiol. 1996, 80, 1880–1890. [Google Scholar] [CrossRef]
- Sugihara, T.; Hildebrandt, J.; Martin, C.J. Viscoelastic properties of alveolar wall. J. Appl. Physiol. 1972, 33, 93–98. [Google Scholar] [CrossRef]
- Bates, J.H.T.; Gaver, D.P.; Habashi, N.M.; Nieman, G.F. Atelectrauma Versus Volutrauma: A Tale of Two Time-Constants. Crit. Care Explor. 2020, 2, e0299. [Google Scholar] [CrossRef]
- Parhar, K.K.S.; Doig, C. Caution-Do Not Attempt This at Home. Airway Pressure Release Ventilation Should Not Routinely Be Used in Patients with or at Risk of Acute Respiratory Distress Syndrome Outside of a Clinical Trial. Crit. Care Med. 2023. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Carsetti, A.; Damiani, E.; Domizi, R.; Scorcella, C.; Pantanetti, S.; Falcetta, S.; Donati, A.; Adrario, E. Airway pressure release ventilation during acute hypoxemic respiratory failure: A systematic review and meta-analysis of randomized controlled trials. Ann. Intensive Care 2019, 9, 44. [Google Scholar] [CrossRef]
- Chen, C.; Zhen, J.; Gong, S.; Yan, J.; Li, L. Efficacy of airway pressure release ventilation for acute respiratory distress syndrome: A systematic review with meta-analysis. Ann. Palliat. Med. 2021, 10, 10349–10359. [Google Scholar] [CrossRef]
- Lim, J.; Litton, E. Airway Pressure Release Ventilation in Adult Patients with Acute Hypoxemic Respiratory Failure: A Systematic Review and Meta-Analysis. Crit. Care Med. 2019, 47, 1794–1799. [Google Scholar] [CrossRef] [PubMed]
- Othman, F.; Alsagami, N.; Alharbi, R.; Almuammer, Y.; Alshahrani, S.; Ismaeil, T. The efficacy of airway pressure release ventilation in acute respiratory distress syndrome adult patients: A meta-analysis of clinical trials. Ann. Thorac. Med. 2021, 16, 245–252. [Google Scholar] [CrossRef]
- Roshdy, A.; Elsayed, A.S.; Saleh, A.S. Airway Pressure Release Ventilation for Acute Respiratory Failure Due to Coronavirus Disease 2019: A Systematic Review and Meta-Analysis. J. Intensive Care Med. 2023, 38, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, Y.; Li, N.; You, D.; Zhao, Y. The safety and efficacy of airway pressure release ventilation in acute respiratory distress syndrome patients: A PRISMA-compliant systematic review and meta-analysis. Medicine 2020, 99, e18586. [Google Scholar] [CrossRef]
- Zhong, X.; Wu, Q.; Yang, H.; Dong, W.; Wang, B.; Zhang, Z.; Liang, G. Airway pressure release ventilation versus low tidal volume ventilation for patients with acute respiratory distress syndrome/acute lung injury: A meta-analysis of randomized clinical trials. Ann. Transl. Med. 2020, 8, 1641. [Google Scholar] [CrossRef]
- Nieman, G.F.; Satalin, J.; Andrews, P.; Aiash, H.; Habashi, N.M.; Gatto, L.A. Personalizing mechanical ventilation according to physiologic parameters to stabilize alveoli and minimize ventilator induced lung injury (VILI). Intensive Care Med. Exp. 2017, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Nieman, G.F.; Gatto, L.A.; Habashi, N.M. Impact of mechanical ventilation on the pathophysiology of progressive acute lung injury. J. Appl. Physiol. 2015, 119, 1245–1261. [Google Scholar] [CrossRef] [Green Version]
- Silva, P.L.; Cruz, F.F.; Samary, C.D.S.; Moraes, L.; de Magalhaes, R.F.; Fernandes, M.V.S.; Bose, R.; Pelegati, V.B.; Carvalho, H.F.; Capelozzi, V.L.; et al. Biological Response to Time-Controlled Adaptive Ventilation Depends on Acute Respiratory Distress Syndrome Etiology. Crit. Care Med. 2018, 46, e609–e617. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Sadowitz, B.; Andrews, P.; Gatto, L.A.; Marx, W.; Ge, L.; Wang, G.; Lin, X.; Dean, D.A.; Kuhn, M.; et al. Early stabilizing alveolar ventilation prevents acute respiratory distress syndrome: A novel timing-based ventilatory intervention to avert lung injury. J. Trauma Acute Care Surg. 2012, 73, 391–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.; Habashi, N.; Sadowitz, B.; Andrews, P.; Ge, L.; Wang, G.; Roy, P.; Ghosh, A.; Kuhn, M.; Satalin, J.; et al. Early airway pressure release ventilation prevents ARDS-a novel preventive approach to lung injury. Shock 2013, 39, 28–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieman, G.F.; Al-Khalisy, H.; Kollisch-Singule, M.; Satalin, J.; Blair, S.; Trikha, G.; Andrews, P.; Madden, M.; Gatto, L.A.; Habashi, N.M. A Physiologically Informed Strategy to Effectively Open, Stabilize, and Protect the Acutely Injured Lung. Front. Physiol. 2020, 11, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollisch-Singule, M.; Satalin, J.; Blair, S.J.; Andrews, P.L.; Gatto, L.A.; Nieman, G.F.; Habashi, N.M. Mechanical Ventilation Lessons Learned From Alveolar Micromechanics. Front. Physiol. 2020, 11, 233. [Google Scholar] [CrossRef] [Green Version]
- Hirshberg, E.L.; Lanspa, M.J.; Peterson, J.; Carpenter, L.; Wilson, E.L.; Brown, S.M.; Dean, N.C.; Orme, J.; Grissom, C.K. Randomized Feasibility Trial of a Low Tidal Volume-Airway Pressure Release Ventilation Protocol Compared with Traditional Airway Pressure Release Ventilation and Volume Control Ventilation Protocols. Crit. Care Med. 2018, 46, 1943–1952. [Google Scholar] [CrossRef]
- Lalgudi Ganesan, S.; Jayashree, M.; Chandra Singhi, S.; Bansal, A. Airway Pressure Release Ventilation in Pediatric Acute Respiratory Distress Syndrome. A Randomized Controlled Trial. Am. J. Respir. Crit. Care Med. 2018, 198, 1199–1207. [Google Scholar] [CrossRef]
- Ibarra-Estrada, M.A.; Garcia-Salas, Y.; Mireles-Cabodevila, E.; Lopez-Pulgarin, J.A.; Chavez-Pena, Q.; Garcia-Salcido, R.; Mijangos-Mendez, J.C.; Aguirre-Avalos, G. Use of Airway Pressure Release Ventilation in Patients with Acute Respiratory Failure Due to COVID-19: Results of a Single-Center Randomized Controlled Trial. Crit. Care Med. 2022, 50, 586–594. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Problem 1: Reducing VT in patients with higher compliance can increase mortality [29]. |
| Problem 2: Using LVT and airway pressure results in atelectasis-induced loss of lung volume, resulting in more stress and strain applied to the remaining normal tissue during ventilation (‘VILI Vortex’) [13]. |
| Problem 3: Areas of atelectasis that remain collapsed, known as collapse induration, can become permanently dysfunctional and fibrotic [50,51,52,53]. |
| Problem 4: Atelectasis shrinks perfusion surface area, causing hypercapnia and hypoxemia resulting in higher FiO2 requirements, which in turn can cause oxygen toxicity and adsorption atelectasis, exacerbating all problems associated with lung collapse. Also, atelectasis is independently associated with loss of surfactant function and carries an increased risk of pneumonia [21,54]. |
| Problem 5: Hypoxemia, hypercapnia, and stretch receptors in the atelectatic tissue cause dyspnea, resulting in patient–ventilator asynchrony [55,56]. |
| Problem 6: To prevent repetitive alveolar collapse and expansion (RACE)-induced atelectrauma, the LVT approach uses changes in oxygenation to guide the settings and adjustments of PEEP. However, oxygenation is a very poor indicator of alveolar instability [57,58,59]. |
| Problem 7: End-expiratory lung volume (EELV) decreases as atelectasis increases, which increases the pulmonary vascular resistance (PVR) [60]; this can lead to right heart failure requiring vasoactive therapy [61]. |
| Problem 8: Low VT and respiratory rate decrease lung lymph, which could exacerbate pulmonary edema accumulation [62]. |
| Problem 9: Lung stretch during ventilation is needed to stimulate exogenous surfactant release [63]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nieman, G.F.; Kaczka, D.W.; Andrews, P.L.; Ghosh, A.; Al-Khalisy, H.; Camporota, L.; Satalin, J.; Herrmann, J.; Habashi, N.M. First Stabilize and then Gradually Recruit: A Paradigm Shift in Protective Mechanical Ventilation for Acute Lung Injury. J. Clin. Med. 2023, 12, 4633. https://doi.org/10.3390/jcm12144633
Nieman GF, Kaczka DW, Andrews PL, Ghosh A, Al-Khalisy H, Camporota L, Satalin J, Herrmann J, Habashi NM. First Stabilize and then Gradually Recruit: A Paradigm Shift in Protective Mechanical Ventilation for Acute Lung Injury. Journal of Clinical Medicine. 2023; 12(14):4633. https://doi.org/10.3390/jcm12144633
Chicago/Turabian StyleNieman, Gary F., David W. Kaczka, Penny L. Andrews, Auyon Ghosh, Hassan Al-Khalisy, Luigi Camporota, Joshua Satalin, Jacob Herrmann, and Nader M. Habashi. 2023. "First Stabilize and then Gradually Recruit: A Paradigm Shift in Protective Mechanical Ventilation for Acute Lung Injury" Journal of Clinical Medicine 12, no. 14: 4633. https://doi.org/10.3390/jcm12144633
APA StyleNieman, G. F., Kaczka, D. W., Andrews, P. L., Ghosh, A., Al-Khalisy, H., Camporota, L., Satalin, J., Herrmann, J., & Habashi, N. M. (2023). First Stabilize and then Gradually Recruit: A Paradigm Shift in Protective Mechanical Ventilation for Acute Lung Injury. Journal of Clinical Medicine, 12(14), 4633. https://doi.org/10.3390/jcm12144633