
Mitochondria: Emerging Consequential in Sickle Cell Disease

 , , , and
, , , and 
Abstract
:1. Introduction
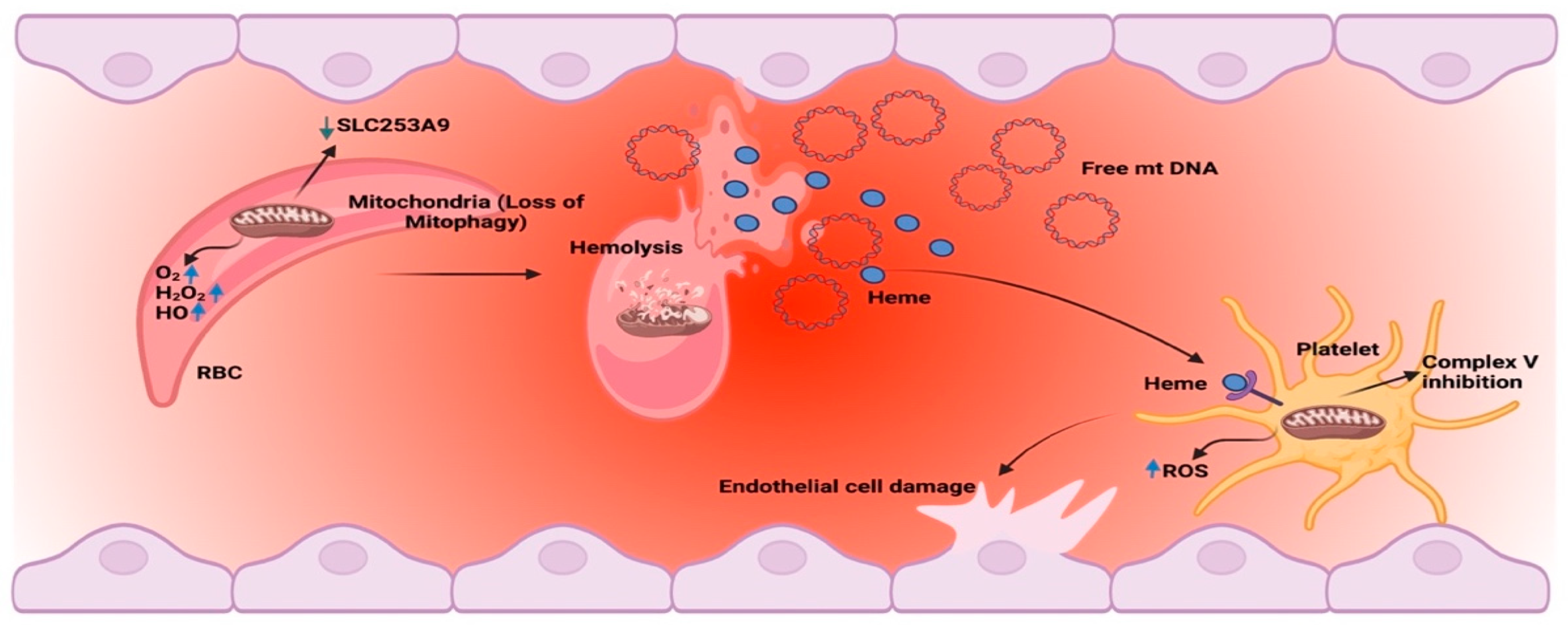
2. Mitochondrial Retention
3. Oxidative Stress
4. Hemolysis
5. mtDNA Variation
6. Circulating Mitochondrial DNA
7. Therapeutic Targeting

{kind=link}
{kind=link}
| Mitochondrial Feature | Increase/Decrease or Presence/Absence | Consequence | Implication | References |
|---|---|---|---|---|
| Cell-free DNA | Increased | Triggers NETs formation | Pathological inflammation | [6] |
| Hypomethylation of mtDNA | Increased | Triggers NETs formation | Increased crises | [7] |
| Complex V activity | Decreased | Increased ROS | Hemolysis | [13,66] |
| PINK1 | Decreased | Impaired mitophagy | Mitochondrial retention in RBC | [25] |
| NIX | Decreased | Impaired mitophagy | Mitochondrial retention in RBC | [25] |
| SOD2V16A | Present | Decreases mitochondrial Complex IV activity | Increased tricuspid regurgitant velocity | [36,66] |
| SOD2 expression | Decreased | Decreased SOD levels | Increased oxidative stress in microenvironment. | [43] |
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xia, M.; Zhang, Y.; Jin, K.; Lu, Z.; Zeng, Z.; Xiong, W. Communication between mitochondria and other organelles: A brand-new perspective on mitochondria in cancer. Cell Biosci. 2019, 9, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audano, M.; Pedretti, S.; Ligorio, S.; Crestani, M.; Caruso, D.; De Fabiani, E.; Mitro, N. “The Loss of Golden Touch”: Mitochondria-Organelle Interactions, Metabolism, and Cancer. Cells 2020, 9, 2519. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, S.; Choi, A.M. Mitochondria in lung disease. J. Clin. Investig. 2016, 126, 809–820. [Google Scholar] [CrossRef] [Green Version]
- Rahman, J.; Rahman, S. Mitochondrial medicine in the omics era. Lancet 2018, 391, 2560–2574. [Google Scholar] [CrossRef] [Green Version]
- Suárez-Rivero, J.M.; Villanueva-Paz, M.; de la Cruz-Ojeda, P.; de la Mata, M.; Cotán, D.; Oropesa-Ávila, M.; de Lavera, I.; Álvarez-Córdoba, M.; Luzón-Hidalgo, R.; Sánchez-Alcázar, J.A. Mitochondrial Dynamics in Mitochondrial Diseases. Diseases 2016, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumburu, L.; Ghosh-Choudhary, S.; Seifuddin, F.T.; Barbu, E.A.; Yang, S.; Ahmad, M.M.; Wilkins, L.H.W.; Tunc, I.; Sivakumar, I.; Nichols, J.S.; et al. Circulating mitochondrial DNA is a proinflammatory DAMP in sickle cell disease. Blood 2021, 137, 3116–3126. [Google Scholar] [CrossRef] [PubMed]
- Tumburu, L.; Ghosh-Choudhary, S.; Barbu, E.A.; Yang, S.; Ware, L.D.H.; Tunc, I.; Seifuddin, F.T.; Pirooznia, M.; Zhu, J.; Thein, S.L. Cell-Free Mitochondrial DNA Is Elevated in Sickle Cell Disease Patients, and Serve As a Potential Proinflammatory DAMP. Blood 2018, 132, 1068. [Google Scholar] [CrossRef]
- Finsterer, J. Hematological Manifestations of Primary Mitochondrial Disorders. Acta Haematol. 2007, 118, 88–98. [Google Scholar] [CrossRef]
- Fontenay, M.; Cathelin, S.; Amiot, M.; Gyan, E.; Solary, E. Mitochondria in hematopoiesis and hematological diseases. Oncogene 2006, 25, 4757–4767. [Google Scholar] [CrossRef] [Green Version]
- Bhagat, V.; Baviskar, S.; Mudey, A.; Goyal, R. Poor health related quality of life among patients of sickle cell disease. Indian J. Palliat. Care 2014, 20, 107–111. [Google Scholar] [CrossRef]
- Aeddula, N.; Bardhan, M.; Baradhi, K. Sickle Cell Nephropathy; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK526017/ (accessed on 13 September 2022).
- Inusa, B.P.D.; Hsu, L.L.; Kohli, N.; Patel, A.; Ominu-Evbota, K.; Anie, K.A.; Atoyebi, W. Sickle Cell Disease—Genetics, Pathophysiology, Clinical Presentation and Treatment. Int. J. Neonatal Screen. 2019, 5, 20. [Google Scholar] [CrossRef] [Green Version]
- Cardenes, N.; Corey, C.; Geary, L.; Jain, S.; Zharikov, S.; Barge, S.; Novelli, E.M.; Shiva, S. Platelet bioenergetic screen in sickle cell patients reveals mitochondrial complex V inhibition, which contributes to platelet activation. Blood 2014, 123, 2864–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Tran, J.; Wang, H.; Guo, C.; Harro, D.; Campbell, A.D.; Eitzman, D.T. mTOR Inhibition improves anaemia and reduces organ damage in a murine model of sickle cell disease. Br. J. Haematol. 2016, 174, 461–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbanera, Y.; Arcioni, F.; Lancioni, H.; La Starza, R.; Cardinali, I.; Matteucci, C.; Nofrini, V.; Roetto, A.; Piga, A.; Grammatico, P.; et al. Comprehensive analysis of mitochondrial and nuclear DNA variations in patients affected by hemoglobinopathies: A pilot study. PLoS ONE 2020, 15, e0240632. [Google Scholar] [CrossRef] [PubMed]
- Liang, R.; Menon, V.; Qiu, J.; Arif, T.; Renuse, S.; Lin, M.; Nowak, R.; Hartmann, B.; Tzavaras, N.; Benson, D.L.; et al. Mitochondrial localization and moderated activity are key to murine erythroid enucleation. Blood Adv. 2021, 5, 2490–2504. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Ibanez, A.M.; Ruiz, L.M.; Jensen, E.; Echeverria, C.A.; Romero, V.; Stiles, L.; Shirihai, O.S.; Elorza, A.A. Erythroid Differentiation and Heme Biosynthesis Are Dependent on a Shift in the Balance of Mitochondrial Fusion and Fission Dynamics. Front. Cell Dev. Biol. 2020, 8, 592035. [Google Scholar] [CrossRef]
- Mortensen, M.; Ferguson, D.; Edelmann, M.; Kessler, B.; Morten, K.; Komatsu, M.; Simon, A. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 832–837. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-W.; Cheng, J.; Xu, F.; Chen, Y.-E.; Du, J.-B.; Yuan, M.; Zhu, F.; Xu, X.-C.; Yuan, S. Red blood cell extrudes nucleus and mitochondria against oxidative stress. IUBMB Life 2011, 63, 560–565. [Google Scholar] [CrossRef]
- Jagadeeswaran, R.; Rivers, A. Evolving treatment paradigms in sickle cell disease. Hematology 2017, 2017, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Hong, L.; Jagadeeswaran, R.; Molokie, R.; Lavelle, D.; Rivers, A.; Diamond, A. Selenium Deficiency in a Mouse Model of Sickle Cell Disease Resulted in Increased Oxygen Consumption and Aberrant Mitochondrial Retention (OR11-05-19). Curr. Dev. Nutr. 2019, 3, nzz044. [Google Scholar] [CrossRef]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef] [Green Version]
- Kundu, M.; Lindsten, T.; Yang, C.-Y.; Wu, J.; Zhao, F.; Zhang, J.; Selak, M.A.; Ney, P.A.; Thompson, C.B. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 2008, 112, 1493–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagadeeswaran, R.; Vazquez, B.A.; Thiruppathi, M.; Ganesh, B.B.; Ibanez, V.; Cui, S.; Engel, J.D.; Diamond, A.M.; Molokie, R.E.; DeSimone, J.; et al. Pharmacological inhibition of LSD1 and mTOR reduces mitochondrial retention and associated ROS levels in the red blood cells of sickle cell disease. Exp. Hematol. 2017, 50, 46–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martino, S.; Arlet, J.; Odièvre, M.; Jullien, V.; Moras, M.; Hattab, C.; Lefebvre, T.; Gouya, L.; Ostuni, M.A.; Lefevre, S.D.; et al. Deficient mitophagy pathways in sickle cell disease. Br. J. Haematol. 2021, 193, 988–993. [Google Scholar] [CrossRef]
- Gallivan, A.; Kanu, A.; Alejandro, M.; Zekaryas, N.; Horneman, H.; Ramasamy, J.; Hong, L.; Lavelle, D.; Diamond, A.; Molokie, R.E.; et al. Reticulocytosis from Stress Erythropoiesis Is a Major Source of Erythrocyte Mitochondrial Retention, Oxygen Consumption and Reactive Oxygen Species in a SCD Mouse Model. Blood 2022, 140, 8239–8240. [Google Scholar] [CrossRef]
- Rai, M.P.; Roy, S.; Konstantinidis, B.D.G.; Ponny, M.S.R.; Mpollo, M.-S.E.M.; Shrestha, A.; Kalfa, T.A.; Malik, P. Angiotensin Signaling Is Essential for Stress Erythropoiesis but Results in Retention of Dysfunctional Mitochondria in Erythrocytes That Generate Excessive Reactive Oxygen Species. Blood 2020, 136, 31–32. [Google Scholar] [CrossRef]
- Ramasamy, J.; Gallivan, A.; Hong, L.; Horneman, H.; Molokie, R.; Diamond, A.; Lavelle, D.; Rivers, A. P103: THE ABNORMAL PRESENCE OF MITOCHONDRIA IN CIRCULATING SCD RED BLOOD CELLS ASSOCIATED WITH STRESS ERYTHROPOIESIS. HemaSphere 2022, 6, 18–19. [Google Scholar] [CrossRef]
- Moras, M.; Hattab, C.; Gonzalez-Menendez, P.; Fader, C.M.; Dussiot, M.; Larghero, J.; Le Van Kim, C.; Kinet, S.; Taylor, N.; Lefevre, S.D.; et al. Human erythroid differentiation requires VDAC1-mediated mitochondrial clearance. Haematologica 2022, 107, 167–177. [Google Scholar] [CrossRef]
- Moriconi, C.; Dzieciatkowska, M.; Roy, M.; D’Alessandro, A.; Roingeard, P.; Lee, J.Y.; Gibb, D.R.; Tredicine, M.; McGill, M.A.; Qiu, A.; et al. Retention of functional mitochondria in mature red blood cells from patients with sickle cell disease. Br. J. Haematol. 2022, 198, 574–586. [Google Scholar] [CrossRef]
- Biswal, S.; Rizwan, H.; Pal, S.; Sabnam, S.; Parida, P.; Pal, A. Oxidative stress, antioxidant capacity, biomolecule damage, and inflammation symptoms of sickle cell disease in children. Hematology 2019, 24, 1–9. [Google Scholar] [CrossRef]
- Vona, R.; Sposi, N.; Mattia, L.; Gambardella, L.; Straface, E.; Pietraforte, D. Sickle Cell Disease: Role of Oxidative Stress and Antioxidant Therapy. Antioxidants 2021, 10, 296. [Google Scholar] [CrossRef]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassa, T.; Jana, S.; Strader, M.B.; Meng, F.; Jia, Y.; Wilson, M.T.; Alayash, A.I. Sickle Cell Hemoglobin in the Ferryl State Promotes βCys-93 Oxidation and Mitochondrial Dysfunction in Epithelial Lung Cells (E10). J. Biol. Chem. 2015, 290, 27939–27958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dosunmu-Ogunbi, A.M.; Wood, K.C.; Novelli, E.M.; Straub, A.C. Decoding the role of SOD2 in sickle cell disease. Blood Adv. 2019, 3, 2679–2687. [Google Scholar] [CrossRef]
- Annarapu, G.K.; Nolfi-Donegan, D.; Reynolds, M.; Wang, Y.; Shiva, S. Mitochondrial reactive oxygen species scavenging attenuates thrombus formation in a murine model of sickle cell disease. J. Thromb. Haemost. 2021, 19, 2256–2262. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Pradhan-Sundd, T.; Pritchard, K.A., Jr.; Hillery, C.A. Redox signaling in sickle cell disease. Curr. Opin. Physiol. 2019, 9, 26–33. [Google Scholar] [CrossRef]
- Wang, Y.; Yen, F.S.; Zhu, X.G.; Timson, R.C.; Weber, R.; Xing, C.; Liu, Y.; Allwein, B.; Luo, H.; Yeh, H.-W.; et al. SLC25A39 is necessary for mitochondrial glutathione import in mammalian cells. Nature 2021, 599, 136–140. [Google Scholar] [CrossRef]
- Zou, X.; Ratti, B.A.; O’Brien, J.G.; Lautenschlager, S.O.; Gius, D.R.; Bonini, M.G.; Zhu, Y. Manganese superoxide dismutase (SOD2): Is there a center in the universe of mitochondrial redox signaling? J. Bioenerg. Biomembr. 2017, 49, 325–333. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, J.; Lin, Y.; Lei, Q.; Guan, K.-L.; Zhao, S.; Xiong, Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011, 12, 534–541. [Google Scholar] [CrossRef]
- Redondo-Horcajo, M.; Romero, N.; Martínez-Acedo, P.; Martínez-Ruiz, A.; Quijano, C.; Lourenço, C.F.; Movilla, N.; Enríquez, J.A.; Rodríguez-Pascual, F.; Rial, E.; et al. Cyclosporine A-induced nitration of tyrosine 34 MnSOD in endothelial cells: Role of mitochondrial superoxide. Cardiovasc. Res. 2010, 87, 356–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armenis, I.; Kalotychou, V.; Tzanetea, R.; Moyssakis, I.; Anastasopoulou, D.; Pantos, C.; Konstantopoulos, K.; Rombos, I. Reduced peripheral blood superoxide dismutase 2 expression in sickle cell disease. Ann. Hematol. 2019, 98, 1561–1572. [Google Scholar] [CrossRef]
- Liang, H.; Van Remmen, H.; Frohlich, V.; Lechleiter, J.; Richardson, A.; Ran, Q. GPX4 protects mitochondrial ATP generation against oxidative damage. Biochem. Biophys. Res. Commun. 2007, 356, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Bryk, A.H.; Wiśniewski, J.R. Quantitative analysis of human red blood cell proteome. J. Proteome Res. 2017, 16, 2752–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canli, Ö.; Alankuş, Y.B.; Grootjans, S.; Vegi, M.N.; Hültner, L.; Hoppe, P.S.; Schroeder, T.; Vandenabeele, P.; Bornkamm, G.W.; Greten, F.R. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood 2016, 127, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Stolwijk, J.M.; Stefely, J.A.; Veling, M.T.; Erve, T.J.V.; Wagner, B.A.; Raife, T.J.; Buettner, G.R. Red blood cells contain enzymatically active GPX4 whose abundance anticorrelates with hemolysis during blood bank storage. Redox Biol. 2021, 46, 102073. [Google Scholar] [CrossRef] [PubMed]
- Santesmasses, D.; Gladyshev, V.N. Selenocysteine Machinery Primarily Supports TXNRD1 and GPX4 Functions and Together They Are Functionally Linked with SCD and PRDX6. Biomolecules 2022, 12, 1049. [Google Scholar] [CrossRef]
- van Vuren, A.J.; van Beers, E.J.; van Wijk, R. A Proposed Concept for Defective Mitophagy Leading to Late Stage Ineffective Erythropoiesis in Pyruvate Kinase Deficiency. Front. Physiol. 2021, 20, 609103. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.; Steinberg, M.H.; Gladwin, M.T. Intravascular hemolysis and the pathophysiology of sickle cell disease. J. Clin. Investig. 2017, 127, 750–760. [Google Scholar] [CrossRef]
- Jana, S.; Meng, F.; Hirsch, R.E.; Friedman, J.M.; Alayash, A.I. Oxidized Mutant Human Hemoglobins S and E Induce Oxidative Stress and Bioenergetic Dysfunction in Human Pulmonary Endothelial Cells. Front. Physiol. 2017, 8, 1082. [Google Scholar] [CrossRef] [PubMed]
- Gbotosho, O.T.; Kapetanaki, M.G.; Kato, G.J. The Worst Things in Life are Free: The Role of Free Heme in Sickle Cell Disease. Front. Immunol. 2021, 11, 561917. [Google Scholar] [CrossRef]
- Chintagari, N.R.; Jana, S.; Alayash, A.I. Oxidized Ferric and Ferryl Forms of Hemoglobin Trigger Mitochondrial Dysfunction and Injury in Alveolar Type I Cells. Am. J. Respir. Cell Mol. Biol. 2016, 55, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Jana, S.; Strader, M.B.; Meng, F.; Hicks, W.; Kassa, T.; Tarandovskiy, I.; De Paoli, S.; Simak, J.; Heaven, M.R.; Belcher, J.D.; et al. Hemoglobin oxidation–dependent reactions promote interactions with band 3 and oxidative changes in sickle cell–derived microparticles. J. Clin. Investig. 2018, 3, e120451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kay, M.M. Band 3 and its alterations in health and disease. Cell. Mol. Biol. 2004, 50, 117–138. [Google Scholar]
- Ostedgaard, L.S.; Jennings, M.L.; Karniski, L.P.; Schuster, V.L. A 45-kDa protein antigenically related to band 3 is selectively expressed in kidney mitochondria. Proc. Natl. Acad. Sci. USA 1991, 88, 981–985. [Google Scholar] [CrossRef] [Green Version]
- Bijur, G.N.; Jope, R.S. Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J. Neurochem. 2003, 87, 1427–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Li, Y.; He, L.; Agarwal, A.R.; Zeng, N.; Cadenas, E.; Stiles, B.L. PI3K/AKT signaling regulates bioenergetics in immortalized hepatocytes. Free. Radic. Biol. Med. 2013, 60, 29–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Slone, J.; Fei, L.; Huang, T. Mitochondrial DNA Variants and Common Diseases: A Mathematical Model for the Diversity of Age-Related mtDNA Mutations. Cells 2019, 8, 608. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Mitochondrial DNA Variation in Human Radiation and Disease. Cell 2015, 163, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Aquadro, C.F.; Greenberg, B.D. Human mitochondrial DNA variation and evolution: Analysis of nucleotide sequences from seven individuals. Genetics 1983, 103, 287–312. [Google Scholar] [CrossRef]
- Bellizzi, D.; D’Aquila, P.; Scafone, T.; Giordano, M.; Riso, V.; Riccio, A.; Passarino, G. The Control Region of Mitochondrial DNA Shows an Unusual CpG and Non-CpG Methylation Pattern. DNA Res. 2013, 20, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, B.M.M.; Tumburu, L.; Liu, C.; Pirooznia, M.; Thein, D.S.L. Mitochondrial DNA Variation in Individuals with Sickle Cell Disease. Blood 2020, 136, 11. [Google Scholar] [CrossRef]
- Gilkerson, R.W.; De Vries, R.L.; Lebot, P.; Wikstrom, J.; Torgyekes, E.; Shirihai, O.S.; Przedborski, S.; Schon, E.A. Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum. Mol. Genet. 2011, 21, 978–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, L.Z.; Figueroa, J.; Leake, D.; Khemani, K.; Kumari, P.; Bakshi, N.; Lane, P.A.; Dampier, C.; Morris, C.R. Safety of intravenous arginine therapy in children with sickle cell disease hospitalized for vaso-occlusive pain: A randomized placebo-controlled trial in progress. Am. J. Hematol. 2021, 97, E21–E24. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.R.; Brown, L.A.S.; Reynolds, M.; Dampier, C.D.; Lane, P.A.; Watt, A.; Kumari, P.; Harris, F.; Manoranjithan, S.; Mendis, R.D.; et al. Impact of arginine therapy on mitochondrial function in children with sickle cell disease during vaso-occlusive pain. Blood 2020, 136, 1402–1406. [Google Scholar] [CrossRef] [PubMed]
- Onalo, R.; Cilliers, A.; Cooper, P.; Morris, C.R. Arginine Therapy and Cardiopulmonary Hemodynamics in Hospitalized Children with Sickle Cell Anemia: A Prospective, Double-blinded, Randomized Placebo-controlled Clinical Trial. Am. J. Respir. Crit. Care Med. 2022, 206, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Dick, M.H.; Abdelgadir, A.; Kulkarni, V.V.; Akram, H.; Chatterjee, A.; Pokhrel, S.; Khan, S. Comparing the Safety and Efficacy of L-Glutamine, Voxelotor, and Crizanlizumab for Reducing the Frequency of Vaso-Occlusive Crisis in Sickle Cell Disease: A Systematic Review. Cureus 2022, 14, 24920. [Google Scholar] [CrossRef]
- Sadaf, A.; Quinn, C.T. L-glutamine for sickle cell disease: Knight or pawn? Exp. Biol. Med. 2020, 245, 146–154. [Google Scholar] [CrossRef] [Green Version]
- Salman, E.K.; Haymond, M.W.; Bayne, E.; Sager, B.K.; Wiisanen, A.; Pitel, P.; Darmaun, D. Protein and Energy Metabolism in Prepubertal Children with Sickle Cell Anemia. Pediatr. Res. 1996, 40, 34–40. [Google Scholar] [CrossRef] [Green Version]
- Niihara, Y.; Miller, S.T.; Kanter, J.; Lanzkron, S.; Smith, W.R.; Hsu, L.L.; Gordeuk, V.R.; Viswanathan, K.; Sarnaik, S.; Osunkwo, I.; et al. A Phase 3 Trial ofl-Glutamine in Sickle Cell Disease. N. Engl. J. Med. 2018, 379, 226–235. [Google Scholar] [CrossRef]
- Walter, P.B.; Hohman, L.S.; Rokeby, A.; Lum, J.J.; Hagar, R.; Lavrisha, L.; Saulys, A.; Kuypers, F.A.; Vichinsky, E.; Morris, C.R. The effects of glutamine supplementation on markers of apoptosis and autophagy in sickle cell disease peripheral blood mononuclear cells. Complement. Ther. Med. 2022, 70, 102856. [Google Scholar] [CrossRef] [PubMed]


| S. No. | Ethnic Origin | Haplotype ID | Haplogroup | GenBank Accession Number |
|---|---|---|---|---|
| 1 | Europe | HT20 | T1a1 + 152 | MT176208 |
| 2 | America | HT29 | B4 | MT176209 |
| 3 | Africa | HT40 | G1a | MT176210 |
| 4 | Africa | HT40 | G1a | MT176211 |
| 5 | Africa | HT32 | L2a1 | MT176212 |
| 6 | Africa | HT32 | L2a1 | MT176213 |
| 7 | Africa | HT09 | L1b2a | MT176214 |
| 8 | Africa | HT21 | L1b1a + 189 | MT176215 |
| 9 | Africa | HT41 | L2a1 + 143 | MT176216 |
| 10 | Europe | HT39 | L3e1b2 | MT176217 |
| 11 | Africa | HT10 | L2a1 | MT176218 |
| 12 | Africa | HT03 | L1c1d | MT176219 |
| 13 | Africa | HT08 | L1c2a1a | MT176220 |
| 14 | Africa | HT04 | L2a1a1 | MT176221 |
| 15 | Africa | HT35 | M30c | MT176222 |
| 16 | America | HT44 | U7a | MT176223 |
| 17 | Africa | HT34 | L2a1 + 16189 + (16192) | MT176224 |
| 18 | Africa | HT13 | L3k1 | MT176225 |
| 19 | Africa | HT26 | L0a1a2 | MT176226 |
| 20 | Africa | HT25 | L0a1a | MT176227 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akhter, M.S.; Hamali, H.A.; Rashid, H.; Dobie, G.; Madkhali, A.M.; Mobarki, A.A.; Oldenburg, J.; Biswas, A. Mitochondria: Emerging Consequential in Sickle Cell Disease. J. Clin. Med. 2023, 12, 765. https://doi.org/10.3390/jcm12030765
Akhter MS, Hamali HA, Rashid H, Dobie G, Madkhali AM, Mobarki AA, Oldenburg J, Biswas A. Mitochondria: Emerging Consequential in Sickle Cell Disease. Journal of Clinical Medicine. 2023; 12(3):765. https://doi.org/10.3390/jcm12030765
Chicago/Turabian StyleAkhter, Mohammad S., Hassan A. Hamali, Hina Rashid, Gasim Dobie, Aymen M. Madkhali, Abdullah A. Mobarki, Johannes Oldenburg, and Arijit Biswas. 2023. "Mitochondria: Emerging Consequential in Sickle Cell Disease" Journal of Clinical Medicine 12, no. 3: 765. https://doi.org/10.3390/jcm12030765
APA StyleAkhter, M. S., Hamali, H. A., Rashid, H., Dobie, G., Madkhali, A. M., Mobarki, A. A., Oldenburg, J., & Biswas, A. (2023). Mitochondria: Emerging Consequential in Sickle Cell Disease. Journal of Clinical Medicine, 12(3), 765. https://doi.org/10.3390/jcm12030765