

Clinical Manifestations and Management of Fibrotic Pulmonary Sarcoidosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Background
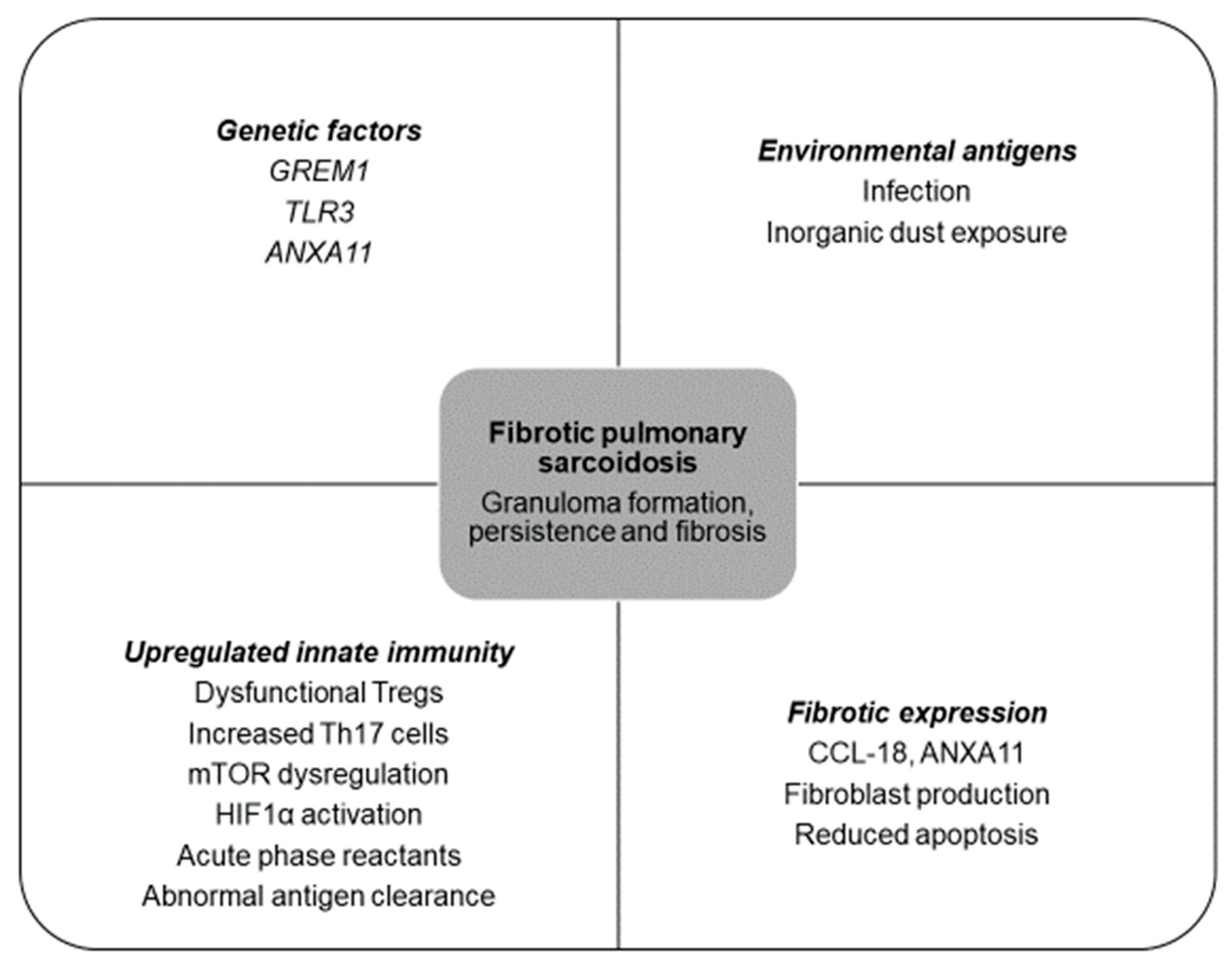
2. Pathobiology
2.1. Basic Pathophysiology: An Interplay between Genetic and Environmental Factors
2.2. Evolving Knowledge of Pathophysiology in Fibrotic Pulmonary Sarcoidosis
3. Clinical Manifestations
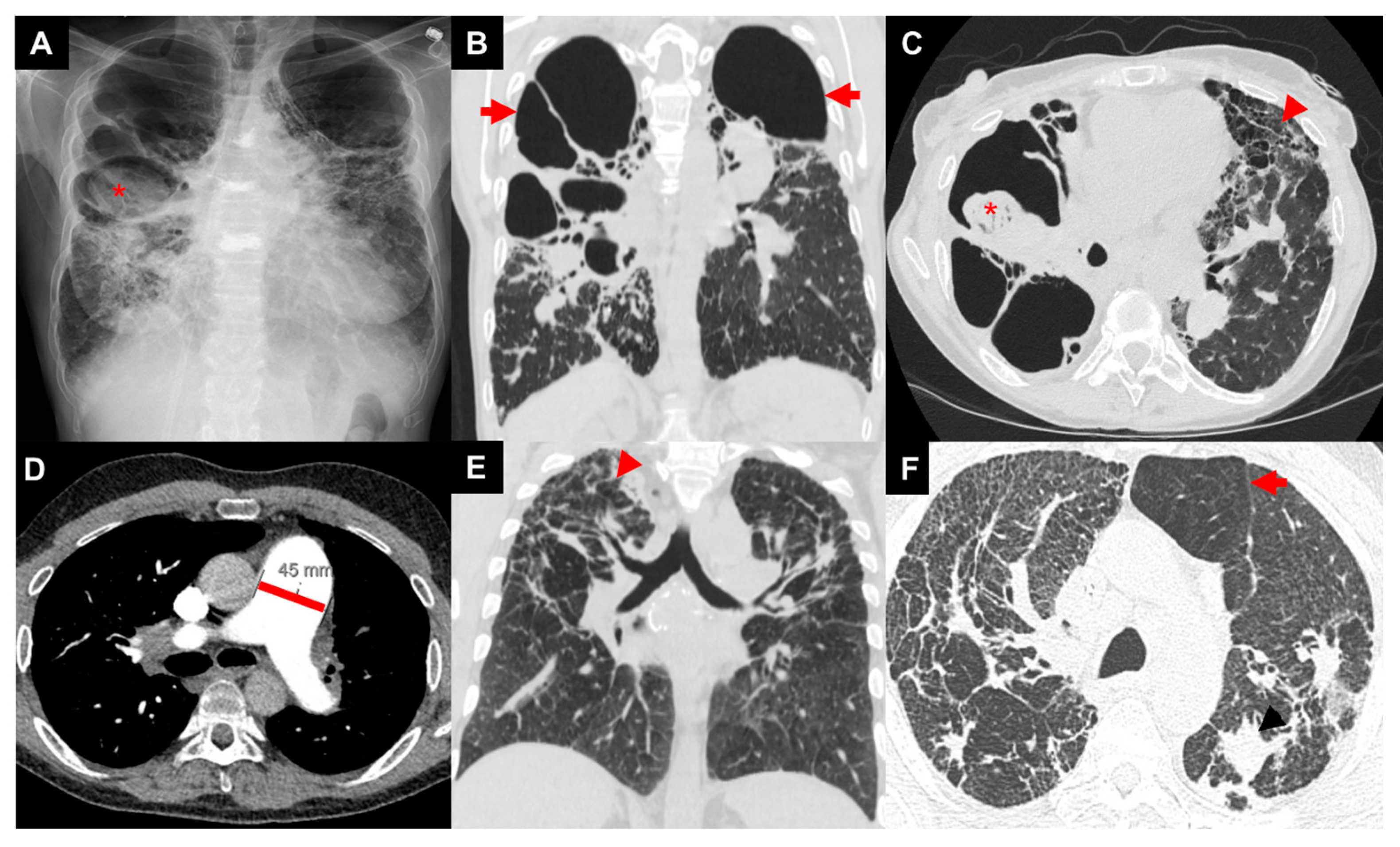
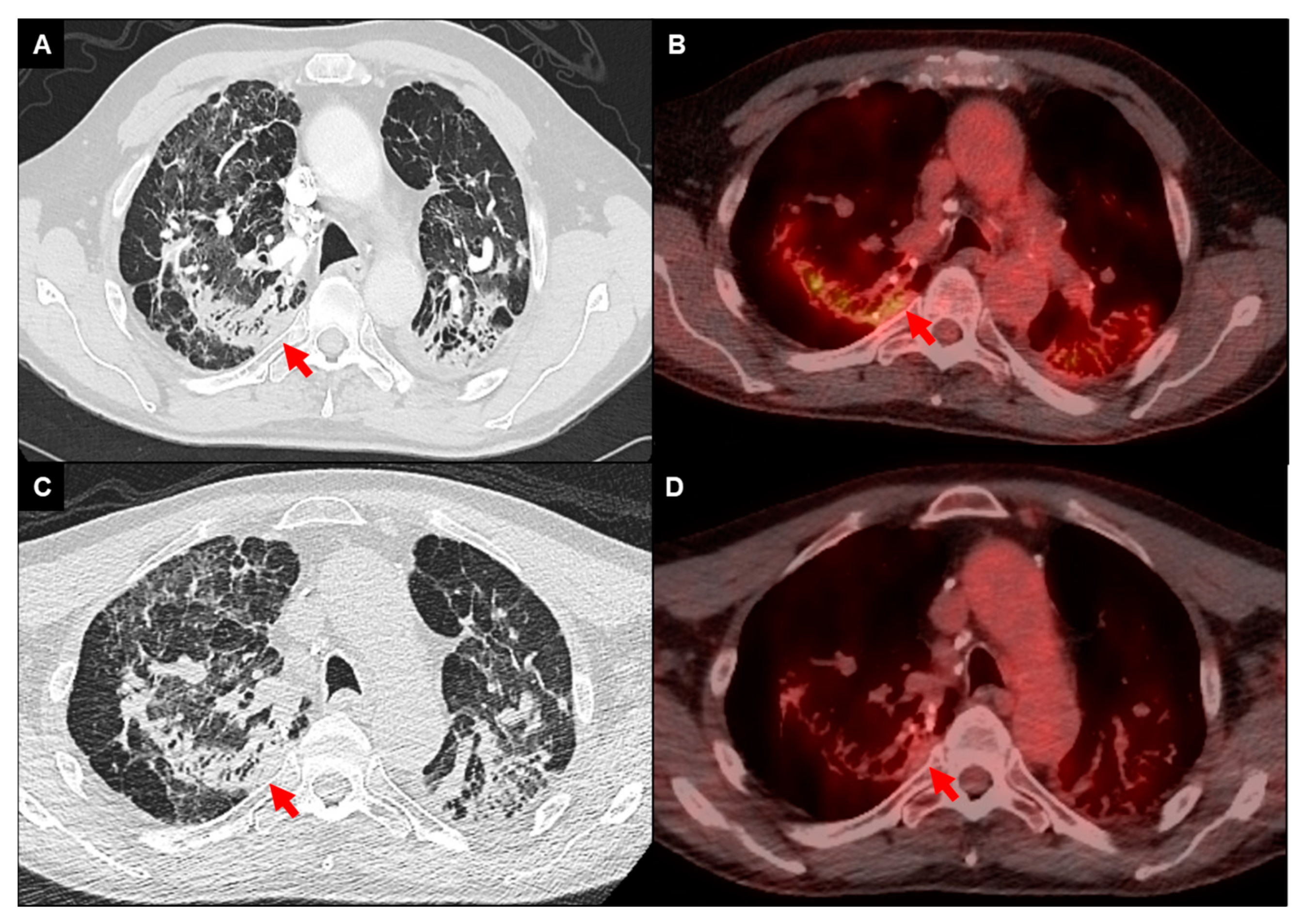
3.1. Imaging
3.2. Pulmonary Function Testing
3.3. Serum Biomarkers
4. Prognosis
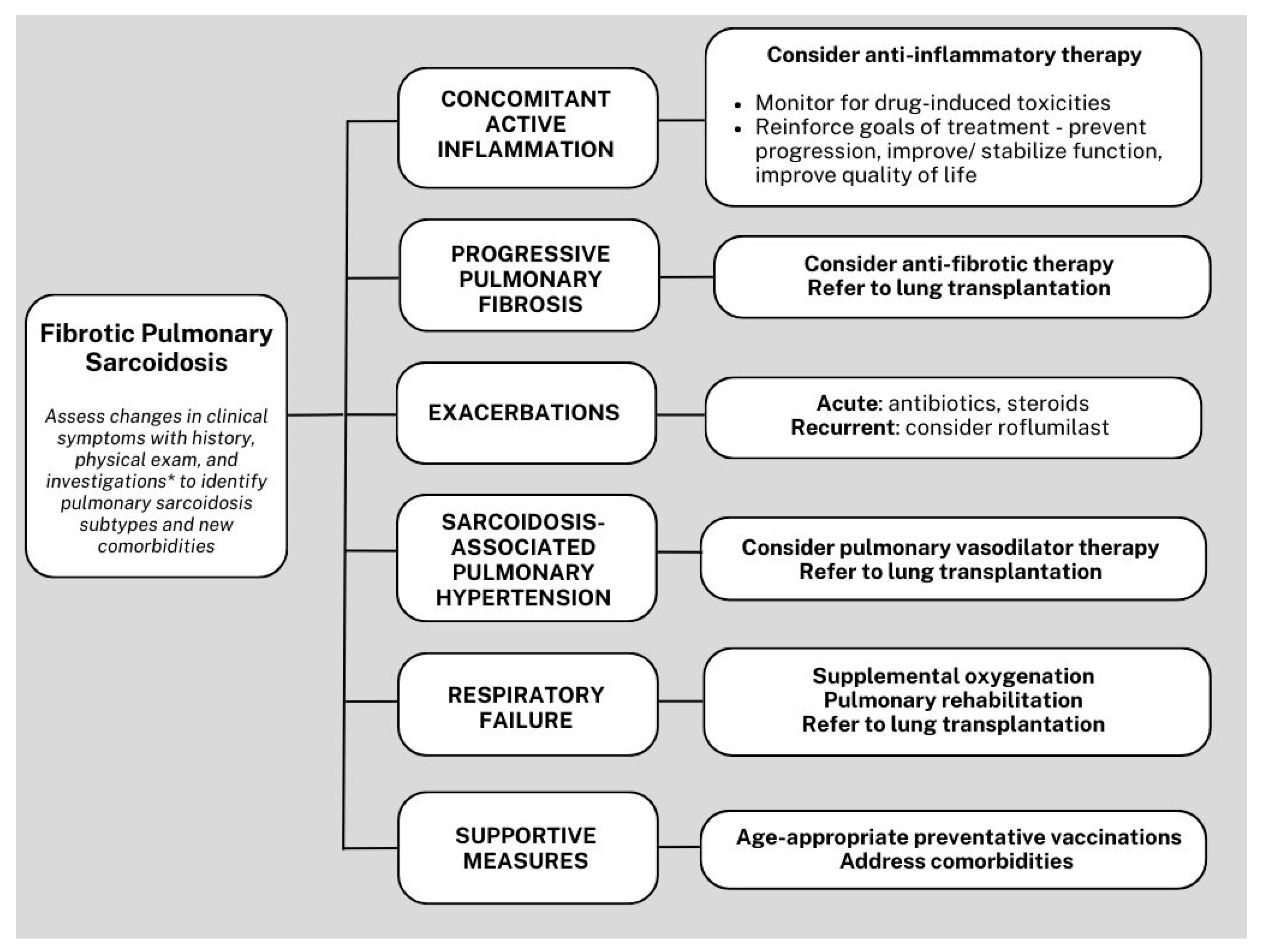
5. Management
5.1. Anti-Inflammatory Therapy
5.2. Sarcoidosis-Associated Bronchiectasis
5.3. Acute Pulmonary Exacerbations of Sarcoidosis
5.4. Infections
5.5. Sarcoidosis-Associated Pulmonary Hypertension
5.6. Antifibrotic Therapy
5.7. Supportive Management
5.8. Lung Transplantation
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hunninghake, G.W.; Costabel, U.; Ando, M.; Baughman, R.; Cordier, J.F.; Bois, R.D.; Eklund, A.; Kitaichi, M.; Lynch, J.; Rizzato, G.; et al. Ats/ers/wasog statement on sarcoidosis. American thoracic society/european respiratory society/world association of sarcoidosis and other granulomatous disorders. Sarcoidosis Vasc. Diffuse Lung Dis. 1999, 16, 149–173. [Google Scholar] [PubMed]
- Judson, M.A.; Thompson, B.W.; Rabin, D.L.; Steimel, J.; Knattereud, G.L.; Lackland, D.T.; Rose, C.; Rand, C.S.; Baughman, R.P.; Teirstein, A.S.; et al. The diagnostic pathway to sarcoidosis. Chest 2003, 123, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Crouser, E.D.; Maier, L.A.; Wilson, K.C.; Bonham, C.A.; Morgenthau, A.S.; Patterson, K.C.; Abston, E.; Bernstein, R.C.; Blankstein, R.; Chen, E.S.; et al. Diagnosis and detection of sarcoidosis. An official american thoracic society clinical practice guideline. Am. J. Respir. Crit. Care Med. 2020, 201, e26–e51. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.P.; Lower, E.E.; Bois, R.M.D. Sarcoidosis. Lancet 2003, 361, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Hena, K.M. Sarcoidosis epidemiology: Race matters. Front. Immunol. 2020, 11, 537382. [Google Scholar] [CrossRef] [PubMed]
- Mirsaeidi, M.; Machado, R.F.; Schraufnagel, D.; Sweiss, N.J.; Baughman, R.P. Racial difference in sarcoidosis mortality in the united states. Chest 2015, 147, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Scadding, J.G. Prognosis of intrathoracic sarcoidosis in england. A review of 136 cases after five years’ observation. Br. Med. J. 1961, 2, 1165–1172. [Google Scholar] [CrossRef]
- Judson, M.A.; Boan, A.D.; Lackland, D.T. The clinical course of sarcoidosis: Presentation, diagnosis, and treatment in a large white and black cohort in the united states. Sarcoidosis Vasc. Diffuse Lung Dis. 2012, 29, 119–127. [Google Scholar]
- Yeager, H.; Rossman, M.D.; Baughman, R.P.; Teirstein, A.S.; Judson, M.A.; Rabin, D.L.; Iannuzzi, M.C.; Rose, C.; Bresnitz, E.A.; DePalo, L.; et al. Pulmonary and psychosocial findings at enrollment in the access study. Sarcoidosis Vasc. Diffuse Lung Dis. 2005, 22, 147–153. [Google Scholar]
- Gupta, R.; Judson, M.A.; Baughman, R.P. Management of advanced pulmonary sarcoidosis. Am. J. Respir. Crit. Care Med. 2022, 205, 495–506. [Google Scholar] [CrossRef]
- Gupta, R.; Baughman, R.P. Advanced pulmonary sarcoidosis. Semin. Respir. Crit. Care Med. 2020, 41, 700–715. [Google Scholar] [CrossRef]
- Nardi, A.; Brillet, P.Y.; Letoumelin, P.; Girard, F.; Brauner, M.; Uzunhan, Y.; Naccache, J.M.; Valeyre, D.; Nunes, H. Stage iv sarcoidosis: Comparison of survival with the general population and causes of death. Eur. Respir. J. 2011, 38, 1368–1373. [Google Scholar] [CrossRef]
- Jeny, F.; Uzunhan, Y.; Lacroix, M.; Gille, T.; Brillet, P.Y.; Nardi, A.; Bouvry, D.; Planes, C.; Nunes, H.; Valeyre, D. Predictors of mortality in fibrosing pulmonary sarcoidosis. Respir. Med. 2020, 169, 105997. [Google Scholar] [CrossRef]
- Shlobin, O.A.; Kouranos, V.; Barnett, S.D.; Alhamad, E.H.; Culver, D.A.; Barney, J.; Cordova, F.C.; Carmona, E.M.; Scholand, M.B.; Wijsenbeek, M.; et al. Physiological predictors of survival in patients with sarcoidosis-associated pulmonary hypertension: Results from an international registry. Eur. Respir. J. 2020, 55, 1901747. [Google Scholar] [CrossRef]
- Shorr, A.F.; Davies, D.B.; Nathan, S.D. Outcomes for patients with sarcoidosis awaiting lung transplantation. Chest 2002, 122, 233–238. [Google Scholar] [CrossRef]
- Ronsmans, S.; De Ridder, J.; Vandebroek, E.; Keirsbilck, S.; Nemery, B.; Hoet, P.H.M.; Vanderschueren, S.; Wuyts, W.A.; Yserbyt, J. Associations between occupational and environmental exposures and organ involvement in sarcoidosis: A retrospective case-case analysis. Respir. Res. 2021, 22, 224. [Google Scholar] [CrossRef]
- Beijer, E.; Veltkamp, M. The emerging role of inorganic elements as potential antigens in sarcoidosis. Curr. Opin. Pulm. Med. 2021, 27, 430–438. [Google Scholar] [CrossRef]
- Beijer, E.; Meek, B.; Bossuyt, X.; Peters, S.; Vermeulen, R.C.H.; Kromhout, H.; Veltkamp, M. Immunoreactivity to metal and silica associates with sarcoidosis in dutch patients. Respir. Res. 2020, 21, 141. [Google Scholar] [CrossRef]
- Grunewald, J.; Grutters, J.C.; Arkema, E.V.; Saketkoo, L.A.; Moller, D.R.; Muller-Quernheim, J. Sarcoidosis. Nat. Rev. Dis. Primers 2019, 5, 45. [Google Scholar] [CrossRef]
- Iannuzzi, M.C.; Maliarik, M.J.; Poisson, L.M.; Rybicki, B.A. Sarcoidosis susceptibility and resistance hla-dqb1 alleles in african americans. Am. J. Respir. Crit. Care Med. 2003, 167, 1225–1231. [Google Scholar] [CrossRef]
- Iannuzzi, M.C.; Iyengar, S.K.; Gray-McGuire, C.; Elston, R.C.; Baughman, R.P.; Donohue, J.F.; Hirst, K.; Judson, M.A.; Kavuru, M.S.; Maliarik, M.J.; et al. Genome-wide search for sarcoidosis susceptibility genes in african americans. Genes Immun. 2005, 6, 509–518. [Google Scholar] [CrossRef]
- Gray-McGuire, C.; Sinha, R.; Iyengar, S.; Millard, C.; Rybicki, B.A.; Elston, R.C.; Iannuzzi, M.C.; Consortium, S.S. Genetic characterization and fine mapping of susceptibility loci for sarcoidosis in african americans on chromosome 5. Hum. Genet. 2006, 120, 420–430. [Google Scholar] [CrossRef]
- Rossman, M.D.; Thompson, B.; Frederick, M.; Maliarik, M.; Iannuzzi, M.C.; Rybicki, B.A.; Pandey, J.P.; Newman, L.S.; Magira, E.; Beznik-Cizman, B.; et al. Hla-drb1*1101: A significant risk factor for sarcoidosis in blacks and whites. Am. J. Hum. Genet. 2003, 73, 720–735. [Google Scholar] [CrossRef]
- Levin, A.M.; Iannuzzi, M.C.; Montgomery, C.G.; Trudeau, S.; Datta, I.; McKeigue, P.; Fischer, A.; Nebel, A.; Rybicki, B.A. Association of anxa11 genetic variation with sarcoidosis in african americans and european americans. Genes Immun. 2013, 14, 13–18. [Google Scholar] [CrossRef]
- Gazzerro, E.; Pereira, R.C.; Jorgetti, V.; Olson, S.; Economides, A.N.; Canalis, E. Skeletal overexpression of gremlin impairs bone formation and causes osteopenia. Endocrinology 2005, 146, 655–665. [Google Scholar] [CrossRef]
- Salez, F.; Gosset, P.; Copin, M.C.; Degros, C.L.; Tonnel, A.B.; Wallaert, B. Transforming growth factor-beta1 in sarcoidosis. Eur. Respir. J. 1998, 12, 913–919. [Google Scholar] [CrossRef]
- Heron, M.; van Moorsel, C.H.; Grutters, J.C.; Huizinga, T.W.; van der Helm-van Mil, A.H.; Nagtegaal, M.M.; Ruven, H.J.; van den Bosch, J.M. Genetic variation in grem1 is a risk factor for fibrosis in pulmonary sarcoidosis. Tissue Antigens 2011, 77, 112–117. [Google Scholar] [CrossRef]
- Kruit, A.; Grutters, J.C.; Ruven, H.J.; van Moorsel, C.H.; Weiskirchen, R.; Mengsteab, S.; van den Bosch, J.M. Transforming growth factor-beta gene polymorphisms in sarcoidosis patients with and without fibrosis. Chest 2006, 129, 1584–1591. [Google Scholar] [CrossRef]
- Sato, H.; Williams, H.R.; Spagnolo, P.; Abdallah, A.; Ahmad, T.; Orchard, T.R.; Copley, S.J.; Desai, S.R.; Wells, A.U.; Bois, R.M.D.; et al. Card15/nod2 polymorphisms are associated with severe pulmonary sarcoidosis. Eur. Respir. J. 2010, 35, 324–330. [Google Scholar] [CrossRef]
- Hill, M.R.; Papafili, A.; Booth, H.; Lawson, P.; Hubner, M.; Beynon, H.; Read, C.; Lindahl, G.; Marshall, R.P.; McAnulty, R.J.; et al. Functional prostaglandin-endoperoxide synthase 2 polymorphism predicts poor outcome in sarcoidosis. Am. J. Respir. Crit. Care Med. 2006, 174, 915–922. [Google Scholar] [CrossRef]
- Gupta, R.; Kim, J.S.; Baughman, R.P. An expert overview of pulmonary fibrosis in sarcoidosis. Expert Rev. Respir. Med. 2023, 17, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Lepzien, R.; Liu, S.; Czarnewski, P.; Nie, M.; Osterberg, B.; Baharom, F.; Pourazar, J.; Rankin, G.; Eklund, A.; Bottai, M.; et al. Monocytes in sarcoidosis are potent tumour necrosis factor producers and predict disease outcome. Eur. Respir. J. 2021, 58, 2003468. [Google Scholar] [CrossRef] [PubMed]
- Weeratunga, P.; Moller, D.R.; Ho, L.P. Immune mechanisms in fibrotic pulmonary sarcoidosis. Eur. Respir. Rev. 2022, 31, 220178. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, D.N.; Armstrong, M.E.; Trujillo, G.; Cooke, G.; Keane, M.P.; Fallon, P.G.; Simpson, A.J.; Millar, A.B.; McGrath, E.E.; Whyte, M.K.; et al. The toll-like receptor 3 l412f polymorphism and disease progression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2013, 188, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Prasse, A.; Probst, C.; Bargagli, E.; Zissel, G.; Toews, G.B.; Flaherty, K.R.; Olschewski, M.; Rottoli, P.; Muller-Quernheim, J. Serum cc-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2009, 179, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Pechkovsky, D.V.; Prasse, A.; Kollert, F.; Engel, K.M.; Dentler, J.; Luttmann, W.; Friedrich, K.; Muller-Quernheim, J.; Zissel, G. Alternatively activated alveolar macrophages in pulmonary fibrosis-mediator production and intracellular signal transduction. Clin. Immunol. 2010, 137, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, R.; Kozhaya, L.; McKevitt, K.; Djuretic, I.M.; Carlson, T.J.; Quintero, M.A.; McCauley, J.L.; Abreu, M.T.; Unutmaz, D.; Sundrud, M.S. Pro-inflammatory human th17 cells selectively express p-glycoprotein and are refractory to glucocorticoids. J. Exp. Med. 2014, 211, 89–104. [Google Scholar] [CrossRef]
- Broos, C.E.; Koth, L.L.; van Nimwegen, M.; In, J.; Paulissen, S.M.J.; van Hamburg, J.P.; Annema, J.T.; Heller-Baan, R.; Kleinjan, A.; Hoogsteden, H.C.; et al. Increased t-helper 17.1 cells in sarcoidosis mediastinal lymph nodes. Eur. Respir. J. 2018, 51, 1701124. [Google Scholar] [CrossRef]
- Sano, T.; Huang, W.; Hall, J.A.; Yang, Y.; Chen, A.; Gavzy, S.J.; Lee, J.Y.; Ziel, J.W.; Miraldi, E.R.; Domingos, A.I.; et al. An il-23r/il-22 circuit regulates epithelial serum amyloid a to promote local effector th17 responses. Cell 2015, 163, 381–393. [Google Scholar] [CrossRef]
- Cho, W.C.; Yip, T.T.; Cheng, W.W.; Au, J.S. Serum amyloid a is elevated in the serum of lung cancer patients with poor prognosis. Br. J. Cancer 2010, 102, 1731–1735. [Google Scholar] [CrossRef]
- Beijer, E.; Roodenburg-Benschop, C.; Schimmelpennink, M.C.; Grutters, J.C.; Meek, B.; Veltkamp, M. Elevated serum amyloid a levels are not specific for sarcoidosis but associate with a fibrotic pulmonary phenotype. Cells 2021, 10, 585. [Google Scholar] [CrossRef]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory t cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef]
- Miyara, M.; Amoura, Z.; Parizot, C.; Badoual, C.; Dorgham, K.; Trad, S.; Kambouchner, M.; Valeyre, D.; Chapelon-Abric, C.; Debre, P.; et al. The immune paradox of sarcoidosis and regulatory t cells. J. Exp. Med. 2006, 203, 359–370. [Google Scholar] [CrossRef]
- Oswald-Richter, K.A.; Richmond, B.W.; Braun, N.A.; Isom, J.; Abraham, S.; Taylor, T.R.; Drake, J.M.; Culver, D.A.; Wilkes, D.S.; Drake, W.P. Reversal of global cd4+ subset dysfunction is associated with spontaneous clinical resolution of pulmonary sarcoidosis. J. Immunol. 2013, 190, 5446–5453. [Google Scholar] [CrossRef]
- Sakthivel, P.; Grunewald, J.; Eklund, A.; Bruder, D.; Wahlstrom, J. Pulmonary sarcoidosis is associated with high-level inducible co-stimulator (icos) expression on lung regulatory t cells—Possible implications for the icos/icos-ligand axis in disease course and resolution. Clin. Exp. Immunol. 2016, 183, 294–306. [Google Scholar] [CrossRef]
- Zhang, H.; Jiang, D.; Zhu, L.; Zhou, G.; Xie, B.; Cui, Y.; Costabel, U.; Dai, H. Imbalanced distribution of regulatory t cells and th17.1 cells in the peripheral blood and balf of sarcoidosis patients: Relationship to disease activity and the fibrotic radiographic phenotype. Front. Immunol. 2023, 14, 1185443. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. Mtor signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Pacheco, Y.; CLim, X.; Weichhart, T.; Valeyre, D.; Bentaher, A.; Calender, A. Sarcoidosis and the mtor, rac1, and autophagy triad. Trends Immunol. 2020, 41, 286–299. [Google Scholar] [CrossRef]
- Vukmirovic, M.; Yan, X.; Gibson, K.F.; Gulati, M.; Schupp, J.C.; DeIuliis, G.; Adams, T.S.; Hu, B.; Mihaljinec, A.; Woolard, T.N.; et al. Transcriptomics of bronchoalveolar lavage cells identifies new molecular endotypes of sarcoidosis. Eur. Respir. J. 2021, 58, 2002950. [Google Scholar] [CrossRef]
- Linke, M.; Pham, H.T.; Katholnig, K.; Schnoller, T.; Miller, A.; Demel, F.; Schutz, B.; Rosner, M.; Kovacic, B.; Sukhbaatar, N.; et al. Chronic signaling via the metabolic checkpoint kinase mtorc1 induces macrophage granuloma formation and marks sarcoidosis progression. Nat. Immunol. 2017, 18, 293–302. [Google Scholar] [CrossRef]
- Jeny, F.; Bernaudin, J.F.; Valeyre, D.; Kambouchner, M.; Pretolani, M.; Nunes, H.; Planes, C.; Besnard, V. Hypoxia promotes a mixed inflammatory-fibrotic macrophages phenotype in active sarcoidosis. Front. Immunol. 2021, 12, 719009. [Google Scholar] [CrossRef]
- Baughman, R.P.; Teirstein, A.S.; Judson, M.A.; Rossman, M.D.; Yeager, H., Jr.; Bresnitz, E.A.; DePalo, L.; Hunninghake, G.; Iannuzzi, M.C.; Johns, C.J.; et al. Clinical characteristics of patients in a case control study of sarcoidosis. Am. J. Respir. Crit. Care Med. 2001, 164, 1885–1889. [Google Scholar] [CrossRef]
- Iannuzzi, M.C.; Rybicki, B.A.; Teirstein, A.S. Sarcoidosis. N. Engl. J. Med. 2007, 357, 2153–2165. [Google Scholar] [CrossRef]
- Lofgren, R.; Meholic, A.; Ketai, L. Fundamentals of Chest Radiology; WB Saunders: Philadelphia, PA, USA, 1996; pp. 120–121. [Google Scholar]
- Abehsera, M.; Valeyre, D.; Grenier, P.; Jaillet, H.; Battesti, J.P.; Brauner, M.W. Sarcoidosis with pulmonary fibrosis: Ct patterns and correlation with pulmonary function. AJR Am. J. Roentgenol. 2000, 174, 1751–1757. [Google Scholar] [CrossRef]
- Udwadia, Z.F.; Pilling, J.R.; Jenkins, P.F.; Harrison, B.D. Bronchoscopic and bronchographic findings in 12 patients with sarcoidosis and severe or progressive airways obstruction. Thorax 1990, 45, 272–275. [Google Scholar] [CrossRef]
- Criado, E.; Sanchez, M.; Ramirez, J.; Arguis, P.; de Caralt, T.M.; Perea, R.J.; Xaubet, A. Pulmonary sarcoidosis: Typical and atypical manifestations at high-resolution ct with pathologic correlation. Radiographics 2010, 30, 1567–1586. [Google Scholar] [CrossRef]
- Walsh, S.L.; Wells, A.U.; Sverzellati, N.; Keir, G.J.; Calandriello, L.; Antoniou, K.M.; Copley, S.J.; Devaraj, A.; Maher, T.M.; Renzoni, E.; et al. An integrated clinicoradiological staging system for pulmonary sarcoidosis: A case-cohort study. Lancet Respir. Med. 2014, 2, 123–130. [Google Scholar] [CrossRef]
- Regis, C.; Benali, K.; Rouzet, F. Fdg pet/ct imaging of sarcoidosis. Semin. Nucl. Med. 2023, 53, 258–272. [Google Scholar] [CrossRef]
- Keijsers, R.G.; Grutters, J.C.; Thomeer, M.; Bois, R.M.D.; Van Buul, M.M.; Lavalaye, J.; Van Den Bosch, J.M.; Verzijlbergen, F.J. Imaging the inflammatory activity of sarcoidosis: Sensitivity and inter observer agreement of 67ga imaging and 18f-fdg pet. Q. J. Nucl. Med. Mol. Imaging 2011, 55, 66–71. [Google Scholar]
- Braun, J.J.; Kessler, R.; Constantinesco, A.; Imperiale, A. 18f-fdg pet/ct in sarcoidosis management: Review and report of 20 cases. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 1537–1543. [Google Scholar] [CrossRef]
- Vender, R.J.; Aldahham, H.; Gupta, R. The role of pet in the management of sarcoidosis. Curr. Opin. Pulm. Med. 2022, 28, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Mostard, R.L.; Verschakelen, J.A.; van Kroonenburgh, M.J.; Nelemans, P.J.; Wijnen, P.A.; Voo, S.; Drent, M. Severity of pulmonary involvement and 18f-fdg pet activity in sarcoidosis. Respir. Med. 2013, 107, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, V.; Zompatori, M.; Fasano, L.; Nanni, C.; Nava, S.; Rubello, D.; Fanti, S. 18f-fdg pet/ct for the assessment of disease extension and activity in patients with sarcoidosis: Results of a preliminary prospective study. Clin. Nucl. Med. 2013, 38, e171–e177. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Jin, R.; Wang, Y.; Li, L.; Li, K.; He, Y. The utility of 18f-fdg pet/ct for monitoring response and predicting prognosis after glucocorticoids therapy for sarcoidosis. BioMed Res. Int. 2018, 2018, 1823710. [Google Scholar] [CrossRef] [PubMed]
- Maturu, V.N.; Rayamajhi, S.J.; Agarwal, R.; Aggarwal, A.N.; Gupta, D.; Mittal, B.R. Role of serial f-18 fdg pet/ct scans in assessing treatment response and predicting relapses in patients with symptomatic sarcoidosis. Sarcoidosis Vasc. Diffuse Lung Dis. 2016, 33, 372–380. [Google Scholar] [PubMed]
- Kouranos, V.; Ward, S.; Kokosi, M.A.; Castillo, D.; Chua, F.; Judge, E.P.; Thomas, S.; Van Tonder, F.; Devaraj, A.; Nicholson, A.G.; et al. Mixed ventilatory defects in pulmonary sarcoidosis: Prevalence and clinical features. Chest 2020, 158, 2007–2014. [Google Scholar] [CrossRef] [PubMed]
- Sharp, M.; Psoter, K.J.; Balasubramanian, A.; Pulapaka, A.V.; Chen, E.S.; Brown, S.W.; Mathai, S.C.; Gilotra, N.A.; Chrispin, J.; Bascom, R.; et al. Heterogeneity of lung function phenotypes in sarcoidosis: Role of race and sex differences. Ann. Am. Thorac. Soc. 2023, 20, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.P.; Sparkman, B.K.; Lower, E.E. Six-minute walk test and health status assessment in sarcoidosis. Chest 2007, 132, 207–213. [Google Scholar] [CrossRef]
- Gupta, R.; Baughman, R.P.; Nathan, S.D.; Wells, A.U.; Kouranos, V.; Alhamad, E.H.; Culver, D.A.; Barney, J.; Carmona, E.M.; Cordova, F.C.; et al. The six-minute walk test in sarcoidosis associated pulmonary hypertension: Results from an international registry. Respir. Med. 2022, 196, 106801. [Google Scholar] [CrossRef]
- Rosen, Y. Pathology of sarcoidosis. Semin. Respir. Crit. Care Med. 2007, 28, 36–52. [Google Scholar] [CrossRef]
- Mathur, R.S.; Kurdikar, V.L.; Shah, J.R. Serum angiotensin converting enzyme in the diagnosis of pulmonary sarcoidosis. J. Assoc. Physicians India 1990, 38, 474–475. [Google Scholar] [PubMed]
- Vorselaars, A.D.; van Moorsel, C.H.; Zanen, P.; Ruven, H.J.; Claessen, A.M.; van Velzen-Blad, H.; Grutters, J.C. Ace and sil-2r correlate with lung function improvement in sarcoidosis during methotrexate therapy. Respir. Med. 2015, 109, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Popevic, S.; Sumarac, Z.; Jovanovic, D.; Babic, D.; Stjepanovic, M.; Jovicic, S.; Sobic-Saranovic, D.; Filipovic, S.; Gvozdenovic, B.; Omcikus, M.; et al. Verifying sarcoidosis activity: Chitotriosidase versus ace in sarcoidosis—A case-control study. J. Med. Biochem. 2016, 35, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Xu, Y.; Zhu, H.; Zhang, H.; Sun, S.; Sun, H.; Wang, W.; Xie, S. Relationship between ct activity score with lung function and the serum angiotensin converting enzyme in pulmonary sarcoidosis on chest hrct. Medicine 2018, 97, e12205. [Google Scholar] [CrossRef] [PubMed]
- Hunninghake, G.W.; Bedell, G.N.; Zavala, D.C.; Monick, M.; Brady, M. Role of interleukin-2 release by lung t-cells in active pulmonary sarcoidosis. Am. Rev. Respir. Dis. 1983, 128, 634–638. [Google Scholar] [PubMed]
- Sweiss, N.J.; Barnathan, E.S.; Lo, K.; A Judson, M.; Baughman, R. C-reactive protein predicts response to infliximab in patients with chronic sarcoidosis. Sarcoidosis Vasc. Diffuse Lung Dis. 2010, 27, 49–56. [Google Scholar] [PubMed]
- McDonnell, M.J.; Saleem, M.I.; Wall, D.; Gilmartin, J.J.; Rutherford, R.M.; O’Regan, A. Predictive value of c-reactive protein and clinically relevant baseline variables in sarcoidosis. Sarcoidosis Vasc. Diffuse Lung Dis. 2016, 33, 331–340. [Google Scholar]
- Kirkil, G.; Lower, E.E.; Baughman, R.P. Predictors of mortality in pulmonary sarcoidosis. Chest 2018, 153, 105–113. [Google Scholar] [CrossRef]
- Baughman, R.P.; Gupta, R.; Judson, M.A.; Lower, E.E.; Birring, S.S.; Stewart, J.; Reeves, R.; Wells, A.U. Value of pulmonary function testing identifying progressive pulmonary disease in fibrotic sarcoidosis: Results of a prospective feasibility study. Sarcoidosis Vasc. Diffuse Lung Dis. 2022, 39, e2022011. [Google Scholar] [CrossRef]
- Obi, O.N.; Alqalyoobi, S.; Maddipati, V.; Lower, E.E.; Baughman, R.P. Hrct fibrotic patterns in stage 4 pulmonary sarcoidosis: Impact on pulmonary function and survival. Chest 2023, 23, S0012-3692(23)05666-0. [Google Scholar] [CrossRef]
- Nunes, H.; Uzunhan, Y.; Gille, T.; Lamberto, C.; Valeyre, D.; Brillet, P.Y. Imaging of sarcoidosis of the airways and lung parenchyma and correlation with lung function. Eur. Respir. J. 2012, 40, 750–765. [Google Scholar] [CrossRef] [PubMed]
- Rahaghi, F.F.; Baughman, R.P.; Saketkoo, L.A.; Sweiss, N.J.; Barney, J.B.; Birring, S.S.; Costabel, U.; Crouser, E.D.; Drent, M.; Gerke, A.K.; et al. Delphi consensus recommendations for a treatment algorithm in pulmonary sarcoidosis. Eur. Respir. Rev. 2020, 29, 190146. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.P.; Valeyre, D.; Korsten, P.; Mathioudakis, A.G.; Wuyts, W.A.; Wells, A.; Rottoli, P.; Nunes, H.; Lower, E.E.; Judson, M.A.; et al. Ers clinical practice guidelines on treatment of sarcoidosis. Eur. Respir. J. 2021, 58, 2004079. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.P.; Winget, D.B.; Lower, E.E. Methotrexate is steroid sparing in acute sarcoidosis: Results of a double blind, randomized trial. Sarcoidosis Vasc. Diffuse Lung Dis. 2000, 17, 60–66. [Google Scholar] [PubMed]
- Baughman, R.P.; Drent, M.; Kavuru, M.; Judson, M.A.; Costabel, U.; Bois, R.D.; Albera, C.; Brutsche, M.; Davis, G.; Donohue, J.F.; et al. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am. J. Respir. Crit. Care Med. 2006, 174, 795–802. [Google Scholar] [CrossRef]
- Rossman, M.D.; Newman, L.S.; Baughman, R.P.; Teirstein, A.; Weinberger, S.E.; Miller, W., Jr.; Sands, B.E. A double-blinded, randomized, placebo-controlled trial of infliximab in subjects with active pulmonary sarcoidosis. Sarcoidosis Vasc. Diffuse Lung Dis. 2006, 23, 201–208. [Google Scholar]
- Baughman, R.P.; Lower, E.E. Frequency of acute worsening events in fibrotic pulmonary sarcoidosis patients. Respir. Med. 2013, 107, 2009–2013. [Google Scholar] [CrossRef]
- Sawahata, M.; Shijubo, N.; Johkoh, T.; Hagiwara, K.; Konno, S.; Yamaguchi, T. Honeycomb lung-like structures resulting from clustering of traction bronchiectasis distally in sarcoidosis. Respirol. Case Rep. 2020, 8, e00539. [Google Scholar] [CrossRef]
- Lewis, M.M.; Mortelliti, M.P.; Yeager, H., Jr.; Tsou, E. Clinical bronchiectasis complicating pulmonary sarcoidosis: Case series of seven patients. Sarcoidosis Vasc. Diffuse Lung Dis. 2002, 19, 154–159. [Google Scholar]
- McKinzie, B.P.; Bullington, W.M.; Mazur, J.E.; Judson, M.A. Efficacy of short-course, low-dose corticosteroid therapy for acute pulmonary sarcoidosis exacerbations. Am. J. Med. Sci. 2010, 339, 1–4. [Google Scholar] [CrossRef]
- Panselinas, E.; Judson, M.A. Acute pulmonary exacerbations of sarcoidosis. Chest 2012, 142, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.P.; Judson, M.A.; Culver, D.A.; Birring, S.S.; Parambil, J.; Zeigler, J.; Lower, E.E. Roflumilast (daliresp(r)) to reduce acute pulmonary events in fibrotic sarcoidosis: A multi-center, double blind, placebo controlled, randomized clinical trial. Sarcoidosis Vasc. Diffuse Lung Dis. 2021, 38, e2021035. [Google Scholar] [CrossRef] [PubMed]
- Girard, C.; Hot, A.; Etienne-Mastroianni, B.; Chidiac, C.; Cordier, J.F. Opportunistic infections and sarcoidosis. Rev. Mal. Respir. 2004, 21, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Jamilloux, Y.; Maucort-Boulch, D.; Kerever, S.; Gerfaud-Valentin, M.; Broussolle, C.; Eb, M.; Valeyre, D.; Seve, P. Sarcoidosis-related mortality in france: A multiple-cause-of-death analysis. Eur. Respir. J. 2016, 48, 1700–1709. [Google Scholar] [CrossRef] [PubMed]
- Uzunhan, Y.; Nunes, H.; Jeny, F.; Lacroix, M.; Brun, S.; Brillet, P.Y.; Martinod, E.; Carette, M.F.; Bouvry, D.; Charlier, C.; et al. Chronic pulmonary aspergillosis complicating sarcoidosis. Eur. Respir. J. 2017, 49, 1602396. [Google Scholar] [CrossRef] [PubMed]
- Rafferty, P.; Biggs, B.A.; Crompton, G.K.; Grant, I.W. What happens to patients with pulmonary aspergilloma? Analysis of 23 cases. Thorax 1983, 38, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Shorr, A.F.; Helman, D.L.; Davies, D.B.; Nathan, S.D. Pulmonary hypertension in advanced sarcoidosis: Epidemiology and clinical characteristics. Eur. Respir. J. 2005, 25, 783–788. [Google Scholar] [CrossRef]
- Shorr, A.F.; Davies, D.B.; Nathan, S.D. Predicting mortality in patients with sarcoidosis awaiting lung transplantation. Chest 2003, 124, 922–928. [Google Scholar] [CrossRef]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.F.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in progressive fibrosing interstitial lung diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef]
- Behr, J.; Prasse, A.; Kreuter, M.; Johow, J.; Rabe, K.F.; Bonella, F.; Bonnet, R.; Grohe, C.; Held, M.; Wilkens, H.; et al. Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (relief): A double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2021, 9, 476–486. [Google Scholar] [CrossRef]
- Holland, A.E.; Cox, N.S.; Houchen-Wolloff, L.; Rochester, C.L.; Garvey, C.; ZuWallack, R.; Nici, L.; Limberg, T.; Lareau, S.C.; Yawn, B.P.; et al. Defining modern pulmonary rehabilitation. An official american thoracic society workshop report. Ann. Am. Thorac. Soc. 2021, 18, e12–e29. [Google Scholar] [CrossRef] [PubMed]
- Puhan, M.A.; Gimeno-Santos, E.; Cates, C.J.; Troosters, T. Pulmonary rehabilitation following exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst. Rev. 2016, 12, CD005305. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, B.; Casey, D.; Devane, D.; Murphy, K.; Murphy, E.; Lacasse, Y. Pulmonary rehabilitation for chronic obstructive pulmonary disease. Cochrane Database Syst. Rev. 2015, 2015, CD003793. [Google Scholar] [CrossRef] [PubMed]
- Hudson, L.D.; Tyler, M.L.; Petty, T.L. Hospitalization needs during an outpatient rehabilitation program for severe chronic airway obstruction. Chest 1976, 70, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Seymour, J.M.; Moore, L.; Jolley, C.J.; Ward, K.; Creasey, J.; Steier, J.S.; Yung, B.; Man, W.D.; Hart, N.; Polkey, M.I.; et al. Outpatient pulmonary rehabilitation following acute exacerbations of copd. Thorax 2010, 65, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Morisset, J.; Dube, B.P.; Garvey, C.; Bourbeau, J.; Collard, H.R.; Swigris, J.J.; Lee, J.S. The unmet educational needs of patients with interstitial lung disease. Setting the stage for tailored pulmonary rehabilitation. Ann. Am. Thorac. Soc. 2016, 13, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Morris, N.R.; Kermeen, F.D.; Holland, A.E. Exercise-based rehabilitation programmes for pulmonary hypertension. Cochrane Database Syst. Rev. 2017, 1, CD011285. [Google Scholar] [CrossRef]
- Li, M.; Mathur, S.; Chowdhury, N.A.; Helm, D.; Singer, L.G. Pulmonary rehabilitation in lung transplant candidates. J. Heart Lung Transplant. 2013, 32, 626–632. [Google Scholar] [CrossRef]
- Strookappe, B.; Swigris, J.; De Vries, J.; Elfferich, M.; Knevel, T.; Drent, M. Benefits of physical training in sarcoidosis. Lung 2015, 193, 701–708. [Google Scholar] [CrossRef]
- Alsina-Restoy, X.; Torres-Castro, R.; Caballeria, E.; Gimeno-Santos, E.; Solis-Navarro, L.; Francesqui, J.; Hernandez-Gonzalez, F.; Ramos-Casals, M.; Blanco, I.; Sellares, J. Pulmonary rehabilitation in sarcoidosis: A systematic review and meta-analysis. Respir. Med. 2023, 219, 107432. [Google Scholar] [CrossRef]
- Syed, H.; Ascoli, C.; Linssen, C.F.; Vagts, C.; Iden, T.; Syed, A.; Kron, J.; Polly, K.; Perkins, D.; Finn, P.W.; et al. Infection prevention in sarcoidosis: Proposal for vaccination and prophylactic therapy. Sarcoidosis Vasc. Diffuse Lung Dis. 2020, 37, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Haraoui, B.; Kumar, D.; Marshall, J.K.; Bissonnette, R.; Bitton, A.; Bressler, B.; Gooderham, M.; Ho, V.; Jamal, S.; et al. Vaccination guidelines for patients with immune-mediated disorders on immunosuppressive therapies. J. Cutan. Med. Surg. 2019, 23, 50–74. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.S.; Krishnan, J.A.; Lederer, D.J.; Ghazipura, M.; Hossain, T.; Tan, A.M.; Carlin, B.; Drummond, M.B.; Ekstrom, M.; Garvey, C.; et al. Home oxygen therapy for adults with chronic lung disease. An official american thoracic society clinical practice guideline. Am. J. Respir. Crit. Care Med. 2020, 202, e121–e141. [Google Scholar] [CrossRef] [PubMed]
- Valapour, M.; Lehr, C.J.; Skeans, M.A.; Smith, J.M.; Uccellini, K.; Lehman, R.; Robinson, A.; Israni, A.K.; Snyder, J.J.; Kasiske, B.L. Optn/srtr 2017 annual data report: Lung. Am. J. Transplant. 2019, 2, 404–484. [Google Scholar] [CrossRef] [PubMed]
- Yusen, R.D.; Edwards, L.B.; Kucheryavaya, A.Y.; Benden, C.; Dipchand, A.I.; Goldfarb, S.B.; Levvey, B.J.; Lund, L.H.; Meiser, B.; Rossano, J.W.; et al. The registry of the international society for heart and lung transplantation: Thirty-second official adult lung and heart-lung transplantation report—2015; focus theme: Early graft failure. J. Heart Lung Transplant. 2015, 34, 1264–1277. [Google Scholar] [CrossRef] [PubMed]
- Weill, D.; Benden, C.; Corris, P.A.; Dark, J.H.; Davis, R.D.; Keshavjee, S.; Lederer, D.J.; Mulligan, M.J.; Patterson, G.A.; Singer, L.G.; et al. A consensus document for the selection of lung transplant candidates: 2014—An update from the pulmonary transplantation council of the international society for heart and lung transplantation. J. Heart Lung Transplant. 2015, 34, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Gupta, R. Lung transplantation in pulmonary sarcoidosis. J. Autoimmun. 2023, 103135. [Google Scholar] [CrossRef]
- Egan, T.M.; Edwards, L.B. Effect of the lung allocation score on lung transplantation in the united states. J. Heart Lung Transplant. 2016, 35, 433–439. [Google Scholar] [CrossRef]
- Fleitas Sosa, D.C.; Gayen, S.; Zheng, M.; Gangemi, A.J.; Zhao, H.; Kim, V.; Sehgal, S.; Criner, G.J.; Gupta, R.; Mamary, A.J. Sarcoidosis lung transplantation waitlist mortality, a national registry database study. ERJ Open Res 2023, 9, 00738–2022. [Google Scholar] [CrossRef]
- Gayen, S.; Sosa, D.F.; Zheng, M.; Gangemi, A.; Sehgal, S.; Zhao, H.; Marchetti, N.; Criner, G.J.; Gupta, R.; Mamary, A.J. Lung transplantation waitlist mortality among sarcoidosis patients by lung allocation score grouping. Transplant. Proc. 2023, 55, 440–445. [Google Scholar] [CrossRef]
- Gangemi, A.J.; Myers, C.N.; Zheng, M.; Brown, J.; Butler-LeBair, M.; Cordova, F.; Marchetti, N.; Criner, G.J.; Gupta, R.; Mamary, A.J. Mortality for sarcoidosis patients on the transplant wait list in the lung allocation score era: Experience from a high volume center. Respir. Med. 2019, 157, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Le Pavec, J.; Valeyre, D.; Gazengel, P.; Holm, A.M.; Schultz, H.H.; Perch, M.; Le Borgne, A.; Reynaud-Gaubert, M.; Knoop, C.; Godinas, L.; et al. Lung transplantation for sarcoidosis: Outcome and prognostic factors. Eur. Respir. J. 2021, 58, 2003358. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.C. Lung transplantation for pulmonary sarcoidosis. Sarcoidosis Vasc. Diffuse Lung Dis. 2019, 36, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Shorr, A.F.; Helman, D.L.; Davies, D.B.; Nathan, S.D. Sarcoidosis, race, and short-term outcomes following lung transplantation. Chest 2004, 125, 990–996. [Google Scholar] [CrossRef]
- Taimeh, Z.; Hertz, M.I.; Shumway, S.; Pritzker, M. Lung transplantation for pulmonary sarcoidosis. Twenty-five years of experience in the USA. Thorax 2016, 71, 378–379. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.S.; Gupta, R. Clinical Manifestations and Management of Fibrotic Pulmonary Sarcoidosis. J. Clin. Med. 2024, 13, 241. https://doi.org/10.3390/jcm13010241
Kim JS, Gupta R. Clinical Manifestations and Management of Fibrotic Pulmonary Sarcoidosis. Journal of Clinical Medicine. 2024; 13(1):241. https://doi.org/10.3390/jcm13010241
Chicago/Turabian StyleKim, Jin Sun, and Rohit Gupta. 2024. "Clinical Manifestations and Management of Fibrotic Pulmonary Sarcoidosis" Journal of Clinical Medicine 13, no. 1: 241. https://doi.org/10.3390/jcm13010241
APA StyleKim, J. S., & Gupta, R. (2024). Clinical Manifestations and Management of Fibrotic Pulmonary Sarcoidosis. Journal of Clinical Medicine, 13(1), 241. https://doi.org/10.3390/jcm13010241