
Advances and Applications of Lung Organoids in the Research on Acute Respiratory Distress Syndrome (ARDS)


Abstract
:1. Basis and Progress in Acute Respiratory Distress Syndrome
1.1. Introduction
1.2. Epithelium Injury in ARDS
1.3. Endothelium Injury in ARDS
1.4. Mechanical Injury in ARDS
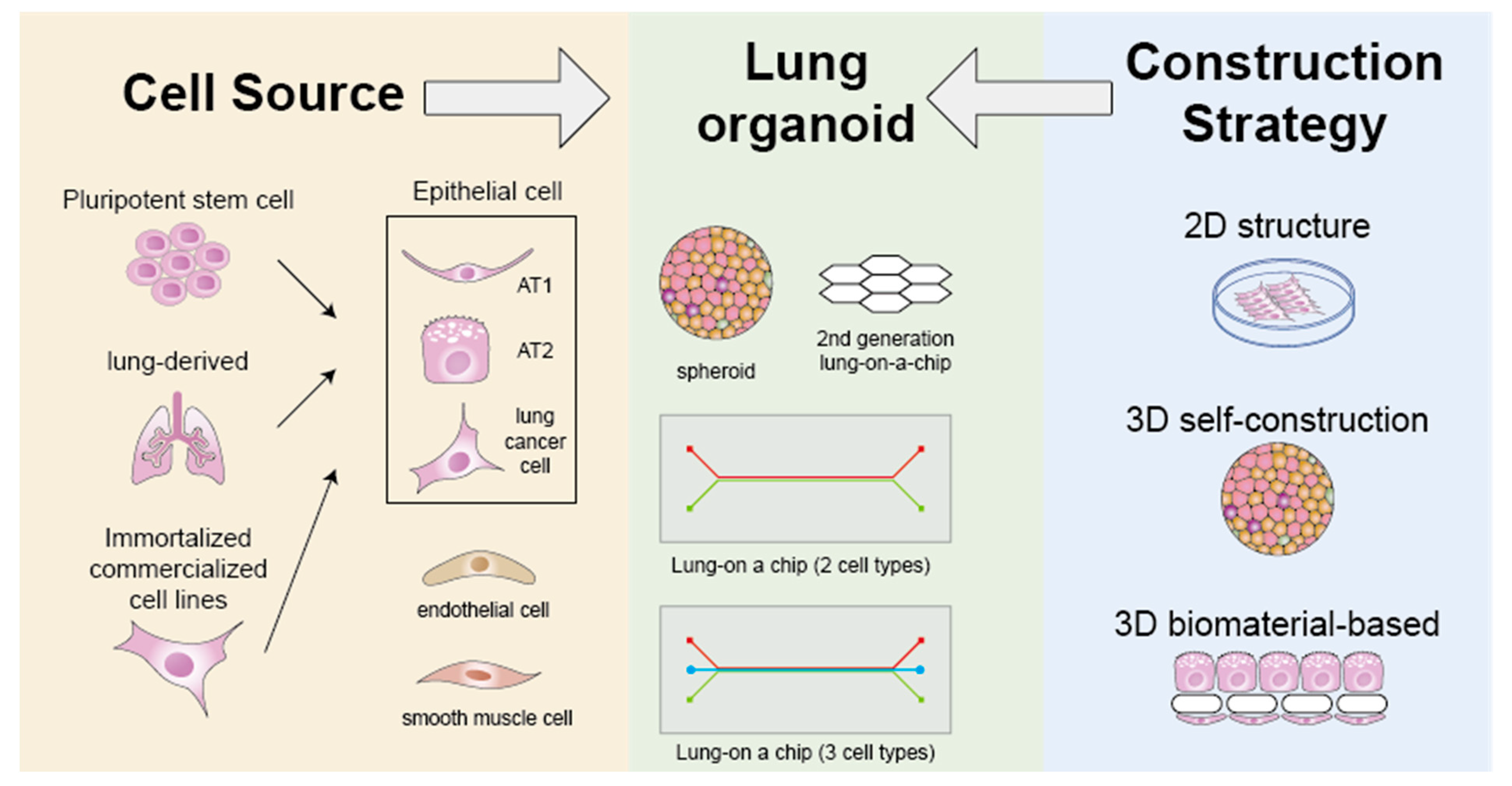
2. Advances in Lung Organoids
2.1. Cell Source and Type
2.2. Construction Strategy
3. Prospects for Future Research
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute respiratory distress syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar] [CrossRef] [PubMed]
- Bellani, G.; Pesenti, A.; Laffey, J.G.; Slutsky, A.S.; Pham, T.; Fan, E.; Wrigge, H.; Brochard, L.; Esteban, A.; Gattinoni, L.; et al. Epidemiology, Patterns of Care, and Mortality for Patients with Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ma, X. Acute respiratory failure in COVID-19: Is it “typical” ARDS? Crit. Care 2020, 24, 198. [Google Scholar] [CrossRef] [PubMed]
- Bain, W.; Yang, H.; Shah, F.A.; Suber, T.; Drohan, C.; Al-Yousif, N.; DeSensi, R.S.; Bensen, N.; Schaefer, C.; Rosborough, B.R.; et al. COVID-19 versus Non-COVID-19 Acute Respiratory Distress Syndrome: Comparison of Demographics, Physiologic Parameters, Inflammatory Biomarkers, and Clinical Outcomes. Ann. Am. Thorac. Soc. 2021, 18, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Bos, L.D.J.; Ware, L.B. Acute respiratory distress syndrome: Causes, pathophysiology, and phenotypes. Lancet 2022, 400, 1145–1156. [Google Scholar] [CrossRef]
- Luo, L.; Shaver, C.M.; Zhao, Z.; Koyama, T.; Calfee, C.S.; Bastarache, J.A.; Ware, L.B. Clinical Predictors of Hospital Mortality Differ between Direct and Indirect ARDS. Chest 2017, 151, 755–763. [Google Scholar] [CrossRef]
- Calfee, C.S.; Janz, D.R.; Bernard, G.R.; May, A.K.; Kangelaris, K.N.; Matthay, M.A.; Ware, L.B. Distinct molecular phenotypes of direct vs indirect ARDS in single-center and multicenter studies. Chest 2015, 147, 1539–1548. [Google Scholar] [CrossRef]
- Liaqat, A.; Mason, M.; Foster, B.J.; Kulkarni, S.; Barlas, A.; Farooq, A.M.; Patak, P.; Liaqat, H.; Basso, R.G.; Zaman, M.S.; et al. Evidence-Based Mechanical Ventilatory Strategies in ARDS. J. Clin. Med. 2022, 11, 319. [Google Scholar] [CrossRef]
- Mercer, B.A.; Lemaître, V.; Powell, C.A.; D’Armiento, J. The Epithelial Cell in Lung Health and Emphysema Pathogenesis. Curr. Respir. Med. Rev. 2006, 2, 101–142. [Google Scholar] [CrossRef]
- Crapo, J.D.; Barry, B.E.; Gehr, P.; Bachofen, M.; Weibel, E.R. Cell number and cell characteristics of the normal human lung. Am. Rev. Respir. Dis. 1982, 126, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, Z.; Huang, H.; Li, J.; Wang, Z.; Yu, Y.; Zhang, C.; Li, J.; Dai, H.; Wang, F.; et al. Pulmonary alveolar type I cell population consists of two distinct subtypes that differ in cell fate. Proc. Natl. Acad. Sci. USA 2018, 115, 2407–2412. [Google Scholar] [CrossRef]
- Wang, S.; Hubmayr, R.D. Type I alveolar epithelial phenotype in primary culture. Am. J. Respir. Cell Mol. Biol. 2011, 44, 692–699. [Google Scholar] [CrossRef]
- Kasper, M.; Barth, K. Potential contribution of alveolar epithelial type I cells to pulmonary fibrosis. Biosci. Rep. 2017, 37, BSR20171301. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ferrari, J.D.; Cao, Y.; Ramirez, M.I.; Jones, M.R.; Quinton, L.J.; Mizgerd, J.P. Type I alveolar epithelial cells mount innate immune responses during pneumococcal pneumonia. J. Immunol. 2012, 189, 2450–2459. [Google Scholar] [CrossRef]
- Lin, W.C.; Gowdy, K.M.; Madenspacher, J.H.; Zemans, R.L.; Yamamoto, K.; Lyons-Cohen, M.; Nakano, H.; Janardhan, K.; Williams, C.J.; Cook, D.N.; et al. Epithelial membrane protein 2 governs transepithelial migration of neutrophils into the airspace. J. Clin. Investig. 2020, 130, 157–170. [Google Scholar] [CrossRef]
- Matthay, M.A.; Folkesson, H.G.; Clerici, C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol. Rev. 2002, 82, 569–600. [Google Scholar] [CrossRef]
- Dobbs, L.G.; Johnson, M.D. Alveolar epithelial transport in the adult lung. Respir. Physiol. Neurobiol. 2007, 159, 283–300. [Google Scholar] [CrossRef]
- Ikegami, M.; Falcone, A.; Whitsett, J.A. STAT-3 regulates surfactant phospholipid homeostasis in normal lung and during endotoxin-mediated lung injury. J. Appl. Physiol. 2008, 104, 1753–1760. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.Y.; Veldhuizen, R.A.; Neumann, A.W.; Petersen, N.O.; Possmayer, F. Current perspectives in pulmonary surfactant--inhibition, enhancement and evaluation. Biochim. Biophys. Acta 2008, 1778, 1947–1977. [Google Scholar] [CrossRef] [PubMed]
- Woods, P.S.; Doolittle, L.M.; Rosas, L.E.; Joseph, L.M.; Calomeni, E.P.; Davis, I.C. Lethal H1N1 influenza A virus infection alters the murine alveolar type II cell surfactant lipidome. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L1160–L1169. [Google Scholar] [CrossRef] [PubMed]
- Peteranderl, C.; Morales-Nebreda, L.; Selvakumar, B.; Lecuona, E.; Vadász, I.; Morty, R.E.; Schmoldt, C.; Bespalowa, J.; Wolff, T.; Pleschka, S.; et al. Macrophage-epithelial paracrine crosstalk inhibits lung edema clearance during influenza infection. J. Clin. Investig. 2016, 126, 1566–1580. [Google Scholar] [CrossRef] [PubMed]
- Kryvenko, V.; Wessendorf, M.; Tello, K.; Herold, S.; Morty, R.E.; Seeger, W.; Vadász, I. Hypercapnia Induces Inositol-Requiring Enzyme 1α-Driven Endoplasmic Reticulum-associated Degradation of the Na,K-ATPase β-Subunit. Am. J. Respir. Cell Mol. Biol. 2021, 65, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Brand, J.D.; Lazrak, A.; Trombley, J.E.; Shei, R.-J.; Adewale, A.T.; Tipper, J.L.; Yu, Z.; Ashtekar, A.R.; Rowe, S.M.; Matalon, S.; et al. Influenza-mediated reduction of lung epithelial ion channel activity leads to dysregulated pulmonary fluid homeostasis. JCI Insight 2018, 3, e123467. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Kasper, J.; van der Aa, S.; Andeweg, A.C.; Zaaraoui-Boutahar, F.; Goeijenbier, M.; Richard, M.; Herold, S.; Becker, C.; Scott, D.P.; et al. Influenza virus damages the alveolar barrier by disrupting epithelial cell tight junctions. Eur. Respir. J. 2016, 47, 954–966. [Google Scholar] [CrossRef] [PubMed]
- Ruan, T.; Sun, J.; Liu, W.; Prinz, R.A.; Peng, D.; Liu, X.; Xu, X. H1N1 Influenza Virus Cross-Activates Gli1 to Disrupt the Intercellular Junctions of Alveolar Epithelial Cells. Cell Rep. 2020, 31, 107801. [Google Scholar] [CrossRef] [PubMed]
- Tolle, L.B.; Standiford, T.J. Danger-associated molecular patterns (DAMPs) in acute lung injury. J. Pathol. 2013, 229, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, A.C.C.; Flori, H.; Dahmer, M.K.; Sim, M.S.; Quasney, M.W.; Curley, M.A.Q.; Matthay, M.A.; Sapru, A. Thrombomodulin is associated with increased mortality and organ failure in mechanically ventilated children with acute respiratory failure: Biomarker analysis from a multicenter randomized controlled trial. Crit. Care 2021, 25, 271. [Google Scholar] [CrossRef]
- Ware, L.B.; Fang, X.; Matthay, M.A. Protein C and thrombomodulin in human acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 285, L514–L521. [Google Scholar] [CrossRef]
- Wang, L.; Bastarache, J.A.; Wickersham, N.; Fang, X.; Matthay, M.A.; Ware, L.B. Novel role of the human alveolar epithelium in regulating intra-alveolar coagulation. Am. J. Respir. Cell Mol. Biol. 2007, 36, 497–503. [Google Scholar] [CrossRef]
- Bastarache, J.A.; Wang, L.; Wang, Z.; Albertine, K.H.; Matthay, M.A.; Ware, L.B. Intra-alveolar tissue factor pathway inhibitor is not sufficient to block tissue factor procoagulant activity. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L874–L881. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Chen, Y.; Chen, M.; Li, J.; Zhang, H.; Yan, S.; Lv, C. Advanced development and mechanism of sepsis-related acute respiratory distress syndrome. Front. Med. 2022, 9, 1043859. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.C.; Wu, Y.K.; Yang, M.C.; Su, W.L.; Kuo, C.Y.; Lan, C.C. Deciphering the role of damage-associated molecular patterns and inflammatory responses in acute lung injury. Life Sci. 2022, 305, 120782. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.C.; Fessler, M.B. Regulatory mechanisms of neutrophil migration from the circulation to the airspace. Cell Mol. Life Sci. 2021, 78, 4095–4124. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Qian, Y.; Li, Z.; Fan, E.K.; Li, Y.; Wu, L.; Billiar, T.R.; Wilson, M.A.; Shi, X.; Fan, J. TLR4-Upregulated IL-1β and IL-1RI Promote Alveolar Macrophage Pyroptosis and Lung Inflammation through an Autocrine Mechanism. Sci. Rep. 2016, 6, 31663. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.P.; Yang, Y.; Janssen, W.J.; Gandjeva, A.; Perez, M.J.; Barthel, L.; Zemans, R.L.; Bowman, J.C.; Koyanagi, D.E.; Yunt, Z.X.; et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat. Med. 2012, 18, 1217–1223. [Google Scholar] [CrossRef]
- Lefrançais, E.; Mallavia, B.; Zhuo, H.; Calfee, C.S.; Looney, M.R. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight 2018, 3, e98178. [Google Scholar] [CrossRef]
- Shaver, C.M.; Wickersham, N.; McNeil, J.B.; Nagata, H.; Miller, A.; Landstreet, S.R.; Kuck, J.L.; Diamond, J.M.; Lederer, D.J.; Kawut, S.M.; et al. Cell-free hemoglobin promotes primary graft dysfunction through oxidative lung endothelial injury. JCI Insight 2018, 3, e98546. [Google Scholar] [CrossRef]
- Hough, R.F.; Islam, M.N.; Gusarova, G.A.; Jin, G.; Das, S.; Bhattacharya, J. Endothelial mitochondria determine rapid barrier failure in chemical lung injury. JCI Insight 2019, 4, e124329. [Google Scholar] [CrossRef]
- Gajic, O.; Dara, S.I.; Mendez, J.L.; Adesanya, A.O.; Festic, E.; Caples, S.M.; Rana, R.; St Sauver, J.L.; Lymp, J.F.; Afessa, B.; et al. Ventilator-associated lung injury in patients without acute lung injury at the onset of mechanical ventilation. Crit. Care Med. 2004, 32, 1817–1824. [Google Scholar] [CrossRef]
- Dreyfuss, D.; Soler, P.; Basset, G.; Saumon, G. High inflation pressure pulmonary edema. Respective effects of high airway pressure, high tidal volume, and positive end-expiratory pressure. Am. Rev. Respir. Dis. 1988, 137, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Ashbaugh, D.G.; Bigelow, D.B.; Petty, T.L.; Levine, B.E. Acute respiratory distress in adults. Lancet 1967, 2, 319–323. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, C.C.; Okutani, D.; Hu, P.; Han, B.; Crimi, E.; He, X.; Keshavjee, S.; Greenwood, C.; Slutsky, A.S.; Zhang, H.; et al. Differential gene profiling in acute lung injury identifies injury-specific gene expression. Crit. Care Med. 2008, 36, 855–865. [Google Scholar] [CrossRef]
- dos Santos, C.C.; Slutsky, A.S. Mechanotransduction, ventilator-induced lung injury and multiple organ dysfunction syndrome. Intensive Care Med. 2000, 26, 638–642. [Google Scholar] [CrossRef]
- Parsons, P.E.; Eisner, M.D.; Thompson, B.T.; Matthay, M.A.; Ancukiewicz, M.; Bernard, G.R.; Wheeler, A.P. Lower tidal volume ventilation and plasma cytokine markers of inflammation in patients with acute lung injury. Crit. Care Med. 2005, 33, 1–6; discussion 230–232. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, V.M.; Suter, P.M.; Tortorella, C.; De Tullio, R.; Dayer, J.M.; Brienza, A.; Bruno, F.; Slutsky, A.S. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: A randomized controlled trial. Jama 1999, 282, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Gross, C.; Desai, A.A.; Zemskov, E.; Wu, X.; Garcia, A.N.; Jacobson, J.R.; Yuan, J.X.; Garcia, J.G.; Black, S.M. Endothelial cell signaling and ventilator-induced lung injury: Molecular mechanisms, genomic analyses, and therapeutic targets. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L452–L476. [Google Scholar] [CrossRef] [PubMed]
- Woods, S.J.; Waite, A.A.; O’Dea, K.P.; Halford, P.; Takata, M.; Wilson, M.R. Kinetic profiling of in vivo lung cellular inflammatory responses to mechanical ventilation. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L912–L921. [Google Scholar] [CrossRef]
- Lin, C.K.; Huang, T.H.; Yang, C.T.; Shi, C.S. Roles of lung-recruited monocytes and pulmonary Vascular Endothelial Growth Factor (VEGF) in resolving Ventilator-Induced Lung Injury (VILI). PLoS ONE 2021, 16, e0248959. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.J.; Xu, D.F.; Zhang, H.; Xu, C.F.; Mao, Y.F.; Lv, Z.; Zhu, X.Y.; Jiang, L. DRD1 downregulation contributes to mechanical stretch-induced lung endothelial barrier dysfunction. Theranostics 2021, 11, 2505–2521. [Google Scholar] [CrossRef]
- Jaecklin, T.; Otulakowski, G.; Kavanagh, B.P. Do soluble mediators cause ventilator-induced lung injury and multi-organ failure? Intensive Care Med. 2010, 36, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Han, Y.; Yang, L.; Lacko, L.A.; Chen, S. Human organoid models to study SARS-CoV-2 infection. Nat. Methods 2022, 19, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Molnar, T.F.; Pongracz, J.E. Tissue engineering and biotechnology in general thoracic surgery. Eur. J. Cardiothorac. Surg. 2010, 37, 1402–1410. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, Y.; Li, D.; Feng, Y.; Xie, Y.; Zhou, Y.; Zhou, M.; Wang, Y.; Qu, J.; Zuo, W. Intrapulmonary distal airway stem cell transplantation repairs lung injury in chronic obstructive pulmonary disease. Cell Prolif. 2021, 54, e13046. [Google Scholar] [CrossRef] [PubMed]
- Green, M.D.; Chen, A.; Nostro, M.C.; d’Souza, S.L.; Schaniel, C.; Lemischka, I.R.; Gouon-Evans, V.; Keller, G.; Snoeck, H.W. Generation of anterior foregut endoderm from human embryonic and induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.X.; Islam, M.N.; O’Neill, J.; Hu, Z.; Yang, Y.G.; Chen, Y.W.; Mumau, M.; Green, M.D.; Vunjak-Novakovic, G.; Bhattacharya, J.; et al. Efficient generation of lung and airway epithelial cells from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 84–91. [Google Scholar] [CrossRef]
- Jacob, A.; Vedaie, M.; Roberts, D.A.; Thomas, D.C.; Villacorta-Martin, C.; Alysandratos, K.D.; Hawkins, F.; Kotton, D.N. Derivation of self-renewing lung alveolar epithelial type II cells from human pluripotent stem cells. Nat. Protoc. 2019, 14, 3303–3332. [Google Scholar] [CrossRef]
- Zhao, L.; Yee, M.; O’Reilly, M.A. Transdifferentiation of alveolar epithelial type II to type I cells is controlled by opposing TGF-β and BMP signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, L409–L418. [Google Scholar] [CrossRef]
- Riemondy, K.A.; Jansing, N.L.; Jiang, P.; Redente, E.F.; Gillen, A.E.; Fu, R.; Miller, A.J.; Spence, J.R.; Gerber, A.N.; Hesselberth, J.R.; et al. Single cell RNA sequencing identifies TGF-β as a key regenerative cue following LPS-induced lung injury. JCI Insight 2019, 5, e123637. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wu, H.; Jiang, K.; Wang, Y.; Zhang, W.; Chu, Q.; Li, J.; Huang, H.; Cai, T.; Ji, H.; et al. MAPK-Mediated YAP Activation Controls Mechanical-Tension-Induced Pulmonary Alveolar Regeneration. Cell Rep. 2016, 16, 1810–1819. [Google Scholar] [CrossRef] [PubMed]
- Nabhan, A.N.; Brownfield, D.G.; Harbury, P.B.; Krasnow, M.A.; Desai, T.J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 2018, 359, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.I.; Bujnis, M.; Barkauskas, C.E.; Kobayashi, Y.; Hogan, B.L.M. Niche-mediated BMP/SMAD signaling regulates lung alveolar stem cell proliferation and differentiation. Development 2018, 145, dev163014. [Google Scholar] [CrossRef] [PubMed]
- Little, D.R.; Gerner-Mauro, K.N.; Flodby, P.; Crandall, E.D.; Borok, Z.; Akiyama, H.; Kimura, S.; Ostrin, E.J.; Chen, J. Transcriptional control of lung alveolar type 1 cell development and maintenance by NK homeobox 2-1. Proc. Natl. Acad. Sci. USA 2019, 116, 20545–20555. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Huang, Z.; Zhang, H.; Posner, C.; Jia, G.; Ramalingam, T.R.; Xu, M.; Brightbill, H.; Egen, J.G.; Dey, A.; et al. TAZ is required for lung alveolar epithelial cell differentiation after injury. JCI Insight 2019, 4, e128674. [Google Scholar] [CrossRef]
- Katsura, H.; Sontake, V.; Tata, A.; Kobayashi, Y.; Edwards, C.E.; Heaton, B.E.; Konkimalla, A.; Asakura, T.; Mikami, Y.; Fritch, E.J.; et al. Human Lung Stem Cell-Based Alveolospheres Provide Insights into SARS-CoV-2-Mediated Interferon Responses and Pneumocyte Dysfunction. Cell Stem Cell 2020, 27, 890–904. [Google Scholar] [CrossRef]
- Kanagaki, S.; Ikeo, S.; Suezawa, T.; Yamamoto, Y.; Seki, M.; Hirai, T.; Hagiwara, M.; Suzuki, Y.; Gotoh, S. Directed induction of alveolar type I cells derived from pluripotent stem cells via Wnt signaling inhibition. Stem Cells 2021, 39, 156–169. [Google Scholar] [CrossRef]
- Burgess, C.L.; Huang, J.; Bawa, P.; Alysandratos, K.D.; Minakin, K.; Morley, M.P.; Babu, A.; Villacorta-Martin, C.; Hinds, A.; Thapa, B.R.; et al. Generation of human alveolar epithelial type I cells from pluripotent stem cells. bioRxiv 2023. [Google Scholar] [CrossRef]
- Patsch, C.; Challet-Meylan, L.; Thoma, E.C.; Urich, E.; Heckel, T.; O’Sullivan, J.F.; Grainger, S.J.; Kapp, F.G.; Sun, L.; Christensen, K.; et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat. Cell Biol. 2015, 17, 994–1003. [Google Scholar] [CrossRef]
- Zhang, F.; Zhu, Y.; Chen, J.; Kuang, W.; Huang, R.; Duan, F.; Li, Y.; Wang, L.; Qiu, H.; Chen, X.; et al. Efficient endothelial and smooth muscle cell differentiation from human pluripotent stem cells through a simplified insulin-free culture system. Biomaterials 2021, 271, 120713. [Google Scholar] [CrossRef]
- Luo, J.; Shi, X.; Lin, Y.; Yuan, Y.; Kural, M.H.; Wang, J.; Ellis, M.W.; Anderson, C.W.; Zhang, S.M.; Riaz, M.; et al. Efficient Differentiation of Human Induced Pluripotent Stem Cells into Endothelial Cells under Xenogeneic-free Conditions for Vascular Tissue Engineering. Acta Biomater. 2021, 119, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, W.J.; Frank, D.B.; Zepp, J.A.; Morley, M.P.; Alkhaleel, F.A.; Kong, J.; Zhou, S.; Cantu, E.; Morrisey, E.E. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 2018, 555, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.J.; Williams, M.C. Type II alveolar cell. Defender of the alveolus. Am. Rev. Respir. Dis. 1977, 115, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J.; et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020, 587, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Adams, T.S.; Marlier, A.; Kaminski, N. Lung Cell Atlases in Health and Disease. Annu. Rev. Physiol. 2023, 85, 47–69. [Google Scholar] [CrossRef] [PubMed]
- Melms, J.C.; Biermann, J.; Huang, H.; Wang, Y.; Nair, A.; Tagore, S.; Katsyv, I.; Rendeiro, A.F.; Amin, A.D.; Schapiro, D.; et al. A molecular single-cell lung atlas of lethal COVID-19. Nature 2021, 595, 114–119. [Google Scholar] [CrossRef]
- Bosáková, V.; De Zuani, M.; Sládková, L.; Garlíková, Z.; Jose, S.S.; Zelante, T.; Hortová Kohoutková, M.; Frič, J. Lung Organoids-The Ultimate Tool to Dissect Pulmonary Diseases? Front. Cell Dev. Biol. 2022, 10, 899368. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Huang, Z.; Tang, Z.; Chen, Y.; Huang, M.; Liu, H.; Huang, W.; Ye, Q.; Jia, B. Research Progress, Challenges, and Breakthroughs of Organoids as Disease Models. Front. Cell Dev. Biol. 2021, 9, 740574. [Google Scholar] [CrossRef]
- Reutershan, J.; Ley, K. Bench-to-bedside review: Acute respiratory distress syndrome—How neutrophils migrate into the lung. Crit. Care 2004, 8, 453–461. [Google Scholar] [CrossRef]
- Duval, K.; Grover, H.; Han, L.H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology 2017, 32, 266–277. [Google Scholar] [CrossRef]
- Tan, Q.; Choi, K.M.; Sicard, D.; Tschumperlin, D.J. Human airway organoid engineering as a step toward lung regeneration and disease modeling. Biomaterials 2017, 113, 118–132. [Google Scholar] [CrossRef]
- Miller, A.J.; Dye, B.R.; Ferrer-Torres, D.; Hill, D.R.; Overeem, A.W.; Shea, L.D.; Spence, J.R. Generation of lung organoids from human pluripotent stem cells in vitro. Nat. Protoc. 2019, 14, 518–540. [Google Scholar] [CrossRef]
- Park, S.E.; Georgescu, A.; Huh, D. Organoids-on-a-chip. Science 2019, 364, 960–965. [Google Scholar] [CrossRef]
- Dye, B.R.; Hill, D.R.; Ferguson, M.A.; Tsai, Y.H.; Nagy, M.S.; Dyal, R.; Wells, J.M.; Mayhew, C.N.; Nattiv, R.; Klein, O.D.; et al. In vitro generation of human pluripotent stem cell derived lung organoids. eLife 2015, 4, e05098. [Google Scholar] [CrossRef]
- Miller, A.J.; Hill, D.R.; Nagy, M.S.; Aoki, Y.; Dye, B.R.; Chin, A.M.; Huang, S.; Zhu, F.; White, E.S.; Lama, V.; et al. In Vitro Induction and In Vivo Engraftment of Lung Bud Tip Progenitor Cells Derived from Human Pluripotent Stem Cells. Stem Cell Rep. 2018, 10, 101–119. [Google Scholar] [CrossRef]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting organ-level lung functions on a chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef]
- Benam, K.H.; Villenave, R.; Lucchesi, C.; Varone, A.; Hubeau, C.; Lee, H.H.; Alves, S.E.; Salmon, M.; Ferrante, T.C.; Weaver, J.C.; et al. Small airway-on-a-chip enables analysis of human lung inflammation and drug responses in vitro. Nat. Methods 2016, 13, 151–157. [Google Scholar] [CrossRef]
- Jain, A.; Barrile, R.; van der Meer, A.D.; Mammoto, A.; Mammoto, T.; De Ceunynck, K.; Aisiku, O.; Otieno, M.A.; Louden, C.S.; Hamilton, G.A.; et al. Primary Human Lung Alveolus-on-a-chip Model of Intravascular Thrombosis for Assessment of Therapeutics. Clin. Pharmacol. Ther. 2018, 103, 332–340. [Google Scholar] [CrossRef]
- Bai, H.; Si, L.; Jiang, A.; Belgur, C.; Zhai, Y.; Plebani, R.; Oh, C.Y.; Rodas, M.; Patil, A.; Nurani, A.; et al. Mechanical control of innate immune responses against viral infection revealed in a human lung alveolus chip. Nat. Commun. 2022, 13, 1928. [Google Scholar] [CrossRef]
- Si, L.; Bai, H.; Rodas, M.; Cao, W.; Oh, C.Y.; Jiang, A.; Moller, R.; Hoagland, D.; Oishi, K.; Horiuchi, S.; et al. A human-airway-on-a-chip for the rapid identification of candidate antiviral therapeutics and prophylactics. Nat. Biomed. Eng. 2021, 5, 815–829. [Google Scholar] [CrossRef]
- Stucki, A.O.; Stucki, J.D.; Hall, S.R.; Felder, M.; Mermoud, Y.; Schmid, R.A.; Geiser, T.; Guenat, O.T. A lung-on-a-chip array with an integrated bio-inspired respiration mechanism. Lab. Chip 2015, 15, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- Zamprogno, P.; Wüthrich, S.; Achenbach, S.; Thoma, G.; Stucki, J.D.; Hobi, N.; Schneider-Daum, N.; Lehr, C.M.; Huwer, H.; Geiser, T.; et al. Second-generation lung-on-a-chip with an array of stretchable alveoli made with a biological membrane. Commun. Biol. 2021, 4, 168. [Google Scholar] [CrossRef] [PubMed]
- Douville, N.J.; Zamankhan, P.; Tung, Y.C.; Li, R.; Vaughan, B.L.; Tai, C.F.; White, J.; Christensen, P.J.; Grotberg, J.B.; Takayama, S. Combination of fluid and solid mechanical stresses contribute to cell death and detachment in a microfluidic alveolar model. Lab. Chip 2011, 11, 609–619. [Google Scholar] [CrossRef]
- Huh, D.; Fujioka, H.; Tung, Y.C.; Futai, N.; Paine, R., 3rd; Grotberg, J.B.; Takayama, S. Acoustically detectable cellular-level lung injury induced by fluid mechanical stresses in microfluidic airway systems. Proc. Natl. Acad. Sci. USA 2007, 104, 18886–18891. [Google Scholar] [CrossRef]
- Sellgren, K.L.; Butala, E.J.; Gilmour, B.P.; Randell, S.H.; Grego, S. A biomimetic multicellular model of the airways using primary human cells. Lab. Chip 2014, 14, 3349–3358. [Google Scholar] [CrossRef]


{kind=link}
| Year | Design and Features | Main Findings | Ref. |
|---|---|---|---|
| 2007 |
|
| [94] |
| 2010 |
|
| [86] |
| 2011 |
|
| [93] |
| 2014 |
|
| [95] |
| 2015 |
|
| [91] |
| 2021 |
|
| [92] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Su, L.; Pan, P. Advances and Applications of Lung Organoids in the Research on Acute Respiratory Distress Syndrome (ARDS). J. Clin. Med. 2024, 13, 346. https://doi.org/10.3390/jcm13020346
Zhang X, Su L, Pan P. Advances and Applications of Lung Organoids in the Research on Acute Respiratory Distress Syndrome (ARDS). Journal of Clinical Medicine. 2024; 13(2):346. https://doi.org/10.3390/jcm13020346
Chicago/Turabian StyleZhang, Xingwu, Longxiang Su, and Pan Pan. 2024. "Advances and Applications of Lung Organoids in the Research on Acute Respiratory Distress Syndrome (ARDS)" Journal of Clinical Medicine 13, no. 2: 346. https://doi.org/10.3390/jcm13020346
APA StyleZhang, X., Su, L., & Pan, P. (2024). Advances and Applications of Lung Organoids in the Research on Acute Respiratory Distress Syndrome (ARDS). Journal of Clinical Medicine, 13(2), 346. https://doi.org/10.3390/jcm13020346