
The Role of Heparin in Postural Orthostatic Tachycardia Syndrome and Other Post-Acute Sequelae of COVID-19

 ,
,  , , , , , , , , and
, , , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
3. Post-Acute Sequelae of COVID
4. Postural Orthostatic Tachycardia Syndrome and Pathological Mechanism: Hypercoagulability, Autoimmunity, and Endothelial Dysfunction
5. Potential Beneficial Effects of Heparin in COVID-19 Post-Acute Sequelae: Immunomodulatory, Anticoagulant, Antiviral, Endothelial, and Vascular Resistance Effects
6. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, C.; Yu, C.; Jing, H.; Wu, X.; Novakovic, V.A.; Xie, R.; Shi, J. Long COVID: The Nature of Thrombotic Sequelae Determines the Necessity of Early Anticoagulation. Front. Cell Infect. Microbiol. 2022, 12, 861703. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Haupert, S.R.; Zimmermann, L.; Shi, X.; Fritsche, L.G.; Mukherjee, B. Global Prevalence of Post-Coronavirus Disease 2019 (COVID-19) Condition or Long COVID: A Meta-Analysis and Systematic Review. J. Infect. Dis. 2022, 226, 1593–1607. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Torre, F.; Mínguez-Olaondo, A.; López-Bravo, A.; Tijero, B.; Grozeva, V.; Walcker, M.; Azkune-Galparsoro, H.; López De Munain, A.; Alcaide, A.B.; Quiroga, J.; et al. Dysautonomia in COVID-19 Patients: A Narrative Review on Clinical Course, Diagnostic and Therapeutic Strategies. Front. Neurol. 2022, 13, 886609. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, R.; Grach, S.L.; Ghosh, A.K.; Bierle, D.M.; Salonen, B.R.; Collins, N.M.; Joshi, A.Y.; Boeder, N.D.; Anstine, C.V.; Mueller, M.R.; et al. The Female-Predominant Persistent Immune Dysregulation of the Post-COVID Syndrome. Mayo Clin. Proc. 2022, 97, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Davitt, E.; Davitt, C.; Mazer, M.B.; Areti, S.S.; Hotchkiss, R.S.; Remy, K.E. COVID-19 Disease and Immune Dysregulation. Best Pract. Res. Clin. Haematol. 2022, 35, 101401. [Google Scholar] [CrossRef] [PubMed]
- Ahamed, J.; Laurence, J. Long COVID Endotheliopathy: Hypothesized Mechanisms and Potential Therapeutic Approaches. J. Clin. Investig. 2022, 132, e161167. [Google Scholar] [CrossRef] [PubMed]
- Pilia, E.; Belletti, A.; Fresilli, S.; Lee, T.C.; Zangrillo, A.; Finco, G.; Landoni, G.; Angelini, M.; Sofia, R.; Full Anticoagulation; et al. The Effect of Heparin Full-Dose Anticoagulation on Survival of Hospitalized, Non-Critically Ill COVID-19 Patients: A Meta-Analysis of High Quality Studies. Lung 2023, 201, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Vitiello, A.; Ferrara, F. Low Molecular Weight Heparin, Anti-Inflammatory/Immunoregulatory and Antiviral Effects, a Short Update. Cardiovasc. Drugs Ther. 2023, 37, 277–281. [Google Scholar] [CrossRef]
- Langer, F.; Kluge, S.; Klamroth, R.; Oldenburg, J. Coagulopathy in COVID-19 and Its Implication for Safe and Efficacious Thromboprophylaxis. Hamostaseologie 2020, 40, 264–269. [Google Scholar] [CrossRef]
- Hernández-Huerta, M.T.; Pérez-Santiago, A.D.; Pérez-Campos Mayoral, L.; Sánchez Navarro, L.M.; Rodal Canales, F.J.; Majluf-Cruz, A.; Matias-Cervantes, C.A.; Pérez-Campos Mayoral, E.; Romero Díaz, C.; Mayoral-Andrade, G.; et al. Mechanisms of Immunothrombosis by SARS-CoV-2. Biomolecules 2021, 11, 1550. [Google Scholar] [CrossRef]
- Dirican, A.; Ildir, S.; Uzar, T.; Karaman, I.; Ozkaya, S. The Role of Endotheliitis in COVID-19: Real-world Experience of 11, 190 Patients and Literature Review for a Pathophysiological Map to Clinical Categorisation. Int. J. Clin. Pract. 2021, 75, 14843. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Yang, K.Y.; Huang, Y.; Lui, K.O. Endothelial Contribution to COVID-19: An Update on Mechanisms and Therapeutic Implications. J. Mol. Cell Cardiol. 2022, 164, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Venter, C.; Laubscher, G.J.; Kotze, M.J.; Oladejo, S.O.; Watson, L.R.; Rajaratnam, K.; Watson, B.W.; Kell, D.B. Prevalence of Symptoms, Comorbidities, Fibrin Amyloid Microclots and Platelet Pathology in Individuals with Long COVID/Post-Acute Sequelae of COVID-19 (PASC). Cardiovasc. Diabetol. 2022, 21, 148. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.; Joseph, P.; Heerdt, P.M.; Cullinan, M.; Lutchmansingh, D.D.; Gulati, M.; Possick, J.D.; Systrom, D.M.; Waxman, A.B. Persistent Exertional Intolerance After COVID-19. Chest 2022, 161, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Peluso, M.J.; Deeks, S.G.; Mustapic, M.; Kapogiannis, D.; Henrich, T.J.; Lu, S.; Goldberg, S.A.; Hoh, R.; Chen, J.Y.; Martinez, E.O.; et al. SARS-CoV-2 and Mitochondrial Proteins in Neural-Derived Exosomes of COVID-19. Ann. Neurol. 2022, 91, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.L.; Villacreses, R.; Nagpal, P.; Guo, J.; Pezzulo, A.A.; Thurman, A.L.; Hamzeh, N.Y.; Blount, R.J.; Fortis, S.; Hoffman, E.A.; et al. Quantitative Chest CT Assessment of Small Airways Disease in Post-Acute SARS-CoV-2 Infection. Radiology 2022, 304, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Guler, S.A.; Ebner, L.; Aubry-Beigelman, C.; Bridevaux, P.-O.; Brutsche, M.; Clarenbach, C.; Garzoni, C.; Geiser, T.K.; Lenoir, A.; Mancinetti, M.; et al. Pulmonary Function and Radiological Features 4 Months after COVID-19: First Results from the National Prospective Observational Swiss COVID-19 Lung Study. Eur. Respir. J. 2021, 57, 2003690. [Google Scholar] [CrossRef] [PubMed]
- Buonsenso, D.; Di Giuda, D.; Sigfrid, L.; Pizzuto, D.A.; Di Sante, G.; De Rose, C.; Lazzareschi, I.; Sali, M.; Baldi, F.; Chieffo, D.P.R.; et al. Evidence of Lung Perfusion Defects and Ongoing Inflammation in an Adolescent with Post-Acute Sequelae of SARS-CoV-2 Infection. Lancet Child Adolesc. Health 2021, 5, 677–680. [Google Scholar] [CrossRef] [PubMed]
- Mentzer, S.J.; Ackermann, M.; Jonigk, D. Endothelialitis, Microischemia, and Intussusceptive Angiogenesis in COVID-19. Cold Spring Harb. Perspect Med. 2022, 12, a041157. [Google Scholar] [CrossRef]
- Poyatos, P.; Luque, N.; Eizaguirre, S.; Sabater, G.; Sebastián, L.; Francisco-Albesa, Í.; Peracaula, M.; Boixadé, M.; Orriols, R.; Tura-Ceide, O. Post-COVID-19 Patients Show an Increased Endothelial Progenitor Cell Production. Transl. Res. 2022, 243, 14–20. [Google Scholar] [CrossRef]
- Dhawan, R.T.; Gopalan, D.; Howard, L.; Vicente, A.; Park, M.; Manalan, K.; Wallner, I.; Marsden, P.; Dave, S.; Branley, H.; et al. Beyond the Clot: Perfusion Imaging of the Pulmonary Vasculature after COVID-19. Lancet Respir. Med. 2021, 9, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Vlok, M.; Venter, C.; Bezuidenhout, J.A.; Laubscher, G.J.; Steenkamp, J.; Kell, D.B. Persistent Clotting Protein Pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) Is Accompanied by Increased Levels of Antiplasmin. Cardiovasc. Diabetol. 2021, 20, 172. [Google Scholar] [CrossRef] [PubMed]
- Renzi, S.; Landoni, G.; Zangrillo, A.; Ciceri, F. MicroCLOTS Pathophysiology in Coronavirus Disease 2019. Korean J. Intern. Med. 2023, 38, 570–571. [Google Scholar] [CrossRef] [PubMed]
- Graham, E.L.; Clark, J.R.; Orban, Z.S.; Lim, P.H.; Szymanski, A.L.; Taylor, C.; DiBiase, R.M.; Jia, D.T.; Balabanov, R.; Ho, S.U.; et al. Persistent Neurologic Symptoms and Cognitive Dysfunction in Non-hospitalized COVID-19 “Long Haulers”. Ann. Clin. Transl. Neurol. 2021, 8, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Batra, A.; Clark, J.R.; LaHaye, K.; Shlobin, N.A.; Hoffman, S.C.; Orban, Z.S.; Colton, K.; Dematte, J.E.; Sorond, F.A.; Koralnik, I.J.; et al. Transcranial Doppler Ultrasound Evidence of Active Cerebral Embolization in COVID-19. J. Stroke Cerebrovasc. Dis. 2021, 30, 105542. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, L.A.; Ancona, M.B.; Filho, R.K.; Tresoldi, M.; Caldas, J.G.; Monti, G.; Carnevale, F.C.; De Cobelli, F.; Moreira De Assis, A.; Ciceri, F.; et al. Microvascular Lung Vessels Obstructive Thromboinflammatory Syndrome in Patients with COVID-19: Insights from Lung Intravascular Optical Coherence Tomography. Front. Med. 2023, 10, 1050531. [Google Scholar] [CrossRef] [PubMed]
- Wadowski, P.P.; Panzer, B.; Józkowicz, A.; Kopp, C.W.; Gremmel, T.; Panzer, S.; Koppensteiner, R. Microvascular Thrombosis as a Critical Factor in Severe COVID-19. IJMS 2023, 24, 2492. [Google Scholar] [CrossRef]
- Elseidy, S.A.; Awad, A.K.; Vorla, M.; Fatima, A.; Elbadawy, M.A.; Mandal, D.; Mohamad, T. Cardiovascular Complications in the Post-Acute COVID-19 Syndrome (PACS). IJC Heart Vasc. 2022, 40, 101012. [Google Scholar] [CrossRef]
- Sherif, Z.A.; Gomez, C.R.; Connors, T.J.; Henrich, T.J.; Reeves, W.B. RECOVER Mechanistic Pathway Task Force Pathogenic Mechanisms of Post-Acute Sequelae of SARS-CoV-2 Infection (PASC). eLife 2023, 12, e86002. [Google Scholar] [CrossRef]
- Sidarta-Oliveira, D.; Jara, C.P.; Ferruzzi, A.J.; Skaf, M.S.; Velander, W.H.; Araujo, E.P.; Velloso, L.A. SARS-CoV-2 Receptor Is Co-Expressed with Elements of the Kinin–Kallikrein, Renin–Angiotensin and Coagulation Systems in Alveolar Cells. Sci. Rep. 2020, 10, 19522. [Google Scholar] [CrossRef]
- Talasaz, A.H.; Kakavand, H.; Van Tassell, B.; Aghakouchakzadeh, M.; Sadeghipour, P.; Dunn, S.; Geraiely, B. Cardiovascular Complications of COVID-19: Pharmacotherapy Perspective. Cardiovasc. Drugs Ther. 2021, 35, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Castiello, T.; Georgiopoulos, G.; Finocchiaro, G.; Claudia, M.; Gianatti, A.; Delialis, D.; Aimo, A.; Prasad, S. COVID-19 and Myocarditis: A Systematic Review and Overview of Current Challenges. Heart Fail. Rev. 2022, 27, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Giustino, G.; Pinney, S.P.; Lala, A.; Reddy, V.Y.; Johnston-Cox, H.A.; Mechanick, J.I.; Halperin, J.L.; Fuster, V. Coronavirus and Cardiovascular Disease, Myocardial Injury, and Arrhythmia. J. Am. Coll. Cardiol. 2020, 76, 2011–2023. [Google Scholar] [CrossRef] [PubMed]
- Escher, R.; Breakey, N.; Lämmle, B. Severe COVID-19 Infection Associated with Endothelial Activation. Thromb. Res. 2020, 190, 62. [Google Scholar] [CrossRef] [PubMed]
- Holter, J.C.; Pischke, S.E.; De Boer, E.; Lind, A.; Jenum, S.; Holten, A.R.; Tonby, K.; Barratt-Due, A.; Sokolova, M.; Schjalm, C.; et al. Systemic Complement Activation Is Associated with Respiratory Failure in COVID-19 Hospitalized Patients. Proc. Natl. Acad. Sci. USA 2020, 117, 25018–25025. [Google Scholar] [CrossRef] [PubMed]
- Soy, M.; Keser, G.; Atagündüz, P.; Tabak, F.; Atagündüz, I.; Kayhan, S. Cytokine Storm in COVID-19: Pathogenesis and Overview of Anti-Inflammatory Agents Used in Treatment. Clin. Rheumatol. 2020, 39, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- De Los Reyes-García, A.M.; Aroca, A.; Arroyo, A.; García-Barbera, N.; Vicente, V.; González-Conejero, R.; Martínez, C. Neutrophil Extracellular Trap Components Increase the Expression of Coagulation Factors. Biomed. Rep. 2019, 10, 195–210. [Google Scholar] [CrossRef] [PubMed]
- Siripanthong, B.; Nazarian, S.; Muser, D.; Deo, R.; Santangeli, P.; Khanji, M.Y.; Cooper, L.T.; Chahal, C.A.A. Recognizing COVID-19–Related Myocarditis: The Possible Pathophysiology and Proposed Guideline for Diagnosis and Management. Heart Rhythm. 2020, 17, 1463–1471. [Google Scholar] [CrossRef]
- Gupta, A.; Jayakumar, M.N.; Saleh, M.A.; Kannan, M.; Halwani, R.; Qaisar, R.; Ahmad, F. SARS-CoV-2 Infection- Induced Growth Factors Play Differential Roles in COVID-19 Pathogenesis. Life Sci. 2022, 304, 120703. [Google Scholar] [CrossRef]
- Chen, R.; Huang, Y.; Quan, J.; Liu, J.; Wang, H.; Billiar, T.R.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. HMGB1 as a Potential Biomarker and Therapeutic Target for Severe COVID-19. Heliyon 2020, 6, e05672. [Google Scholar] [CrossRef]
- Hajjo, R.; Sabbah, D.A.; Bardaweel, S.K.; Tropsha, A. Shedding the Light on Post-Vaccine Myocarditis and Pericarditis in COVID-19 and Non-COVID-19 Vaccine Recipients. Vaccines 2021, 9, 1186. [Google Scholar] [CrossRef] [PubMed]
- Molina-Ramos, A.; Gómez-Moyano, E.; Rodríguez-Capitán, J.; Angullo-Gómez, M.; Gallardo-Jiménez, P.; Pérez De Pedro, I.; Valiente De Santis, L.; Pérez-Villardón, B.; Piñero-Uribe, I.; Mora-Robles, J.; et al. Myocarditis Related to COVID-19 and SARS-CoV-2 Vaccination. JCM 2022, 11, 6999. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, H.; Townsend, L.; Morrin, H.; Ahmad, A.; Comerford, C.; Karampini, E.; Englert, H.; Byrne, M.; Bergin, C.; O’Sullivan, J.M.; et al. Persistent Endotheliopathy in the Pathogenesis of Long COVID Syndrome. J. Thromb. Haemost. 2021, 19, 2546–2553. [Google Scholar] [CrossRef] [PubMed]
- Morganstein, T.; Haidar, Z.; Trivlidis, J.; Azuelos, I.; Huang, M.J.; Eidelman, D.H.; Baglole, C.J. Involvement of the ACE2/Ang-(1–7)/MasR Axis in Pulmonary Fibrosis: Implications for COVID-19. IJMS 2021, 22, 12955. [Google Scholar] [CrossRef] [PubMed]
- Zechendorf, E.; Schröder, K.; Stiehler, L.; Frank, N.; Beckers, C.; Kraemer, S.; Dreher, M.; Kersten, A.; Thiemermann, C.; Marx, G.; et al. The Potential Impact of Heparanase Activity and Endothelial Damage in COVID-19 Disease. JCM 2022, 11, 5261. [Google Scholar] [CrossRef]
- Halushka, M.K.; Vander Heide, R.S. Myocarditis Is Rare in COVID-19 Autopsies: Cardiovascular Findings across 277 Postmortem Examinations. Cardiovasc. Pathol. 2021, 50, 107300. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial Cell Infection and Endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Reis Carneiro, D.; Rocha, I.; Habek, M.; Helbok, R.; Sellner, J.; Struhal, W.; Wenning, G.; Fanciulli, A. Clinical Presentation and Management Strategies of Cardiovascular Autonomic Dysfunction Following a COVID-19 Infection—A Systematic Review. Eur. J. Neurol. 2023, 30, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
- Quan, W.; Wang, Y.; Chen, S.; Du, J. Orthostatic Intolerance and Coagulation Abnormalities: An Update. Neurosci. Bull. 2019, 35, 171–177. [Google Scholar] [CrossRef]
- Johansson, M.; Yan, H.; Welinder, C.; Végvári, Á.; Hamrefors, V.; Bäck, M.; Sutton, R.; Fedorowski, A. Plasma Proteomic Profiling in Postural Orthostatic Tachycardia Syndrome (POTS) Reveals New Disease Pathways. Sci. Rep. 2022, 12, 20051. [Google Scholar] [CrossRef]
- Masoud, M.; Sarig, G.; Brenner, B.; Jacob, G. Orthostatic Hypercoagulability: A Novel Physiological Mechanism to Activate the Coagulation System. Hypertension 2008, 51, 1545–1551. [Google Scholar] [CrossRef] [PubMed]
- Cvirn, G.; Kneihsl, M.; Rossmann, C.; Paar, M.; Gattringer, T.; Schlagenhauf, A.; Leschnik, B.; Koestenberger, M.; Tafeit, E.; Reibnegger, G.; et al. Orthostatic Challenge Shifts the Hemostatic System of Patients Recovered from Stroke toward Hypercoagulability. Front. Physiol. 2017, 8, 12. [Google Scholar] [CrossRef]
- Masoud, M.; Sarig, G.; Brenner, B.; Jacob, G. Hydration Does Not Prevent Orthostatic Hypercoagulability. Thromb. Haemost 2010, 103, 284–290. [Google Scholar] [CrossRef] [PubMed]
- De Candia, E.; De Cristofaro, R.; Landolfi, R. Thrombin-Induced Platelet Activation Is Inhibited by High- and Low-Molecular-Weight Heparin. Circulation 1999, 99, 3308–3314. [Google Scholar] [CrossRef] [PubMed]
- Grasser, E.K.; Goswami, N.; Rössler, A.; Vrecko, K.; Hinghofer-Szalkay, H. Hemodynamic and Neurohormonal Responses to Extreme Orthostatic Stress in Physically Fit Young Adults. Acta Astronaut. 2009, 64, 688–696. [Google Scholar] [CrossRef]
- Amiral, J.; Seghatchian, J. Autoimmune Complications of COVID-19 and Potential Consequences for Long-Lasting Disease Syndromes. Transfus. Apher. Sci. 2023, 62, 103625. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Kem, D.C.; Reim, S.; Khan, M.; Vanderlinde-Wood, M.; Zillner, C.; Collier, D.; Liles, C.; Hill, M.A.; Cunningham, M.W.; et al. Agonistic Autoantibodies as Vasodilators in Orthostatic Hypotension: A New Mechanism. Hypertension 2012, 59, 402–408. [Google Scholar] [CrossRef]
- Fedorowski, A.; Li, H.; Yu, X.; Koelsch, K.A.; Harris, V.M.; Liles, C.; Murphy, T.A.; Quadri, S.M.S.; Scofield, R.H.; Sutton, R.; et al. Antiadrenergic Autoimmunity in Postural Tachycardia Syndrome. EP Eur. 2017, 19, 1211–1219. [Google Scholar] [CrossRef]
- Li, H.; Yu, X.; Liles, C.; Khan, M.; Vanderlinde-Wood, M.; Galloway, A.; Zillner, C.; Benbrook, A.; Reim, S.; Collier, D.; et al. Autoimmune Basis for Postural Tachycardia Syndrome. JAHA 2014, 3, e000755. [Google Scholar] [CrossRef]
- Yu, X.; Stavrakis, S.; Hill, M.A.; Huang, S.; Reim, S.; Li, H.; Khan, M.; Hamlett, S.; Cunningham, M.W.; Kem, D.C. Autoantibody Activation of Beta-Adrenergic and Muscarinic Receptors Contributes to an “Autoimmune” Orthostatic Hypotension. J. Am. Soc. Hypertens. 2012, 6, 40–47. [Google Scholar] [CrossRef]
- Ruzieh, M.; Batizy, L.; Dasa, O.; Oostra, C.; Grubb, B. The Role of Autoantibodies in the Syndromes of Orthostatic Intolerance: A Systematic Review. Scand. Cardiovasc. J. 2017, 51, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Wallukat, G.; Hohberger, B.; Wenzel, K.; Fürst, J.; Schulze-Rothe, S.; Wallukat, A.; Hönicke, A.-S.; Müller, J. Functional Autoantibodies against G-Protein Coupled Receptors in Patients with Persistent Long-COVID-19 Symptoms. J. Transl. Autoimmun. 2021, 4, 100100. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Kitazawa, T.; Somlyo, A.V.; Somlyo, A.P. Cytosolic Heparin Inhibits Muscarinic and Alpha-Adrenergic Ca2+ Release in Smooth Muscle. Physiological Role of Inositol 1,4,5-Trisphosphate in Pharmacomechanical Coupling. J. Biol. Chem. 1989, 264, 17997–18004. [Google Scholar] [CrossRef] [PubMed]
- Schofield, J. Persistent Antiphospholipid Antibodies, Mast Cell Activation Syndrome, Postural Orthostatic Tachycardia Syndrome and Post-COVID Syndrome: 1 Year On. Eur. J. Case Rep. Intern. Med. 2021, 8, 002378. [Google Scholar] [CrossRef]
- Chopoorian, A.H.; Wahba, A.; Celedonio, J.; Nwazue, V.; Smith, E.C.; Garland, E.M.; Paranjape, S.; Okamoto, L.E.; Black, B.K.; Biaggioni, I.; et al. Impaired Endothelial Function in Patients with Postural Tachycardia Syndrome. Hypertension 2021, 77, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Novak, P.; Mukerji, S.S.; Alabsi, H.S.; Systrom, D.; Marciano, S.P.; Felsenstein, D.; Mullally, W.J.; Pilgrim, D.M. Multisystem Involvement in Post-Acute Sequelae of Coronavirus Disease 19. Ann. Neurol. 2022, 91, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Cieślik-Guerra, U.I.; Fila, M.; Kamiński, M.; Kotas, R.; Wróblewski, J.; Trzos, E.; Uznańska-Loch, B.; Rechciński, T.; Wierzbowska-Drabik, K.; Kasprzak, J.D.; et al. Correlation between the Activity of the Autonomic Nervous System and Endothelial Function in Patients with Acute Coronary Syndrome. Pol. Arch. Intern. Med. 2014, 124, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, U.; Li, J. Heparin—An Old Drug with Multiple Potential Targets in COVID-19 Therapy. J. Thromb. Haemost. 2020, 18, 2422–2424. [Google Scholar] [CrossRef] [PubMed]
- Eilts, F.; Bauer, S.; Fraser, K.; Dordick, J.S.; Wolff, M.W.; Linhardt, R.J.; Zhang, F. The Diverse Role of Heparan Sulfate and Other GAGs in SARS-CoV-2 Infections and Therapeutics. Carbohydr. Polym. 2023, 299, 120167. [Google Scholar] [CrossRef]
- Xu, K.; Jin, L. The Role of Heparin/Heparan Sulphate in the IFN-γ-Led Arena. Biochimie 2020, 170, 1–9. [Google Scholar] [CrossRef]
- Salas, A. Heparin Attenuates TNF-Alpha Induced Inflammatory Response through a CD11b Dependent Mechanism. Gut 2000, 47, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Litov, L.; Petkov, P.; Rangelov, M.; Ilieva, N.; Lilkova, E.; Todorova, N.; Krachmarova, E.; Malinova, K.; Gospodinov, A.; Hristova, R.; et al. Molecular Mechanism of the Anti-Inflammatory Action of Heparin. IJMS 2021, 22, 10730. [Google Scholar] [CrossRef] [PubMed]
- Xie-Zukauskas, H.; Das, J.; Short, B.L.; Gutkind, J.S.; Ray, P.E. Heparin Inhibits Angiotensin II-Induced Vasoconstriction on Isolated Mouse Mesenteric Resistance Arteries through Rho-A- and PKA-Dependent Pathways. Vasc. Pharmacol. 2013, 58, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Sumitomo-Ueda, Y.; Aihara, K.; Ise, T.; Yoshida, S.; Ikeda, Y.; Uemoto, R.; Yagi, S.; Iwase, T.; Ishikawa, K.; Hirata, Y.; et al. Heparin Cofactor II Protects Against Angiotensin II-Induced Cardiac Remodeling Via Attenuation of Oxidative Stress in Mice. Hypertension 2010, 56, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Hogwood, J.; Pitchford, S.; Mulloy, B.; Page, C.; Gray, E. Heparin and Non-Anticoagulant Heparin Attenuate Histone-Induced Inflammatory Responses in Whole Blood. PLoS ONE 2020, 15, e0233644. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Jiang, J.; Zhang, Z.; Ma, X. Heparin reduces endothelial cell damage induced by neutrophil extracellular traps. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2017, 29, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Kotani, T.; Suzuki, K. Antifibrotic Therapy by Sustained Release of Low Molecular Weight Heparin from Poly(Lactic-Co-Glycolic Acid) Microparticles on Bleomycin-Induced Pulmonary Fibrosis in Mice. Sci. Rep. 2020, 10, 19019. [Google Scholar] [CrossRef] [PubMed]
- Toro, L.; Michea, L.; Parra-Lucares, A.; Mendez-Valdes, G.; Villa, E.; Bravo, I.; Pumarino, C.; Ayala, P.; Sanhueza, M.E.; Torres, R.; et al. High Plasma Levels of Fibroblast Growth Factor 23 Are Associated with Increased Risk of COVID-19 in End-Stage Renal Disease Patients on Hemodialysis: Results of a Prospective Cohort. Toxins 2023, 15, 97. [Google Scholar] [CrossRef]
- Levine, A.; Kenet, G.; Bruck, R.; Avni, Y.; Avinoach, I.; Aeed, H.; Matas, Z.; David, M.; Yayon, A. Effect of Heparin on Tissue Binding Activity of Fibroblast Growth Factor and Heparin-Binding Epidermal Growth Factor in Experimental Colitis in Rats. Pediatr. Res. 2002, 51, 635–640. [Google Scholar] [CrossRef]
- Mousavi, S.; Moradi, M.; Khorshidahmad, T.; Motamedi, M. Anti-Inflammatory Effects of Heparin and Its Derivatives: A Systematic Review. Adv. Pharmacol. Sci. 2015, 2015, 507151. [Google Scholar] [CrossRef]
- Buijsers, B.; Yanginlar, C.; Maciej-Hulme, M.L.; De Mast, Q.; Van Der Vlag, J. Beneficial Non-Anticoagulant Mechanisms Underlying Heparin Treatment of COVID-19 Patients. eBioMedicine 2020, 59, 102969. [Google Scholar] [CrossRef]
- Longstaff, C.; Hogwood, J.; Gray, E.; Komorowicz, E.; Varjú, I.; Varga, Z.; Kolev, K. Neutralisation of the Anti-Coagulant Effects of Heparin by Histones in Blood Plasma and Purified Systems. Thromb. Haemost 2016, 115, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Kinaneh, S.; Khamaysi, I.; Karram, T.; Hamoud, S. Heparanase as a Potential Player in SARS-CoV-2 Infection and Induced Coagulopathy. Biosci. Rep. 2021, 41, BSR20210290. [Google Scholar] [CrossRef]
- Kumano, N.; Ikeda, S.; Arimori, Y.; Ono, T.; Saeki, S. Heparin resistance associated with elevated factor VIII. Masui 2008, 57, 471–473. [Google Scholar] [PubMed]
- Paiardi, G.; Richter, S.; Oreste, P.; Urbinati, C.; Rusnati, M.; Wade, R.C. The Binding of Heparin to Spike Glycoprotein Inhibits SARS-CoV-2 Infection by Three Mechanisms. J. Biol. Chem. 2022, 298, 101507. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.Y.Y.; Suryadinata, R.; McCafferty, C.; Ignjatovic, V.; Purcell, D.F.J.; Robinson, P.; Morton, C.J.; Parker, M.W.; Anderson, G.P.; Monagle, P.; et al. Heparin Inhibits SARS-CoV-2 Replication in Human Nasal Epithelial Cells. Viruses 2022, 14, 2620. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057.e15. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Pang, H.; Li, S.J. Heparin Interacts with the Main Protease of SARS-CoV-2 and Inhibits Its Activity. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2022, 267, 120595. [Google Scholar] [CrossRef]
- Wettstein, L.; Immenschuh, P.; Weil, T.; Conzelmann, C.; Almeida-Hernández, Y.; Hoffmann, M.; Kempf, A.; Nehlmeier, I.; Lotke, R.; Petersen, M.; et al. Native and Activated Antithrombin Inhibits TMPRSS2 Activity and SARS-CoV-2 Infection. J. Med. Virol. 2023, 95, e28124. [Google Scholar] [CrossRef]
- Oikonomou, E.; Souvaliotis, N.; Lampsas, S.; Siasos, G.; Poulakou, G.; Theofilis, P.; Papaioannou, T.G.; Haidich, A.-B.; Tsaousi, G.; Ntousopoulos, V.; et al. Endothelial Dysfunction in Acute and Long Standing COVID−19: A Prospective Cohort Study. Vasc. Pharmacol. 2022, 144, 106975. [Google Scholar] [CrossRef]
- Kondashevskaya, M.V. Horizons of Heparin Therapy in COVID-19 and Pandemic-Related Diseases. J. Evol. Biochem. Phys. 2022, 58, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Ilyas, I.; Weng, J. Endothelial Dysfunction in COVID-19: An Overview of Evidence, Biomarkers, Mechanisms and Potential Therapies. Acta Pharmacol. Sin. 2023, 44, 695–709. [Google Scholar] [CrossRef] [PubMed]
- Buijsers, B.; Yanginlar, C.; De Nooijer, A.; Grondman, I.; Maciej-Hulme, M.L.; Jonkman, I.; Janssen, N.A.F.; Rother, N.; De Graaf, M.; Pickkers, P.; et al. Increased Plasma Heparanase Activity in COVID-19 Patients. Front. Immunol. 2020, 11, 575047. [Google Scholar] [CrossRef] [PubMed]
- Drost, C.C.; Rovas, A.; Osiaevi, I.; Rauen, M.; Van Der Vlag, J.; Buijsers, B.; Salmenov, R.; Lukasz, A.; Pavenstädt, H.; Linke, W.A.; et al. Heparanase Is a Putative Mediator of Endothelial Glycocalyx Damage in COVID-19—A Proof-of-Concept Study. Front. Immunol. 2022, 13, 916512. [Google Scholar] [CrossRef] [PubMed]
- Calabretta, E.; Moraleda, J.M.; Iacobelli, M.; Jara, R.; Vlodavsky, I.; O’Gorman, P.; Pagliuca, A.; Mo, C.; Baron, R.M.; Aghemo, A.; et al. COVID-19-induced Endotheliitis: Emerging Evidence and Possible Therapeutic Strategies. Br. J. Haematol. 2021, 193, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Yu, X. Potential of Heparin in the Treatment of COVID-19–Associated Myocarditis. Pediatr. Emer. Care 2022, 38, e504. [Google Scholar] [CrossRef]
- Lindner, V.; Olson, N.E.; Clowes, A.W.; Reidy, M.A. Inhibition of Smooth Muscle Cell Proliferation in Injured Rat Arteries. Interaction of Heparin with Basic Fibroblast Growth Factor. J. Clin. Investig. 1992, 90, 2044–2049. [Google Scholar] [CrossRef]
- Paredes-Gamero, E.J.; Medeiros, V.P.; Farias, E.H.C.; Justo, G.Z.; Trindade, E.S.; Andrade-Lopes, A.L.; Godinho, R.O.; De Miranda, A.; Ferreira, A.T.; Tersariol, I.L.S.; et al. Heparin Induces Rat Aorta Relaxation via Integrin-Dependent Activation of Muscarinic M3 Receptors. Hypertension 2010, 56, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, A.; Popov, D.; Capraru, M.; Simionescu, M. Enoxaparin—A Low Molecular Weight Heparin, Restores the Altered Vascular Reactivity of Resistance Arteries in Aged and Aged–Diabetic Hamsters. Vasc. Pharmacol. 2003, 40, 167–174. [Google Scholar] [CrossRef]
- Al-Awaida, W.J.; Jawabrah Al Hourani, B.; Swedan, S.; Nimer, R.; Alzoughool, F.; Al-Ameer, H.J.; Al Tamam, S.E.; Alashqar, R.; Al Bawareed, O.; Gushchina, Y.; et al. Correlates of SARS-CoV-2 Variants on Deaths, Case Incidence and Case Fatality Ratio among the Continents for the Period of 1 December 2020 to 15 March 2021. Genes 2021, 12, 1061. [Google Scholar] [CrossRef]
- Uaprasert, N.; Tangcheewinsirikul, N.; Rojnuckarin, P.; Patell, R.; Zwicker, J.I.; Chiasakul, T. Heparin-Induced Thrombocytopenia in Patients with COVID-19: A Systematic Review and Meta-Analysis. Blood Adv. 2021, 5, 4521–4534. [Google Scholar] [CrossRef] [PubMed]
- Courtney, L.A.; Trujillo, T.C.; Saseen, J.J.; Wright, G.; Palkimas, S. Evaluation of the Clinical Impact of Thromboprophylaxis in Patients With COVID-19 Following Hospital Discharge. Ann. Pharmacother. 2022, 56, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Rachina, S.; Belkova, Y.; Shchendrygina, A.; Suvorov, A.; Bourgeois, D.; Karuk, M.; Sitnikova, V.; Dyatlov, N. Safety and Efficacy of Different Anticoagulant Doses for Patients with COVID-19 in the ICU: A Systematic Review and Meta-Analysis. JCM 2023, 12, 2222. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, M.; Bertinato, E.M.; Birocchi, S.; Brizio, C.; Malavolta, D.; Manzoni, M.; Muscarella, G.; Orlandi, M. Pulmonary Embolism or Pulmonary Thrombosis in COVID-19? Is the Recommendation to Use High-Dose Heparin for Thromboprophylaxis Justified? Thromb. Haemost 2020, 120, 1230–1232. [Google Scholar] [CrossRef] [PubMed]
- Montorfano, M.; Leoni, O.; Andreassi, A.; Ludergnani, M.; Moroni, F.; Ancona, M.B.; Landoni, G.; Ciceri, F.; Zangrillo, A. Chronic Anticoagulant Treatment and Risk of Mortality in SARS-Cov2 Patients: A Large Population-Based Study. Minerva. Med. 2023, 114, 628–633. [Google Scholar] [CrossRef]
- Spyropoulos, A.C.; Bonaca, M.P. Studying the Coagulopathy of COVID-19. Lancet 2022, 399, 118–119. [Google Scholar] [CrossRef]


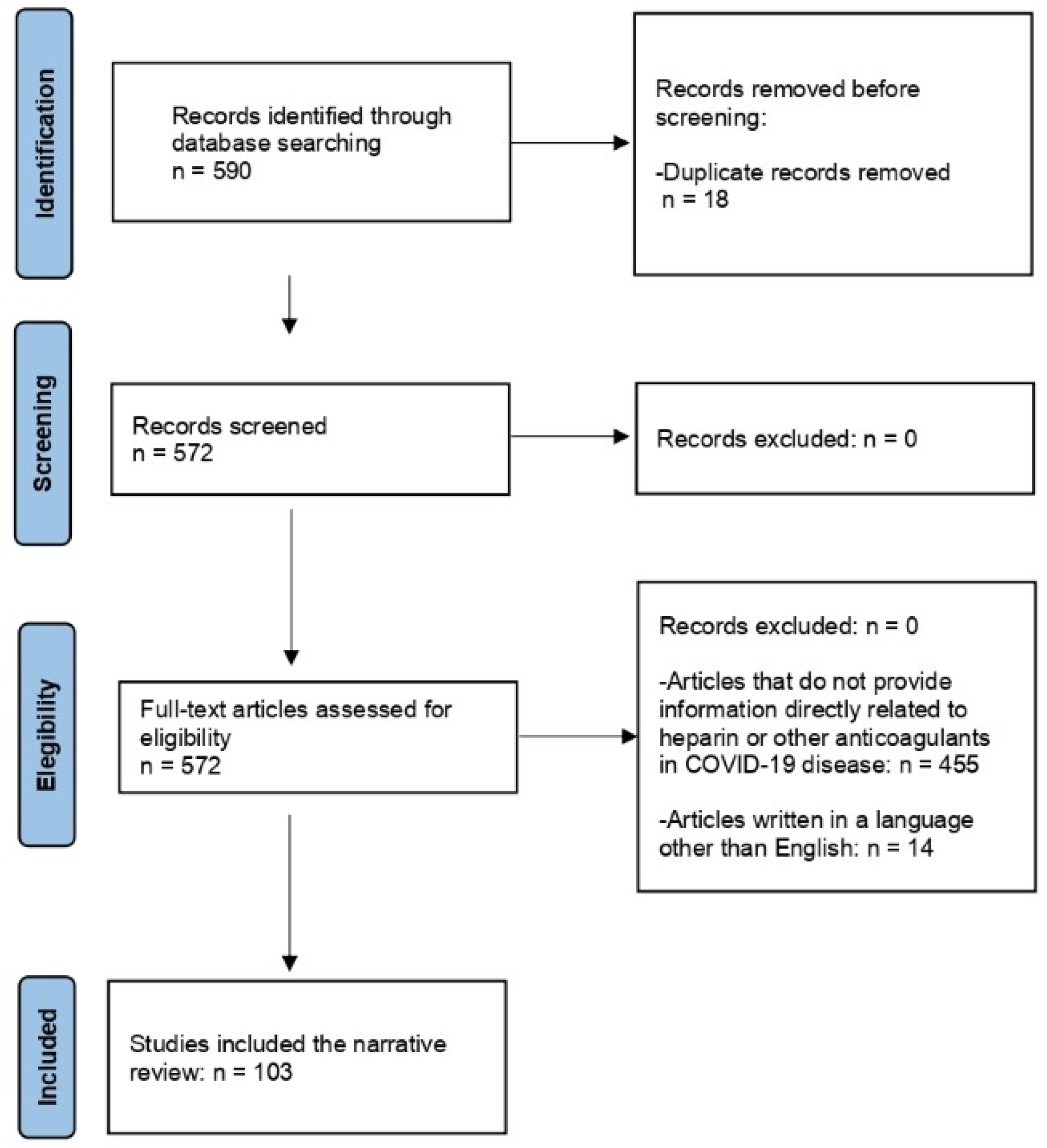{kind=link}
| Mechanisms | SARS-CoV-2 | Heparin |
|---|---|---|
| Antiviral effects | Association between PASC and the viral persistence in the tissues. | Binding to spike glycoprotein. Competition with SARS-CoV-2 for the binding to heparan sulfate. |
| TMPRSS2 | Necessary to provide the virion cellular access. | Activation of antithrombin, and antithrombin binds inhibition. |
| Interleukin-6 | Increased release. | Inhibition. |
| Interferon-gamma | Increased levels. | Prevention of the interferon-gamma—interferon-gamma receptor interaction. |
| TNF-alpha | Production. | Attenuation through a CD11b-dependent mechanism. |
| Complement | Activation. | Inhibition of activation. |
| Angiotensin 2 | Negative regulation of ACE2 activity. Protection against angiotensin-2-induced cardiac remodeling. Vasoconstriction. | Inhibition of angiotensin-2-induced vasoconstriction. |
| HMGB-lipopolysaccharide | Elevation. | Inhibition. |
| Heparanase | Increase. | Inhibition. |
| Heparan sulfate | Interaction of spike glycoprotein’s receptor-binding domain with heparan sulfate. | Binding of heparan sulfate, inhibition of the virus attachment to the cell surface and the viral entry. |
| Histones | Increase. | Neutralization. |
| Pulmonary fibrosis | Increased concentration of the fibrosis-promoting angiotensin 2. Participation of TGF-beta pathways in the development process of pulmonary fibrosis. | Antifibrotic activity, mediated by cellular secretion of hepatocyte growth factor. Activation of TGF-beta by dissociating it from alpha 2-macroglobulin. |
| Endothelial function | Endothelial dysfunction. | Reduction in endothelial adhesiveness. Restoration of the altered membrane’s electronegative potential. Inhibition of the activation of cellular and plasma coagulation factors. Glycocalyx-stabilizing effect. |
| Neutrophil extracellular traps | Increased. | Reduction in endothelial cell damage. |
| Muscarinic and adrenergic receptors | Presence of muscarinic acetylcholine receptor antibody. | Inhibition of muscarinic and α-adrenergic calcium release in smooth muscle. |
| Hepatocyte growth factor | Upregulation. | Antifibrotic activity, mediated by cellular secretion of hepatocyte growth factor. |
| Glycoprotein 1B | Upregulation. | Inhibition of thrombin ligation to glycoprotein Ib. |
| Coagulation | Hypercoagulation, platelet hyperactivity, and abnormal fibrinolysis. Increase in vWF antigen, vWF pro-peptide, and factor VIII. | Anti-Xa activity. Factor IIa inhibition. Decrease in vWF synthesis. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Moyano, E.; Pavón-Morón, J.; Rodríguez-Capitán, J.; Bardán-Rebollar, D.; Ramos-Carrera, T.; Villalobos-Sánchez, A.; Pérez de Pedro, I.; Ruiz-García, F.J.; Mora-Robles, J.; López-Sampalo, A.; et al. The Role of Heparin in Postural Orthostatic Tachycardia Syndrome and Other Post-Acute Sequelae of COVID-19. J. Clin. Med. 2024, 13, 2405. https://doi.org/10.3390/jcm13082405
Gómez-Moyano E, Pavón-Morón J, Rodríguez-Capitán J, Bardán-Rebollar D, Ramos-Carrera T, Villalobos-Sánchez A, Pérez de Pedro I, Ruiz-García FJ, Mora-Robles J, López-Sampalo A, et al. The Role of Heparin in Postural Orthostatic Tachycardia Syndrome and Other Post-Acute Sequelae of COVID-19. Journal of Clinical Medicine. 2024; 13(8):2405. https://doi.org/10.3390/jcm13082405
Chicago/Turabian StyleGómez-Moyano, Elisabeth, Javier Pavón-Morón, Jorge Rodríguez-Capitán, Daniel Bardán-Rebollar, Teresa Ramos-Carrera, Aurora Villalobos-Sánchez, Iván Pérez de Pedro, Francisco J. Ruiz-García, Javier Mora-Robles, Almudena López-Sampalo, and et al. 2024. "The Role of Heparin in Postural Orthostatic Tachycardia Syndrome and Other Post-Acute Sequelae of COVID-19" Journal of Clinical Medicine 13, no. 8: 2405. https://doi.org/10.3390/jcm13082405
APA StyleGómez-Moyano, E., Pavón-Morón, J., Rodríguez-Capitán, J., Bardán-Rebollar, D., Ramos-Carrera, T., Villalobos-Sánchez, A., Pérez de Pedro, I., Ruiz-García, F. J., Mora-Robles, J., López-Sampalo, A., Pérez-Velasco, M. A., Bernal-López, M. -R., Gómez-Huelgas, R., Jiménez-Navarro, M., Romero-Cuevas, M., Costa, F., Trenas, A., & Pérez-Belmonte, L. M. (2024). The Role of Heparin in Postural Orthostatic Tachycardia Syndrome and Other Post-Acute Sequelae of COVID-19. Journal of Clinical Medicine, 13(8), 2405. https://doi.org/10.3390/jcm13082405