
Hypothalamic Obesity in Craniopharyngioma Patients: Disturbed Energy Homeostasis Related to Extent of Hypothalamic Damage and Its Implication for Obesity Intervention

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Clinical Manifestations and Risk Factors
2.1. Hypothalamic Obesity Syndrome
2.2. Hypothalamic Obesity in Patients with Craniopharyngioma
2.3. Cardiometabolic Risk Factors
2.4. Risk Factors for Excessive Weight Gain
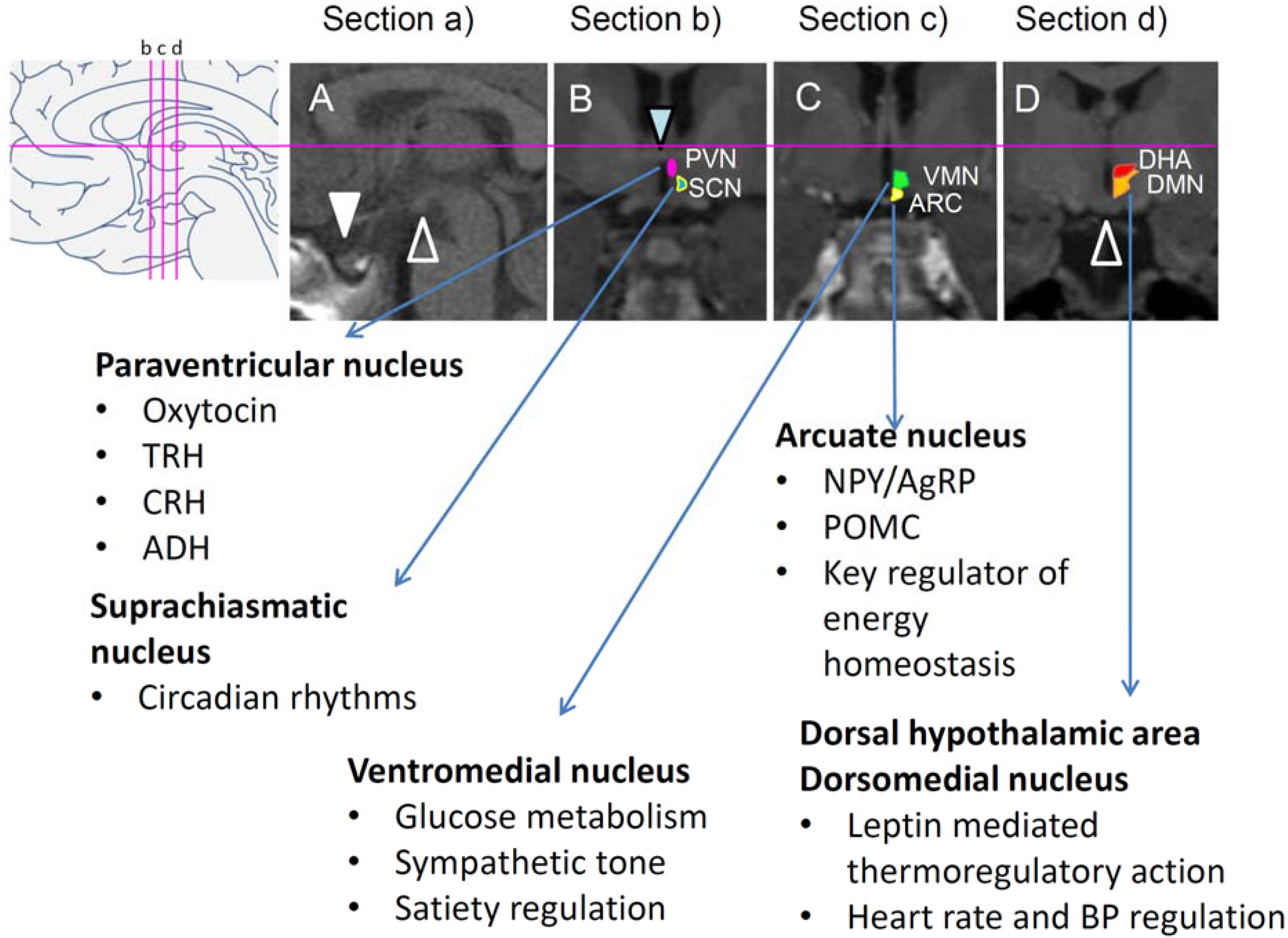
2.5. Key Brain Areas Involved in Disrupted Energy Homeostasis
2.6. Functional Neuroimaging in Patients with CP
2.7. Structural Neuroimaging in Patients with CP

3. Studying Hypothalamic Obesity in Rats
3.1. Hypothalamic Lesion-Induced Obesity
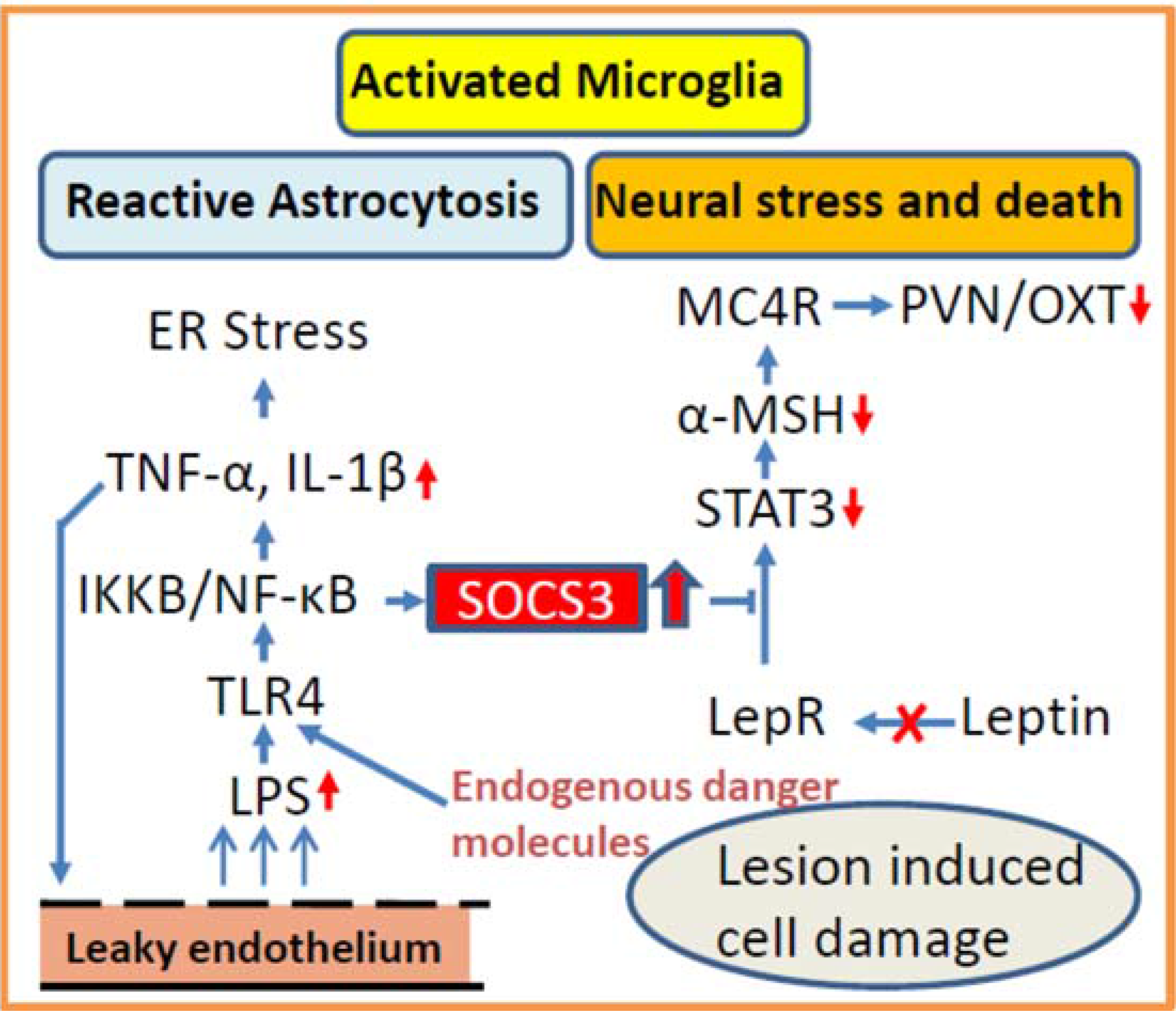
3.2. Inflammation as Potential Contributor for Disturbed Hypothalamic Signaling

4. Pharmacological Interventions
4.1. Rodent Studies
4.2. Studies in Humans
5. Summary
Conflicts of Interest
List of Abbreviations
| α-MSH | alpha-melanocyte-stimulating hormone |
| ARC | arcuate nucleus |
| BMI | body mass index |
| CMHL | combined medial hypothalamic lesions |
| CP | craniopharyngioma |
| CVD | cardiovascular disease |
| DHA | dorsal hypothalamic area |
| DMN | dorsomedial nucleus |
| fMRI | functional magnetic resonance imaging |
| GLP-1 | glucagon-like peptide-1 |
| HLS | hypothalamic lesion scoring |
| HO | hypothalamic obesity |
| IL | interleukin |
| IKKВ | inhibitor of nuclear factor kappa-B kinase subunit beta |
| MC | melanocortin |
| MC4R | melanocortin-4 receptor |
| MSG | monosodium l-glutamate |
| MTII | melanotan 2 |
| NF-κB | nuclear factor kappa B |
| OXT | oxytocin |
| PVN | paraventricular nucleus |
| SOCS-3 | suppressor of cytokine signaling-3 |
| TLR4 | toll-like receptor-4 |
| TNF-α | Tumor necrosis factor-α |
| T2DM | type 2 diabetes mellitus |
| T3 | tri-iodothyronine |
| T4 | thyroxine |
| VMN | ventromedial nucleus |
References
- Steinberger, J.; Daniels, S.R. Obesity, insulin resistance, diabetes, and cardiovascular risk in children: An american heart association scientific statement from the atherosclerosis, hypertension, and obesity in the young committee (council on cardiovascular disease in the young) and the diabetes committee (council on nutrition, physical activity, and metabolism). Circulation 2003, 107, 1448–1453. [Google Scholar] [PubMed]
- Ogden, C.L.; Carroll, M.D.; Kit, B.K.; Flegal, K.M. Prevalence of obesity and trends in body mass index among us children and adolescents, 1999–2010. JAMA 2012, 307, 483–490. [Google Scholar] [CrossRef] [PubMed]
- May, A.L.; Kuklina, E.V.; Yoon, P.W. Prevalence of cardiovascular disease risk factors among us adolescents, 1999–2008. Pediatrics 2012, 129, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Freedman, D.S.; Mei, Z.; Srinivasan, S.R.; Berenson, G.S.; Dietz, W.H. Cardiovascular risk factors and excess adiposity among overweight children and adolescents: The bogalusa heart study. J. Pediatr. 2007, 150, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Heymsfield, S.B.; Avena, N.M.; Baier, L.; Brantley, P.; Bray, G.A.; Burnett, L.C.; Butler, M.G.; Driscoll, D.J.; Egli, D.; Elmquist, J.; et al. Hyperphagia: Current concepts and future directions proceedings of the 2nd international conference on hyperphagia. Obesity (Silver Spring) 2014, 22, S1–S17. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.L. Hypothalamic obesity in patients with craniopharyngioma: Profound changes of several weight regulatory circuits. Front. Endocrinol. (Lausanne) 2011, 2, 49. [Google Scholar] [CrossRef] [PubMed]
- Muller, H.L. Craniopharyngioma. Endocr. Rev. 2014, er20131115. [Google Scholar]
- Goldstone, A.P. The hypothalamus, hormones, and hunger: Alterations in human obesity and illness. Prog. Brain Res. 2006, 153, 57–73. [Google Scholar] [PubMed]
- Zegers, D.; van Hul, W.; van Gaal, L.F.; Beckers, S. Monogenic and complex forms of obesity: Insights from genetics reveal the leptin-melanocortin signaling pathway as a common player. Crit. Rev. Eukaryot. Gene Expr. 2012, 22, 325–343. [Google Scholar] [CrossRef] [PubMed]
- Cai, D. Neuroinflammation and neurodegeneration in overnutrition-induced diseases. Trends Endocrinol. Metab. 2013, 24, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, I.S. Monogenic human obesity syndromes. Prog. Brain Res. 2006, 153, 119–125. [Google Scholar] [PubMed]
- Harwood-Nash, D.C. Neuroimaging of childhood craniopharyngioma. Pediatr. Neurosurg. 1994, 21, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Dolecek, T.A.; Propp, J.M.; Stroup, N.E.; Kruchko, C. Cbtrus statistical report: Primary brain and central nervous system tumors diagnosed in the united states in 2005–2009. Neuro Oncol. 2012, 14, v1–v49. [Google Scholar] [CrossRef] [PubMed]
- Hankinson, T.C.; Fields, E.C.; Torok, M.R.; Beaty, B.L.; Handler, M.H.; Foreman, N.K.; O’Neill, B.R.; Liu, A.K. Limited utility despite accuracy of the national seer dataset for the study of craniopharyngioma. J. Neurooncol. 2012, 110, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. Cbtrus statistical report: Primary brain and central nervous system tumors diagnosed in the united states in 2006–2010. Neuro Oncol. 2013, 15, ii1–ii56. [Google Scholar] [CrossRef] [PubMed]
- Muller, H.L. Consequences of craniopharyngioma surgery in children. J. Clin. Endocrinol. Metab. 2011, 96, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Crom, D.B.; Smith, D.; Xiong, Z.; Onar, A.; Hudson, M.M.; Merchant, T.E.; Morris, E.B. Health status in long-term survivors of pediatric craniopharyngiomas. J. Neurosci. Nurs. 2010, 42, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.A.; Wilkening, G.N.; Filley, C.M.; Reardon, M.S.; Kleinschmidt-DeMasters, B.K. Neurobehavioral outcome in pediatric craniopharyngioma. Pediatr. Neurosurg. 1997, 26, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Muller, H.L.; Bruhnken, G.; Emser, A.; Faldum, A.; Etavard-Gorris, N.; Gebhardt, U.; Kolb, R.; Sorensen, N. Longitudinal study on quality of life in 102 survivors of childhood craniopharyngioma. Childs Nerv. Syst. 2005, 21, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Erfurth, E.M. Endocrine aspects and sequel in patients with craniopharyngioma. J. Pediatr. Endocrinol. Metab. 2015, 28, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Olsson, D.S.; Andersson, E.; Bryngelsson, I.L.; Nilsson, A.G.; Johannsson, G. Excess mortality and morbidity in patients with craniopharyngioma, especially in patients with childhood onset: A population-based study in sweden. J. Clin. Endocrinol. Metab. 2015, 100, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Sorva, R. Children with craniopharyngioma. Early growth failure and rapid postoperative weight gain. Acta Paediatr. Scand. 1988, 77, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Muller, H.L.; Emser, A.; Faldum, A.; Bruhnken, G.; Etavard-Gorris, N.; Gebhardt, U.; Oeverink, R.; Kolb, R.; Sorensen, N. Longitudinal study on growth and body mass index before and after diagnosis of childhood craniopharyngioma. J. Clin. Endocrinol. Metab. 2004, 89, 3298–3305. [Google Scholar] [CrossRef] [PubMed]
- Muller, H.L.; Heinrich, M.; Bueb, K.; Etavard-Gorris, N.; Gebhardt, U.; Kolb, R.; Sorensen, N. Perioperative dexamethasone treatment in childhood craniopharyngioma—Influence on short-term and long-term weight gain. Exp. Clin. Endocrinol. Diabetes 2003, 111, 330–334. [Google Scholar] [PubMed]
- Roth, C.L.; Eslamy, H.; Werny, D.; Elfers, C.; Shaffer, M.L.; Pihoker, C.; Ojemann, J.; Dobyns, W.B. Semiquantitative analysis of hypothalamic damage on mri predicts risk for hypothalamic obesity. Obesity (Silver Spring) 2015, 23, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Daubenbuchel, A.M.; Hoffmann, A.; Gebhardt, U.; Warmuth-Metz, M.; Sterkenburg, A.S.; Muller, H.L. Hydrocephalus and hypothalamic involvement in pediatric patients with craniopharyngioma or cysts of rathke’s pouch: Impact on long-term prognosis. Eur. J. Endocrinol. 2015, 172, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Holmer, H.; Pozarek, G.; Wirfalt, E.; Popovic, V.; Ekman, B.; Bjork, J.; Erfurth, E.M. Reduced energy expenditure and impaired feeding-related signals but not high energy intake reinforces hypothalamic obesity in adults with childhood onset craniopharyngioma. J. Clin. Endocrinol. Metab. 2010, 95, 5395–5402. [Google Scholar] [CrossRef] [PubMed]
- Harz, K.J.; Muller, H.L.; Waldeck, E.; Pudel, V.; Roth, C. Obesity in patients with craniopharyngioma: Assessment of food intake and movement counts indicating physical activity. J. Clin. Endocrinol. Metab. 2003, 88, 5227–5231. [Google Scholar] [CrossRef] [PubMed]
- Lustig, R.H.; Hinds, P.S.; Ringwald-Smith, K.; Christensen, R.K.; Kaste, S.C.; Schreiber, R.E.; Rai, S.N.; Lensing, S.Y.; Wu, S.; Xiong, X. Octreotide therapy of pediatric hypothalamic obesity: A double-blind, placebo-controlled trial. J. Clin. Endocrinol. Metab. 2003, 88, 2586–2592. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.; Wilken, B.; Hanefeld, F.; Schroter, W.; Leonhardt, U. Hyperphagia in children with craniopharyngioma is associated with hyperleptinaemia and a failure in the downregulation of appetite. Eur. J. Endocrinol. 1998, 138, 89–91. [Google Scholar] [CrossRef] [PubMed]
- Patel, L.; Cooper, C.D.; Quinton, N.D.; Butler, G.E.; Gill, M.S.; Jefferson, I.G.; Kibirige, M.S.; Price, D.A.; Shalet, S.M.; Wales, J.K.; et al. Serum leptin and leptin binding activity in children and adolescents with hypothalamic dysfunction. J. Pediatr. Endocrinol. Metab. 2002, 15, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, M.G.; Grundy, R.G.; Kirk, J.M. Hyperleptinaemia rather than fasting hyperinsulinaemia is associated with obesity following hypothalamic damage in children. Eur. J. Endocrinol. 2008, 159, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Guran, T.; Turan, S.; Bereket, A.; Akcay, T.; Unluguzel, G.; Bas, F.; Gunoz, H.; Saka, N.; Bundak, R.; Darendeliler, F.; et al. The role of leptin, soluble leptin receptor, resistin, and insulin secretory dynamics in the pathogenesis of hypothalamic obesity in children. Eur. J. Pediatr. 2009, 168, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Simoneau-Roy, J.; O’Gorman, C.; Pencharz, P.; Adeli, K.; Daneman, D.; Hamilton, J. Insulin sensitivity and secretion in children and adolescents with hypothalamic obesity following treatment for craniopharyngioma. Clin. Endocrinol. (Oxf.) 2010, 72, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.L.; Enriori, P.J.; Gebhardt, U.; Hinney, A.; Muller, H.L.; Hebebrand, J.; Reinehr, T.; Cowley, M.A. Changes of peripheral alpha-melanocyte-stimulating hormone in childhood obesity. Metabolism 2010, 59, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.L.; Hunneman, D.H.; Gebhardt, U.; Stoffel-Wagner, B.; Reinehr, T.; Muller, H.L. Reduced sympathetic metabolites in urine of obese patients with craniopharyngioma. Pediatr. Res. 2007, 61, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.L.; Gebhardt, U.; Muller, H.L. Appetite-regulating hormone changes in patients with craniopharyngioma. Obesity (Silver Spring) 2011, 19, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Muller, H.L.; Bueb, K.; Bartels, U.; Roth, C.; Harz, K.; Graf, N.; Korinthenberg, R.; Bettendorf, M.; Kuhl, J.; Gutjahr, P.; et al. Obesity after childhood craniopharyngioma—German multicenter study on pre-operative risk factors and quality of life. Klin. Padiatr. 2001, 213, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, M.G.; Grundy, R.G.; Kirk, J.M. Reductions in basal metabolic rate and physical activity contribute to hypothalamic obesity. J. Clin. Endocrinol. Metab. 2008, 93, 2588–2593. [Google Scholar] [CrossRef] [PubMed]
- Coutant, R.; Maurey, H.; Rouleau, S.; Mathieu, E.; Mercier, P.; Limal, J.M.; le Bouil, A. Defect in epinephrine production in children with craniopharyngioma: Functional or organic origin? J. Clin. Endocrinol. Metab. 2003, 88, 5969–5975. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.L.; Blevins, J.E.; Ralston, M.; Elfers, C.; Ogimoto, K.; Kaiyala, K.J.; Morton, G.J. A novel rodent model that mimics the metabolic sequelae of obese craniopharyngioma patients. Pediatr. Res. 2011, 69, 230–236. [Google Scholar] [CrossRef] [PubMed]
- O’Gorman, C.S.; Simoneau-Roy, J.; Pencharz Mb, P.; Adeli, K.; Hamilton, J. Delayed ghrelin suppression following oral glucose tolerance test in children and adolescents with hypothalamic injury secondary to craniopharyngioma compared with obese controls. Int. J. Pediatr. Obes. 2011, 6, 285–288. [Google Scholar] [CrossRef] [PubMed]
- An, J.J.; Rhee, Y.; Kim, S.H.; Kim, D.M.; Han, D.H.; Hwang, J.H.; Jin, Y.J.; Cha, B.S.; Baik, J.H.; Lee, W.T.; et al. Peripheral effect of alpha-melanocyte-stimulating hormone on fatty acid oxidation in skeletal muscle. J. Biol. Chem. 2007, 282, 2862–2870. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Ogle, G.D.; Garnett, S.P.; Briody, J.N.; Lee, J.W.; Cowell, C.T. Features of the metabolic syndrome after childhood craniopharyngioma. J. Clin. Endocrinol. Metab. 2004, 89, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Bulow, B.; Attewell, R.; Hagmar, L.; Malmstrom, P.; Nordstrom, C.H.; Erfurth, E.M. Postoperative prognosis in craniopharyngioma with respect to cardiovascular mortality, survival, and tumor recurrence. J. Clin. Endocrinol. Metab. 1998, 83, 3897–3904. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.M.; Schmid, E.M.; Schutte, P.J.; Voormolen, J.H.; Biermasz, N.R.; van Thiel, S.W.; Corssmit, E.P.; Smit, J.W.; Roelfsema, F.; Romijn, J.A. High prevalence of long-term cardiovascular, neurological and psychosocial morbidity after treatment for craniopharyngioma. Clin. Endocrinol. (Oxf.) 2005, 62, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Mong, S.; Pomeroy, S.L.; Cecchin, F.; Juraszek, A.; Alexander, M.E. Cardiac risk after craniopharyngioma therapy. Pediatr. Neurol. 2008, 38, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Van Gaal, L.F.; Mertens, I.L.; de Block, C.E. Mechanisms linking obesity with cardiovascular disease. Nature 2006, 444, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Greenway, F.L.; Bray, G.A. Treatment of hypothalamic obesity with caffeine and ephedrine. Endocr. Pract. 2008, 14, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.K.; Conwell, L.S.; Syme, C.; Ahmet, A.; Jeffery, A.; Daneman, D. Hypothalamic obesity following craniopharyngioma surgery: Results of a pilot trial of combined diazoxide and metformin therapy. Int. J. Pediatr. Endocrinol. 2011, 2011, 417949. [Google Scholar] [CrossRef] [PubMed]
- Bereket, A.; Kiess, W.; Lustig, R.H.; Muller, H.L.; Goldstone, A.P.; Weiss, R.; Yavuz, Y.; Hochberg, Z. Hypothalamic obesity in children. Obes. Rev. 2012, 13, 780–798. [Google Scholar] [CrossRef] [PubMed]
- Lustig, R.H. Hypothalamic obesity after craniopharyngioma: Mechanisms, diagnosis, and treatment. Front. Endocrinol. (Lausanne) 2011, 2, 60. [Google Scholar] [CrossRef] [PubMed]
- Lustig, R.H. Hypothalamic obesity: Causes, consequences, treatment. Pediatr. Endocrinol. Rev. 2008, 6, 220–227. [Google Scholar] [PubMed]
- Danielsson, P.; Janson, A.; Norgren, S.; Marcus, C. Impact sibutramine therapy in children with hypothalamic obesity or obesity with aggravating syndromes. J. Clin. Endocrinol. Metab. 2007, 92, 4101–4106. [Google Scholar] [CrossRef] [PubMed]
- Mason, P.W.; Krawiecki, N.; Meacham, L.R. The use of dextroamphetamine to treat obesity and hyperphagia in children treated for craniopharyngioma. Arch. Pediatr. Adolesc. Med. 2002, 156, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Ismail, D.; O’Connell, M.A.; Zacharin, M.R. Dexamphetamine use for management of obesity and hypersomnolence following hypothalamic injury. J. Pediatr. Endocrinol. Metab. 2006, 19, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Elowe-Gruau, E.; Beltrand, J.; Brauner, R.; Pinto, G.; Samara-Boustani, D.; Thalassinos, C.; Busiah, K.; Laborde, K.; Boddaert, N.; Zerah, M.; et al. Childhood craniopharyngioma: Hypothalamus-sparing surgery decreases the risk of obesity. J. Clin. Endocrinol. Metab. 2013, 98, 2376–2382. [Google Scholar] [CrossRef] [PubMed]
- Muller, H.L.; Gebhardt, U.; Teske, C.; Faldum, A.; Zwiener, I.; Warmuth-Metz, M.; Pietsch, T.; Pohl, F.; Sorensen, N.; Calaminus, G. Post-operative hypothalamic lesions and obesity in childhood craniopharyngioma: Results of the multinational prospective trial kraniopharyngeom 2000 after 3-year follow-up. Eur. J. Endocrinol. 2011, 165, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.L.; Eslamy, H.; Pihoker, C.; Elfers, C.; Ojemann, J.; Dobyns, W.B. Semi-quantitative analysis of hypothalamic damage on mri predicts risk for hypothalamic obesity. Obesity 2014, 23, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Muller, H.L. Childhood craniopharyngioma—Current concepts in diagnosis, therapy and follow-up. Nat. Rev. Endocrinol. 2010, 6, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Muller, H.L. Childhood craniopharyngioma. Recent advances in diagnosis, treatment and follow-up. Horm. Res. 2008, 69, 193–202. [Google Scholar] [PubMed]
- Batterham, R.L.; ffytche, D.H.; Rosenthal, J.M.; Zelaya, F.O.; Barker, G.J.; Withers, D.J.; Williams, S.C. Pyy modulation of cortical and hypothalamic brain areas predicts feeding behaviour in humans. Nature 2007, 450, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Figlewicz, D.P.; Benoit, S.C. Insulin, leptin, and food reward: Update 2008. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R9–R19. [Google Scholar] [CrossRef] [PubMed]
- Morton, G.J.; Cummings, D.E.; Baskin, D.G.; Barsh, G.S.; Schwartz, M.W. Central nervous system control of food intake and body weight. Nature 2006, 443, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Bouret, S.G.; Draper, S.J.; Simerly, R.B. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science 2004, 304, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Glavas, M.M.; Joachim, S.E.; Draper, S.J.; Smith, M.S.; Grove, K.L. Melanocortinergic activation by melanotan ii inhibits feeding and increases uncoupling protein 1 messenger ribonucleic acid in the developing rat. Endocrinology 2007, 148, 3279–3287. [Google Scholar] [CrossRef] [PubMed]
- Mihaly, E.; Fekete, C.; Legradi, G.; Lechan, R.M. Hypothalamic dorsomedial nucleus neurons innervate thyrotropin-releasing hormone-synthesizing neurons in the paraventricular nucleus. Brain Res. 2001, 891, 20–31. [Google Scholar] [CrossRef]
- van Swieten, M.M.; Pandit, R.; Adan, R.A.; van der Plasse, G. The neuroanatomical function of leptin in the hypothalamus. J. Chem. Neuroanat. 2014, 61–62, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.J.; Johnstone, L.E.; Leng, G. Neuroendocrine mechanisms of change in food intake during pregnancy: A potential role for brain oxytocin. Physiol. Behav. 2007, 91, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Matarazzo, V.; Schaller, F.; Nedelec, E.; Benani, A.; Penicaud, L.; Muscatelli, F.; Moyse, E.; Bauer, S. Inactivation of socs3 in the hypothalamus enhances the hindbrain response to endogenous satiety signals via oxytocin signaling. J. Neurosci. 2012, 32, 17097–17107. [Google Scholar] [CrossRef] [PubMed]
- Munzberg, H.; Flier, J.S.; Bjorbaek, C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology 2004, 145, 4880–4889. [Google Scholar] [CrossRef] [PubMed]
- Maejima, Y.; Sedbazar, U.; Suyama, S.; Kohno, D.; Onaka, T.; Takano, E.; Yoshida, N.; Koike, M.; Uchiyama, Y.; Fujiwara, K.; et al. Nesfatin-1-regulated oxytocinergic signaling in the paraventricular nucleus causes anorexia through a leptin-independent melanocortin pathway. Cell Metab. 2009, 10, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Holder, J.L., Jr.; Butte, N.F.; Zinn, A.R. Profound obesity associated with a balanced translocation that disrupts the sim1 gene. Hum. Mol. Genet. 2000, 9, 101–108. [Google Scholar] [PubMed]
- Swaab, D.F.; Purba, J.S.; Hofman, M.A. Alterations in the hypothalamic paraventricular nucleus and its oxytocin neurons (putative satiety cells) in prader-willi syndrome: A study of five cases. J. Clin. Endocrinol. Metab. 1995, 80, 573–579. [Google Scholar] [PubMed]
- Hochberg, I.; Hochberg, Z. Expanding the definition of hypothalamic obesity. Obes. Rev. 2010, 11, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.; Syme, C.; McCrindle, B.W.; Hamilton, J. Autonomic nervous system balance in children and adolescents with craniopharyngioma and hypothalamic obesity. Eur. J. Endocrinol. 2013, 168, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Kitano, M.; Taneda, M. Extended transsphenoidal surgery for suprasellar craniopharyngiomas: Infrachiasmatic radical resection combined with or without a suprachiasmatic trans-lamina terminalis approach. Surg. Neurol. 2009, 71, 290–298, discussion 298. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, I.; Amar, A.P.; Couldwell, W.; Weiss, M.H. Long-term neurological, visual, and endocrine outcomes following transnasal resection of craniopharyngioma. J. Neurosurg. 2005, 102, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.L.; Aylward, E.; Liang, O.; Kleinhans, N.M.; Pauley, G.; Schur, E.A. Functional neuroimaging in craniopharyngioma: A useful tool to better understand hypothalamic obesity? Obes. Facts 2012, 5, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Ozyurt, J.; Lorenzen, A.; Gebhardt, U.; Warmuth-Metz, M.; Muller, H.L.; Thiel, C.M. Remote effects of hypothalamic lesions in the prefrontal cortex of craniopharygioma patients. Neurobiol. Learn. Mem. 2014, 111, 71–80. [Google Scholar] [CrossRef] [PubMed]
- DeVile, C.J.; Grant, D.B.; Hayward, R.D.; Stanhope, R. Growth and endocrine sequelae of craniopharyngioma. Arch. Dis. Child. 1996, 75, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Pascual, J.M.; Carrasco, R.; Prieto, R.; Gonzalez-Llanos, F.; Alvarez, F.; Roda, J.M. Craniopharyngioma classification. J. Neurosurg. 2008, 109, 1180–1182. [Google Scholar] [CrossRef] [PubMed]
- Daousi, C.; Dunn, A.J.; Foy, P.M.; MacFarlane, I.A.; Pinkney, J.H. Endocrine and neuroanatomic features associated with weight gain and obesity in adult patients with hypothalamic damage. Am. J. Med. 2005, 118, 45–50. [Google Scholar] [CrossRef] [PubMed]
- De Vile, C.J.; Grant, D.B.; Hayward, R.D.; Kendall, B.E.; Neville, B.G.; Stanhope, R. Obesity in childhood craniopharyngioma: Relation to post-operative hypothalamic damage shown by magnetic resonance imaging. J. Clin. Endocrinol. Metable 1996, 81, 2734–2737. [Google Scholar]
- Van Gompel, J.J.; Nippoldt, T.B.; Higgins, D.M.; Meyer, F.B. Magnetic resonance imaging-graded hypothalamic compression in surgically treated adult craniopharyngiomas determining postoperative obesity. Neurosurg. Focus 2010, 28, E3. [Google Scholar] [CrossRef] [PubMed]
- Puget, S.; Garnett, M.; Wray, A.; Grill, J.; Habrand, J.L.; Bodaert, N.; Zerah, M.; Bezerra, M.; Renier, D.; Pierre-Kahn, A.; et al. Pediatric craniopharyngiomas: Classification and treatment according to the degree of hypothalamic involvement. J. Neurosurg. 2007, 106, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Ahmet, A.; Blaser, S.; Stephens, D.; Guger, S.; Rutkas, J.T.; Hamilton, J. Weight gain in craniopharyngioma—A model for hypothalamic obesity. J. Pediatr. Endocrinol. Metab. 2006, 19, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Elfers, C.; Ralston, M.; Roth, C.L. Studies of different female rat models of hypothalamic obesity. J. Pediatr. Endocrinol. Metab. 2011, 24, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, K.; Matsuzawa, Y.; Fujioka, S.; Kobatake, T.; Keno, Y.; Odaka, H.; Matsuo, T.; Tarui, S. Pvn-lesioned obese rats maintain ambulatory activity and its circadian rhythm. Brain Res. Bull. 1991, 26, 393–396. [Google Scholar] [CrossRef]
- Tokunaga, K.; Bray, G.A.; Matsuzawa, Y. Improved yield of obese rats using a double coordinate system to locate the ventromedial or paraventricular nucleus. Brain Res. Bull. 1993, 32, 191–194. [Google Scholar] [CrossRef]
- Olney, J.W. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science 1969, 164, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Schoelch, C.; Hubschle, T.; Schmidt, I.; Nuesslein-Hildesheim, B. Msg lesions decrease body mass of suckling-age rats by attenuating circadian decreases of energy expenditure. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E604–E611. [Google Scholar] [CrossRef] [PubMed]
- Kubota, A.; Nakagawa, Y.; Igarashi, Y. Studies of gene expression in liver of insulin-like growth factor (igf)-i, igf binding protein-3 and growth hormone (gh) receptor/gh binding protein in rats treated neonatally with monosodium glutamate. Horm. Metab. Res. 1994, 26, 497–503. [Google Scholar] [CrossRef] [PubMed]
- De Souza, C.T.; Nunes, W.M.; Gobatto, C.A.; de Mello, M.A. Insulin secretion in monosodium glutamate (msg) obese rats submitted to aerobic exercise training. Physiol. Chem. Phys. Med. NMR 2003, 35, 43–53. [Google Scholar] [PubMed]
- Perello, M.; Moreno, G.; Gaillard, R.C.; Spinedi, E. Glucocorticoid-dependency of increased adiposity in a model of hypothalamic obesity. Neuro Endocrinol. Lett. 2004, 25, 119–126. [Google Scholar] [PubMed]
- Balbo, S.L.; Bonfleur, M.L.; Carneiro, E.M.; Amaral, M.E.; Filiputti, E.; Mathias, P.C. Parasympathetic activity changes insulin response to glucose and neurotransmitters. Diabetes Metab. 2002, 28, 3S13-17, discussion 13S108–112. [Google Scholar] [PubMed]
- Bonfleur, M.L.; Borck, P.C.; Ribeiro, R.A.; Caetano, L.C.; Soares, G.M.; Carneiro, E.M.; Balbo, S.L. Improvement in the expression of hepatic genes involved in fatty acid metabolism in obese rats supplemented with taurine. Life Sci. 2015, 135, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Leite, N.C.; Montes, E.G.; Fisher, S.V.; Cancian, C.R.; de Oliveira, J.C.; Martins-Pinge, M.C.; Kanunfre, C.C.; Souza, K.L.; Grassiolli, S. Splenectomy attenuates obesity and decreases insulin hypersecretion in hypothalamic obese rats. Metabolism 2015, 64, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Tomankova, V.; Liskova, B.; Skalova, L.; Bartikova, H.; Bousova, I.; Jourova, L.; Anzenbacher, P.; Ulrichova, J.; Anzenbacherova, E. Altered cytochrome p450 activities and expression levels in the liver and intestines of the monosodium glutamate-induced mouse model of human obesity. Life Sci. 2015, 133, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Poletto, A.C.; David-Silva, A.; Yamamoto, A.P.; Machado, U.F.; Furuya, D.T. Reduced slc2a4/glut4 expression in subcutaneous adipose tissue of monosodium glutamate obese mice is recovered after atorvastatin treatment. Diabetol. Metab. Syndr. 2015, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Lubaczeuski, C.; Balbo, S.L.; Ribeiro, R.A.; Vettorazzi, J.F.; Santos-Silva, J.C.; Carneiro, E.M.; Bonfleur, M.L. Vagotomy ameliorates islet morphofunction and body metabolic homeostasis in msg-obese rats. Braz. J. Med. Biol. Res. 2015, 48, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, E.D.; de Caires Junior, L.C.; Musso, C.M.; Macedo de Almeida, M.; Goncalves, C.F.; Pettersen, K.G.; Paes, S.T.; Gonzalez Garcia, R.M.; de Freitas Mathias, P.C.; Torrezan, R.; et al. Altered behavior of adult obese rats by monosodium l-glutamate neonatal treatment is related to hypercorticosteronemia and activation of hypothalamic erk1 and erk2. Nutr. Neurosci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Dolnikoff, M.; Martin-Hidalgo, A.; Machado, U.F.; Lima, F.B.; Herrera, E. Decreased lipolysis and enhanced glycerol and glucose utilization by adipose tissue prior to development of obesity in monosodium glutamate (msg) treated-rats. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 426–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, D.R.; Newhouse, J.P. The toxic effect of sodium l-glutamate on the inner layers of the retina. AMA Arch. Ophthalmol. 1957, 58, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Smyth, C.E.; Natarajan, M.; Wilkinson, M. Neonatal hypothalamic c-fos expression in an excitotoxicity-induced model of precocious puberty. Brain Res. Dev. Brain Res. 1997, 104, 153–162. [Google Scholar] [CrossRef]
- Young, E.; Olney, J.; Akil, H. Selective alterations of opiate receptor subtypes in monosodium glutamate-treated rats. J. Neurochem. 1983, 40, 1558–1564. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, L.K.; Woiciechowsky, C.; Bechmann, I. Inflammatory response after neurosurgery. Best Pract. Res. Clin. Anaesthesiol. 2004, 18, 407–424. [Google Scholar] [CrossRef] [PubMed]
- Martelli, C.; Iavarone, F.; Vincenzoni, F.; Rossetti, D.V.; D’Angelo, L.; Tamburrini, G.; Caldarelli, M.; di Rocco, C.; Messana, I.; Castagnola, M.; et al. Proteomic characterization of pediatric craniopharyngioma intracystic fluid by lc-ms top-down/bottom-up integrated approaches. Electrophoresis 2014, 35, 2172–2183. [Google Scholar] [CrossRef] [PubMed]
- Pettorini, B.L.; Inzitari, R.; Massimi, L.; Tamburrini, G.; Caldarelli, M.; Fanali, C.; Cabras, T.; Messana, I.; Castagnola, M.; di Rocco, C. The role of inflammation in the genesis of the cystic component of craniopharyngiomas. Childs Nerv. Syst. 2010, 26, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Takeshima, H.; Kuratsu, J. Expression of interleukin-6 in human craniopharyngiomas: A possible inducer of tumor-associated inflammation. Int. J. Mol. Med. 2004, 14, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Sparks, R.; Clay, M.; Dallman, M.F. Rats with hypothalamic obesity are insensitive to central leptin injections. Endocrinology 1999, 140, 4426–4433. [Google Scholar] [CrossRef] [PubMed]
- Milanski, M.; Degasperi, G.; Coope, A.; Morari, J.; Denis, R.; Cintra, D.E.; Tsukumo, D.M.; Anhe, G.; Amaral, M.E.; Takahashi, H.K.; et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of tlr4 signaling in hypothalamus: Implications for the pathogenesis of obesity. J. Neurosci. 2009, 29, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.P.; Schwartz, M.W. Minireview: Inflammation and obesity pathogenesis: The hypothalamus heats up. Endocrinology 2010, 151, 4109–4115. [Google Scholar] [CrossRef] [PubMed]
- Oh, I.S.; Thaler, J.P.; Ogimoto, K.; Wisse, B.E.; Morton, G.J.; Schwartz, M.W. Central administration of interleukin-4 exacerbates hypothalamic inflammation and weight gain during high-fat feeding. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E47–E53. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.P.; Choi, S.J.; Schwartz, M.W.; Wisse, B.E. Hypothalamic inflammation and energy homeostasis: Resolving the paradox. Front. Neuroendocrinol. 2010, 31, 79–84. [Google Scholar] [CrossRef] [PubMed]
- El-Haschimi, K.; Pierroz, D.D.; Hileman, S.M.; Bjorbaek, C.; Flier, J.S. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J. Clin. Investig. 2000, 105, 1827–1832. [Google Scholar] [CrossRef] [PubMed]
- Savastano, D.M.; Covasa, M. Adaptation to a high-fat diet leads to hyperphagia and diminished sensitivity to cholecystokinin in rats. J. Nutr. 2005, 135, 1953–1959. [Google Scholar] [PubMed]
- Posey, K.A.; Clegg, D.J.; Printz, R.L.; Byun, J.; Morton, G.J.; Vivekanandan-Giri, A.; Pennathur, S.; Baskin, D.G.; Heinecke, J.W.; Woods, S.C.; et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1003–E1012. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Scarpace, P.J. Circumventing central leptin resistance: Lessons from central leptin and pomc gene delivery. Peptides 2006, 27, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.P.; Choi, S.J.; Sajan, M.P.; Ogimoto, K.; Nguyen, H.T.; Matsen, M.; Benoit, S.C.; Wisse, B.E.; Farese, R.V.; Schwartz, M.W. Atypical protein kinase c activity in the hypothalamus is required for lipopolysaccharide-mediated sickness responses. Endocrinology 2009, 150, 5362–5372. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.; Yi, C.X.; Schur, E.; Guyenet, S.; Hwang, B.; Dietrich, M.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.; et al. Evidence that obesity is associated with hypothalamic injury in rodent models and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Wisse, B.E.; Schwartz, M.W. Does hypothalamic inflammation cause obesity? Cell Metab. 2009, 10, 241–242. [Google Scholar] [CrossRef] [PubMed]
- Velloso, L.A. The brain is the conductor: Diet-induced inflammation overlapping physiological control of body mass and metabolism. Arq. Bras. Endocrinol. Metabol. 2009, 53, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Horvath, T.L.; Sarman, B.; Garcia-Caceres, C.; Enriori, P.J.; Sotonyi, P.; Shanabrough, M.; Borok, E.; Argente, J.; Chowen, J.A.; Perez-Tilve, D.; et al. Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proc. Natl. Acad. Sci. USA 2010, 107, 14875–14880. [Google Scholar] [CrossRef] [PubMed]
- Cazettes, F.; Cohen, J.I.; Yau, P.L.; Talbot, H.; Convit, A. Obesity-mediated inflammation may damage the brain circuit that regulates food intake. Brain Res. 2011, 1373, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.; Torres, R. Role of interleukin-1beta during pain and inflammation. Brain Res. Rev. 2009, 60, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Csuka, E.; Morganti-Kossmann, M.C.; Lenzlinger, P.M.; Joller, H.; Trentz, O.; Kossmann, T. Il-10 levels in cerebrospinal fluid and serum of patients with severe traumatic brain injury: Relationship to il-6, tnf-alpha, tgf-beta1 and blood-brain barrier function. J. Neuroimmunol. 1999, 101, 211–221. [Google Scholar] [CrossRef]
- Kossmann, T.; Hans, V.H.; Imhof, H.G.; Stocker, R.; Grob, P.; Trentz, O.; Morganti-Kossmann, C. Intrathecal and serum interleukin-6 and the acute-phase response in patients with severe traumatic brain injuries. Shock 1995, 4, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Beamer, N.B.; Coull, B.M.; Clark, W.M.; Hazel, J.S.; Silberger, J.R. Interleukin-6 and interleukin-1 receptor antagonist in acute stroke. Ann. Neurol 1995, 37, 800–805. [Google Scholar] [CrossRef] [PubMed]
- Denis, R.G.; Arruda, A.P.; Romanatto, T.; Milanski, M.; Coope, A.; Solon, C.; Razolli, D.S.; Velloso, L.A. Tnf-alpha transiently induces endoplasmic reticulum stress and an incomplete unfolded protein response in the hypothalamus. Neuroscience 2010, 170, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Mallard, C. Innate immune regulation by toll-like receptors in the brain. ISRN Neurol 2012, 2012, 701950. [Google Scholar] [CrossRef] [PubMed]
- Gautron, L.; Lee, C.; Funahashi, H.; Friedman, J.; Lee, S.; Elmquist, J. Melanocortin-4 receptor expression in a vago-vagal circuitry involved in postprandial functions. J. Comp. Neurol. 2010, 518, 6–24. [Google Scholar] [CrossRef] [PubMed]
- Borowsky, B.; Durkin, M.M.; Ogozalek, K.; Marzabadi, M.R.; DeLeon, J.; Lagu, B.; Heurich, R.; Lichtblau, H.; Shaposhnik, Z.; Daniewska, I.; et al. Antidepressant, anxiolytic and anorectic effects of a melanin-concentrating hormone-1 receptor antagonist. Nat. Med. 2002, 8, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.G.; Adams-Deutsch, T.; Pickens, C.L.; Smith, D.G.; Shaham, Y. Effects of the mch1 receptor antagonist snap 94847 on high-fat food-reinforced operant responding and reinstatement of food seeking in rats. Psychopharmacology (Berl.) 2009, 205, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Billes, S.K.; Cowley, M.A. Inhibition of dopamine and norepinephrine reuptake produces additive effects on energy balance in lean and obese mice. Neuropsychopharmacology 2007, 32, 822–834. [Google Scholar] [CrossRef] [PubMed]
- Arch, J.R. The discovery of drugs for obesity, the metabolic effects of leptin and variable receptor pharmacology: Perspectives from beta3-adrenoceptor agonists. Naunyn Schmiedebergs Arch. Pharmacol. 2008, 378, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Cettour-Rose, P.; Rohner-Jeanrenaud, F. The leptin-like effects of 3-d peripheral administration of a melanocortin agonist are more marked in genetically obese zucker (fa/fa) than in lean rats. Endocrinology 2002, 143, 2277–2283. [Google Scholar] [CrossRef] [PubMed]
- Elfers, C.T.; Simmons, J.H.; Roth, C.L. Glucagon-like peptide-1 agonist exendin-4 leads to reduction of weight and caloric intake in a rat model of hypothalamic obesity. Horm. Res. Paediatr. 2012, 78, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Elfers, C.T.; Roth, C.L. Effects of methylphenidate on weight gain and food intake in hypothalamic obesity. Front. Endocrinol. (Lausanne) 2011, 2, 78. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.L.; Elfers, C.T.; Morton, G.J. A Model of Disturbed Weight Regulatory Pathways in Hypothalamic Obesity. In Proceedings of the Abstract NIH NIDDK Scientific Workshop- Rare Syndromic Body Fat Disorders, Bethesda, MD, USA, 1–2 March 2012.
- Li, G.M.; Sun, X.J.; Shao, P. Postoperative pituitary hormonal disturbances and hormone replacement therapy time and dosage in children with craniopharyngiomas. Chin. Med. J. (Engl.) 2008, 121, 2077–2082. [Google Scholar] [PubMed]
- Fernandes, J.K.; Klein, M.J.; Ater, J.L.; Kuttesch, J.F.; Vassilopoulou-Sellin, R. Triiodothyronine supplementation for hypothalamic obesity. Metabolism 2002, 51, 1381–1383. [Google Scholar] [CrossRef] [PubMed]
- Page-Wilson, G.; Wardlaw, S.L.; Khandji, A.G.; Korner, J. Hypothalamic obesity in patients with craniopharyngioma: Treatment approaches and the emerging role of gastric bypass surgery. Pituitary 2012, 15, 84–92. [Google Scholar] [CrossRef] [PubMed]
- van Santen, H.M.; Schouten-Meeteren, A.Y.; Serlie, M.; Meijneke, R.W.; van Trotsenburg, A.S.; Verberne, H.; Holleman, F.; Fliers, E. Effects of t3 treatment on brown adipose tissue and energy expenditure in a patient with craniopharyngioma and hypothalamic obesity. J. Pediatr. Endocrinol. Metab. 2015, 28, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Simmons, J.H.; Shoemaker, A.H.; Roth, C.L. Treatment with glucagon-like peptide-1 agonist exendin-4 in a patient with hypothalamic obesity secondary to intracranial tumor. Horm. Res. Paediatr. 2012, 78, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.S.; Rudser, K.D.; Nathan, B.M.; Fox, C.K.; Metzig, A.M.; Coombes, B.J.; Fitch, A.K.; Bomberg, E.M.; Abuzzahab, M.J. The effect of glucagon-like peptide-1 receptor agonist therapy on body mass index in adolescents with severe obesity: A randomized, placebo-controlled, clinical trial. JAMA Pediatr. 2013, 167, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Zoicas, F.; Droste, M.; Mayr, B.; Buchfelder, M.; Schofl, C. Glp-1 analogues as a new treatment option for hypothalamic obesity in adults: Report of nine cases. Eur. J. Endocrinol. 2013, 168, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Holst, J.J. Incretin hormones and the satiation signal. Int. J. Obes. (Lond.) 2013, 37, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lu, J.; Willars, G.B. Allosteric modulation of the activity of the glucagon-like peptide-1 (glp-1) metabolite glp-1 9–36 amide at the glp-1 receptor. PLoS ONE 2012, 7, e47936. [Google Scholar] [CrossRef] [PubMed]
- Secher, A.; Jelsing, J.; Baquero, A.F.; Hecksher-Sorensen, J.; Cowley, M.A.; Dalboge, L.S.; Hansen, G.; Grove, K.L.; Pyke, C.; Raun, K.; et al. The arcuate nucleus mediates glp-1 receptor agonist liraglutide-dependent weight loss. J. Clin. Investig. 2014, 124, 4473–4488. [Google Scholar] [CrossRef] [PubMed]
- Bloom, S.; Wynne, K.; Chaudhri, O. Gut feeling—The secret of satiety? Clin. Med. 2005, 5, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Bojanowska, E. Physiology and pathophysiology of glucagon-like peptide-1 (glp-1): The role of glp-1 in the pathogenesis of diabetes mellitus, obesity, and stress. Med. Sci. Monit. 2005, 11, RA271–RA278. [Google Scholar] [PubMed]
- Gutzwiller, J.P.; Degen, L.; Heuss, L.; Beglinger, C. Glucagon-like peptide 1 (glp-1) and eating. Physiol. Behav. 2004, 82, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Bruning, J.; Ashcroft, F.M. Glucagon-like peptide 1 stimulates hypothalamic proopiomelanocortin neurons. J. Neurosci. 2007, 27, 7125–7129. [Google Scholar] [CrossRef] [PubMed]
- Van Bloemendaal, L.; RG, I.J.; Ten Kulve, J.S.; Barkhof, F.; Konrad, R.J.; Drent, M.L.; Veltman, D.J.; Diamant, M. Glp-1 receptor activation modulates appetite- and reward-related brain areas in humans. Diabetes 2014, 63, 4186–4196. [Google Scholar] [CrossRef] [PubMed]
- Ottney, A. Glucagon-like peptide-1 receptor agonists for weight loss in adult patients without diabetes. Am. J. Health Syst. Pharm. 2013, 70, 2097–2103. [Google Scholar] [CrossRef] [PubMed]
- Vilsboll, T.; Christensen, M.; Junker, A.E.; Knop, F.K.; Gluud, L.L. Effects of glucagon-like peptide-1 receptor agonists on weight loss: Systematic review and meta-analyses of randomised controlled trials. BMJ 2012, 344, d7771. [Google Scholar] [CrossRef] [PubMed]
- Gedulin, B.R.; Nikoulina, S.E.; Smith, P.A.; Gedulin, G.; Nielsen, L.L.; Baron, A.D.; Parkes, D.G.; Young, A.A. Exenatide (exendin-4) improves insulin sensitivity and {beta}-cell mass in insulin-resistant obese fa/fa zucker rats independent of glycemia and body weight. Endocrinology 2005, 146, 2069–2076. [Google Scholar] [CrossRef] [PubMed]
- Buse, J.B.; Nauck, M.; Forst, T.; Sheu, W.H.; Shenouda, S.K.; Heilmann, C.R.; Hoogwerf, B.J.; Gao, A.; Boardman, M.K.; Fineman, M.; et al. Exenatide once weekly versus liraglutide once daily in patients with type 2 diabetes (duration-6): A randomised, open-label study. Lancet 2013, 381, 117–124. [Google Scholar] [CrossRef]
- Aroda, V.R.; Ratner, R. The safety and tolerability of glp-1 receptor agonists in the treatment of type 2 diabetes: A review. Diabetes Metab. Res. Rev. 2011, 27, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Pinelli, N.R.; Hurren, K.M. Efficacy and safety of long-acting glucagon-like peptide-1 receptor agonists compared with exenatide twice daily and sitagliptin in type 2 diabetes mellitus: A systematic review and meta-analysis. Ann. Pharm. 2011, 45, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Stolar, M.W.; Grimm, M.; Chen, S. Comparison of extended release glp-1 receptor agonist therapy versus sitagliptin in the management of type 2 diabetes. Diabetes Metab. Syndr. Obes. 2013, 6, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Senda, M.; Ogawa, S.; Nako, K.; Okamura, M.; Sakamoto, T.; Ito, S. The glucagon-like peptide-1 analog liraglutide suppresses ghrelin and controls diabetes in a patient with prader-willi syndrome. Endocr. J. 2012, 59, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Linnebjerg, H.; Kothare, P.A.; Skrivanek, Z.; de la Pena, A.; Atkins, M.; Ernest, C.S.; Trautmann, M.E. Exenatide: Effect of injection time on postprandial glucose in patients with type 2 diabetes. Diabet Med. 2006, 23, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Norwood, P.; Liutkus, J.F.; Haber, H.; Pintilei, E.; Boardman, M.K.; Trautmann, M.E. Safety of exenatide once weekly in patients with type 2 diabetes mellitus treated with a thiazolidinedione alone or in combination with metformin for 2 years. Clin. Ther. 2012, 34, 2082–2090. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, N.; Atsumi, Y.; Oura, T.; Saito, H.; Imaoka, T. Efficacy and safety profile of exenatide once weekly compared with insulin once daily in japanese patients with type 2 diabetes treated with oral antidiabetes drug(s): Results from a 26-week, randomized, open-label, parallel-group, multicenter, noninferiority study. Clin. Ther. 2012, 34, 1892–1908. [Google Scholar] [PubMed]
- Jensterle, M.; Kocjan, T.; Kravos, N.A.; Pfeifer, M.; Janez, A. Short-term intervention with liraglutide improved eating behavior in obese women with polycystic ovary syndrome. Endocr. Res. 2015, 40, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Jensterle Sever, M.; Kocjan, T.; Pfeifer, M.; Kravos, N.A.; Janez, A. Short-term combined treatment with liraglutide and metformin leads to significant weight loss in obese women with polycystic ovary syndrome and previous poor response to metformin. Eur. J. Endocrinol. 2014, 170, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Yu, D.M.; Chen, L.M.; Chang, B.C.; Ji, Q.D.; Li, S.Y.; Zhu, M.; Ding, S.H.; Zhang, B.Z.; Wang, S.L.; et al. Liraglutide reduces the body weight and waist circumference in chinese overweight and obese type 2 diabetic patients. Acta Pharmacol. Sin. 2015, 36, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Yuan, G.; Feng, Y.; Zhang, J.; Guo, X. Early liraglutide treatment is better in glucose control, beta-cell function improvement and mass preservation in db/db mice. Peptides 2014, 52, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.T.; Labate, A.M.; Giandalia, A.; Romeo, E.L.; Villari, P.; Alibrandi, A.; Perdichizzi, G.; Cucinotta, D. Twelve-month treatment with liraglutide ameliorates visceral adiposity index and common cardiovascular risk factors in type 2 diabetes outpatients. J. Endocrinol. Investig. 2015, 38, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Maeda, N.; Fujishima, Y.; Fukuda, S.; Nagao, H.; Yamaoka, M.; Hirata, A.; Nishizawa, H.; Funahashi, T.; Shimomura, I. Long-term impact of liraglutide, a glucagon-like peptide-1 (glp-1) analogue, on body weight and glycemic control in japanese type 2 diabetes: An observational study. Diabetol. Metab. Syndr. 2014, 6, 95. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, P.; Civantos, S.; Vega, B.; Pavon, I.; Guijarro, G.; Monereo, S. Clinical effectiveness of exenatide in diabetic patients waiting for bariatric surgery. Obes. Surg. 2015, 25, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Flint, A.; Raben, A.; Ersboll, A.K.; Holst, J.J.; Astrup, A. The effect of physiological levels of glucagon-like peptide-1 on appetite, gastric emptying, energy and substrate metabolism in obesity. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Bradley, D.P.; Kulstad, R.; Racine, N.; Shenker, Y.; Meredith, M.; Schoeller, D.A. Alterations in energy balance following exenatide administration. Appl. Physiol. Nutr. Metab. 2012, 37, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Pannacciulli, N.; Bunt, J.C.; Koska, J.; Bogardus, C.; Krakoff, J. Higher fasting plasma concentrations of glucagon-like peptide 1 are associated with higher resting energy expenditure and fat oxidation rates in humans. Am. J. Clin. Nutr. 2006, 84, 556–560. [Google Scholar] [PubMed]
- Flint, A.; Raben, A.; Rehfeld, J.F.; Holst, J.J.; Astrup, A. The effect of glucagon-like peptide-1 on energy expenditure and substrate metabolism in humans. Int. J. Obes. Relat. Metab. Disord. 2000, 24, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.E.; Kim, D.D.; Marjason, J.; Proietto, J.; Whitehead, J.P.; Vath, J.E. Ascending dose-controlled trial of beloranib, a novel obesity treatment for safety, tolerability, and weight loss in obese women. Obesity (Silver Spring) 2013, 21, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roth, C.L. Hypothalamic Obesity in Craniopharyngioma Patients: Disturbed Energy Homeostasis Related to Extent of Hypothalamic Damage and Its Implication for Obesity Intervention. J. Clin. Med. 2015, 4, 1774-1797. https://doi.org/10.3390/jcm4091774
Roth CL. Hypothalamic Obesity in Craniopharyngioma Patients: Disturbed Energy Homeostasis Related to Extent of Hypothalamic Damage and Its Implication for Obesity Intervention. Journal of Clinical Medicine. 2015; 4(9):1774-1797. https://doi.org/10.3390/jcm4091774
Chicago/Turabian StyleRoth, Christian L. 2015. "Hypothalamic Obesity in Craniopharyngioma Patients: Disturbed Energy Homeostasis Related to Extent of Hypothalamic Damage and Its Implication for Obesity Intervention" Journal of Clinical Medicine 4, no. 9: 1774-1797. https://doi.org/10.3390/jcm4091774
APA StyleRoth, C. L. (2015). Hypothalamic Obesity in Craniopharyngioma Patients: Disturbed Energy Homeostasis Related to Extent of Hypothalamic Damage and Its Implication for Obesity Intervention. Journal of Clinical Medicine, 4(9), 1774-1797. https://doi.org/10.3390/jcm4091774