
Omega-3 Fatty Acids and Cancer Cell Cytotoxicity: Implications for Multi-Targeted Cancer Therapy


Abstract
:1. Introduction
2. Induction of Cancer Cell Apoptosis by n-3 Polyunsaturated Fatty Acids (PUFAs) and Triggering of the Intrinsic and Extrinsic Apoptotic Pathways
2.1. In Vitro and in Vivo Induction of Cancer Cell Apoptosis by n-3 PUFAs

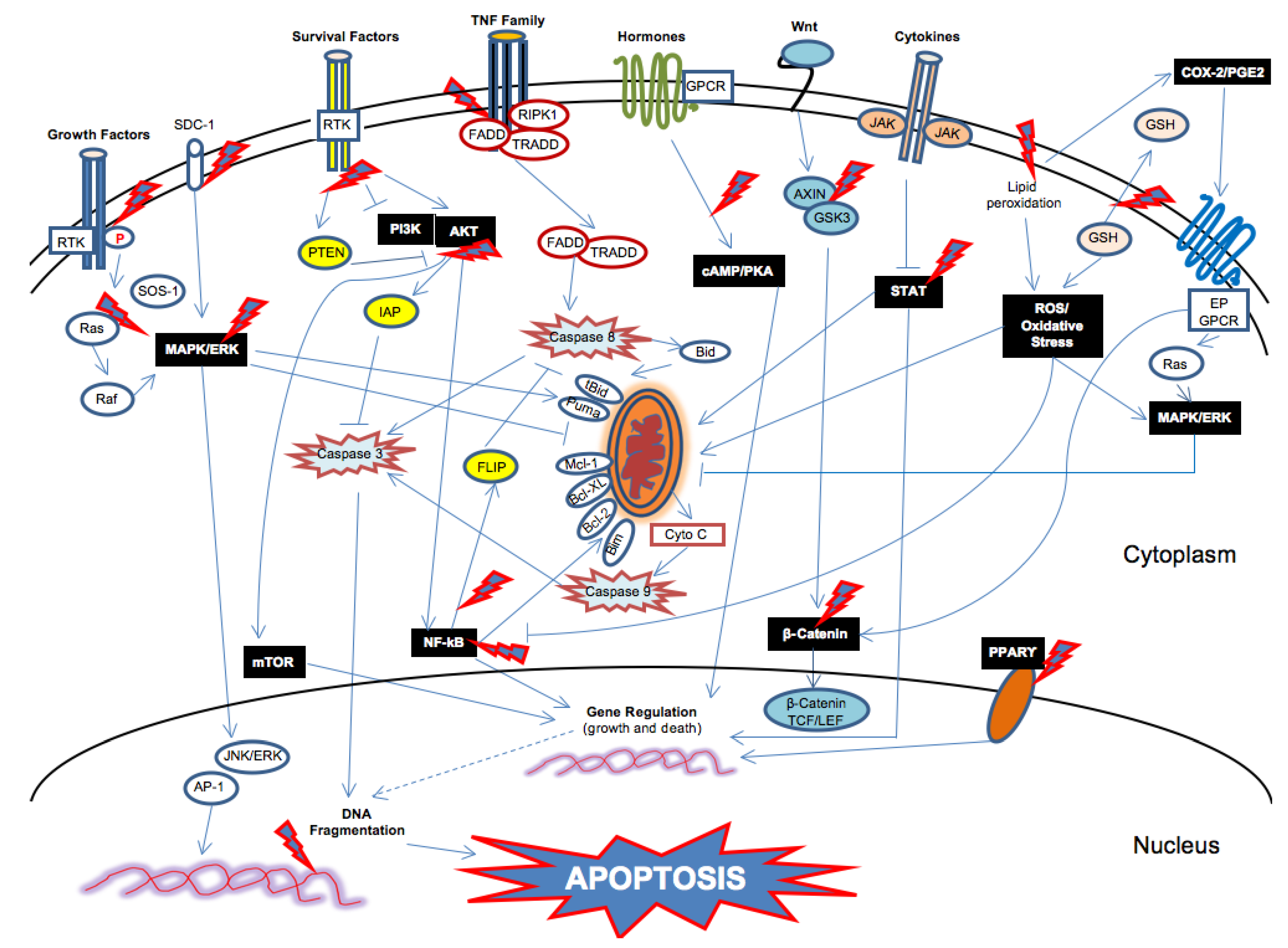{kind=link}
| Cancer Type | Study Type | Enrolled Subjects | Pts (n) | FA/Daily | Objectives | Outcomes | Ref. |
|---|---|---|---|---|---|---|---|
| CRC | Phase II double-blind RCT | Patients under-going liver resection surgery for CRCLM | 43 (T) 45 (C) | EPA (2 g) | To evaluate: ki67 proliferation index; safety and tolerability; tumor FA content; CD31-positive vascularity. | No difference in Ki67 proliferation index. Treatment was safe and well tolerated. EPA was incorporated into CRC liver metastasis tissue. Treatment reduced vascularity of CRC liver metastases. In the first 18 months after CRCLM resection, EPA-treated patients obtained OS benefit compared with control, although early CRC recurrence rates were similar. | [95] |
| CRC | Systematic review and meta-analysis: 9 trials published until September 2014 | Patients with CRC undergoing concomitant surgery (5 trials) or chemotherapy (3 trials) | 242 (T) 233(C) | EPA + DHA (2.2 g: median daily dose (range 0.6-4.8) | To evaluate the effects of n-3 PUFAs on inflammatory mediators (cytokines and acute phase proteins): IL-6 and IL-1β, TNF, CRP and CRP/albumin ratio. | Benefits on some inflammatory mediators, but they are specific for some supplementation protocols (duration, dose, route) and concomitant anti-cancer treatment: reduction in IL-6 occurs in surgical patients that received 0.2 g/kg of FO parenterally at postoperative period (p = 0.002); increase in albumin occurs in surgical patients that received >2.5 g/d of EPA+DHA orally at preoperative period (p = 0.038); in patients undergoing chemo- therapy, the supplementation of 0.6 g/d of EPA+DHA during 9 week reduces CRP levels (p = 0.017), and CRP/albumin ratio (p = 0.016). | [101] |
| CRC | RCT with two arms, parallelgroups,open label | Patients with advanced CRC never submitted to chemotherapy | 17 (T) 13 (C) | FO (2 g); (0.6g/day EPA + DHA) | To evaluate clinical outcomes during and after chemotherapy in individuals with CRC who received FO in the first 9 week of treatment. Outcomes assessed were: number of chemotherapy cycles administered; days undergoing chemotherapy; number of delays and interruptions in the admi-nistration of chemotherapy; number of hospitalizations during chemothery; tumor progression; values of CEA; days until events (death and progression); and 3-year survival. | Time to tumor progression was significantly longer in treated (593 days ±211.5) vs control (330 days ±135.1) patients (P = 0.04); treated patients presented also lower CEA values after chemotherapy (however these differences were not statistically significant); other outcomes did not differ between groups. | [90] |
| Breast cancer | Open-label, one-arm phase II study | Metastatic breast cancer patients undergoing anthracycline-based chemotherapy (5-FU, epirubicin, cyclophosphamide) at first-line treatment for metastases | 25 (T) | DHA (1.8 g) | To investigate the efficacy and safety of adding DHA to an oral supplement ROS generating chemotherapy treatment, by measuring response rate and OS. | No adverse effects. Higher plasma DHA concentrations were associated to greater median time to progression (8.7 months) and OS (34 months) compared to patients with low plasma DHA levels (3.5 and 18 months, respectively). | [91] |
| Breast cancer | A population-based follow-up study (using resources from the Long Island Breast Cancer Study Project) | Women newly diagnosed with first primary in situ (16%) or invasive (84%) breast cancer | 1463 | Variable dietary fish intake | To investigate whether dietary n-3 PUFA intake benefits survival after breast cancer. | All cause mortality was reduced by 16% to 34% among women with breast cancer who reported a high intake of fish and n-3 PUFAs. | [100] |
| NSCLC | Two-arm, non-randomized phase II study | Patients with advanced NSCLC undergoing platinum-based chemotherapy (carboplatin with vinorelbine or gemcitabine) as first-line treatment | 15 (T) 31 (C) | EPA + DHA (2.5 g) | To evaluate whether the combination of FO and chemotherapy provided a benefit over standard of care on response rate and clinical benefit from chemotherapy. | Plasma EPA and DHA were higher in treated patients (p < 0.001 and p = 0.004, respectively). Treated patients had an increased response rate and greater clinical benefit compared with the control group (60.0% vs 25.8%, p = 0.008; 80.0% vs 41.9%, p = 0.02, respectively). The incidence of dose-limiting toxicity did not differ between groups (p = 0.46). One-year survival tended to be greater in treated patients (60.0% vs 38.7%; p = 0.15). | [93] |
| NSCLC | Prospective RCT | Adva-ced NSCLC receiving paclitaxel and cisplatin/carboplatin treatment | 46 (T) 46 (C) | EPA (2 g) | To compare the effect of an oral EPA enriched supplement with an isocaloric diet on nutritional, clinical and inflammatory parameters and health-related quality of life. Response to chemotherapy and survival were also evaluated. | Improvement of energy and protein intake, body composition, and decreased fatigue, loss of appetite and neuropathy. There was no difference in response rate or OS between control and EPA group. | [96] |
| Pancreatic Cancer | A systematic evaluation of results of 11 prospective cohort RCTs | Unresectable pancreatic cancer patients | 602 (T) 765 (C) | EPA (range 1-6 g) and/or DHA (range 0.96-1 g) | To systematically evaluate results of trials examining the effects of n-3 PUFA consumption on body weight, lean body mass, resting energy expenditure, and OS. | A significant increase in body weight (p < 0.00001) and lean body mass (p < 0.00001), a significant decrease in resting energy expenditure (p = 0.03), and an increase in OS (130–259 days vs 63–130 days) in patients who consumed an oral nutrition supplement enriched with n-3 PUFAs compared to those who consumed conventional nutrition. | [98] |
2.2. Triggering of the Intrinsic and Extrinsic Apoptotic Pathways by n-3 PUFAs
3. Molecules, Signals and Networks Targeted by n-3 PUFAs: Upstream Events in the Triggering of the Apoptotic Pathways
3.1. Cell Membrane Enrichment in n-3 PUFAs and Changes in the Distribution and Function of Key Survival and Death Signals in Cancer Cells
3.1.1. Changes in Lipid Raft-Associated Onco-Proteins by n-3 PUFAs
3.1.2. Inhibition of the Wnt/β-Catenin Pathway by n-3 PUFAs
3.1.3. Modulation of the Mitogen-Activated Protein Kinase (MAPK)/ERK (or Ras/Raf/MEK/ERK) Pathway by n-3 PUFAs
3.1.4. Inhibition of the PI3K/Akt/mTOR Pathway by n-3 PUFAs
3.1.5. Inhibition of the JAK-STAT Pathway by n-3 PUFAs
3.1.6. Inhibition of the NF-κB Pathway by n-3 PUFAs
3.2. Cell Membrane Enrichment in n-3 PUFAs and Increased Oxidative Stress in Tumor Cells
Increased Oxidative Stress in Cancer Cells by n-3 PUFAs and Induction of Apoptosis
3.3. Cell Membrane Enrichment in n-3 PUFAs and Changes in the Level and Quality of Eicosanoid Metabolites
Modulation of Eicosanoid Bioproducts by n-3 PUFAs and Induction of Cancer Cell Apoptosis
3.4. Binding of Nuclear Receptors by n-3 PUFAs and Changes in Gene Expression
| Cell Lines | Cancer Type | Fatty Acid | Anti-Cancer Drug | Molecular Targets | Ref. |
|---|---|---|---|---|---|
| Caco-2, HT-29 | Colorectal | FO | - | ↓COX-2 signaling:↓Bcl-2 expression | [103] |
| COLO 201 | Colorectal | DHA | - | Bcl-2 family proteins:↑Bak and Bcl-xS;↓Bcl-xL and Bcl-2 | [104] |
| LS-174, Colo 320 (p53-wild-type), HT-29 and Colo 205 (p53-mutant) | Colorectal | DHA | ↑Susceptibility to 5-FU | Bcl-2 family proteins:↓Bcl-xL and Bcl-2 | [105] |
| SW620 | Colorectal | DHA | - | ↑ER stress genes (ERK-ATF4-CHOP pathway); ↑eIF2α, ↑cytosolic Ca2+; Bcl-2 family proteins: ↑Bid; ↓Bad and Bik | [106] |
| Caco-2 | Colorectal | DHA | - | Modulation of apoptotic genes: caspase-9 and -8 activation; pro-apoptotic Bcl-2 family, PG family, LOX, PPARα and γ | [108] |
| Caco-2, HT-29, HCT116, LoVo, SW480 | Colorectal | DHA, EPA | - | ↓FLIP, ↓XIAP | [39] |
| SW480, HCT116 | Colorectal | DHA | - | ↑Proteosomal degradation of β-catenin: ↓TCF-β-catenin target genes expression (survivin) | [124] |
| Caco-2 | Colorectal | DHA | - | ↓PI3K and↓p38 MAPK/Akt pathway | [130], [40] |
| HT-29 | Colorectal | DHA | ↑ Susceptibility to 5-FU, OX and irinotecan | Caspase-9 activation | [145] |
| HT-29, Caco-2 | Colorectal | EPA, DHA | - | ↑Lipid peroxidation, ↓Bcl-2 levels | [146] |
| HCA-7 | Colorectal | EPA | - | ↑COX-2-dependent PGE2/PGE3 switch | [153] |
| LoVo | Colorectal | EPA(1), DHA(2) | - | (1)↓PGE2, LTB4, COX-2, ALOX and mPGEs; (2)↑LXA4, ↓LTB4, COX-2, ALOX5 and mPGES; ↑PGE2 and LXA4 | [113] |
| MDA-MB-231 | Breast | n-3 PUFAs | - | Lipid raft composition: ↑EGFR onco-protein; ↑EGFR and p38 MAPK signaling | [116] |
| A549, WiDr, MDA-MB-231 | Lung, Colorectal, Breast | DHA | - | Lipid raft composition: ↓EGFR onco-protein; ↓EGFR and ERK signaling | [117] |
| MDA-MB-231, MCF-7 | Breast | EPA, DHA | - | ↓EGFR signaling; ↓Bcl-2; caspase-8 activation | [118] |
| MDA-MB-231 | Breast | DHA | - | Lipid raft internalization: ↓lipid-raft-associated onco-proteins (EGFR, Hsp90, Akt, Src) | [119] |
| HB4aC5.2 | Breast | EPA | - | Lipid raft diruption : ↓HER-2 onco-protein-mediated Akt and ERK1/2 signaling | [43] |
| BT-474 | Breast | DHA | - | ↓HER-2 onco-protein-mediated Akt and ERK1/2 signaling | [120] |
| MCF-7, T47D | Breast | DHA, EPA | - | ↑Estrogen-mediated GPER1-cAMP-PKA signaling | [121] |
| MDA-MB-231 | Breast | DHA | ↑Susceptibility to doxorubicin | ↑CD95-induced apoptosis | [122] |
| MCF-7 | Breast | DHA | - | ↓Wnt/β-catenin pathway | [126] |
| MCF-7, SK-BR-3 | Breast | DHA | - | ↑SDC-1 expression: ↓MEK/ERK/Bad signaling | [127] |
| MDA-MB-231 | Breast | n-3 PUFAs | - | ↓PIK3/Akt/NF-κB signaling | [128] |
| MDA-MB-231 | Breast | DHA, EPA | - | ↑PTEN: ↓PIK3/Akt/NF-κB signaling and ↓transcription of Bcl-2 and Bcl-XL genes | [129] |
| SK-BR-3 | Breast | DHA | - | ↓ERK1/2 and STAT3 signaling | [134] |
| TIC | Breast | DHA | - | ↑SHP-1: ↓STAT3 phosphorylation | [85] |
| MDA-MB-231 | Breast | DHA | ↑Susceptibility to doxorubicin | ↓GPx-1 | [143] |
| MCF-7 | Breast | DHA | - | ↑ROS production and capspase-8 activation | [149] |
| MCF-7 | Breast | DHA | - | PPARγ activation: ↑SDC-1 expression | [162] |
| PC3, LNCaP | Prostate | DHA | - | ↓PIP3 and Akt localization: ↓Akt signaling | [123] |
| PC3, LNCaP, DU145 | Prostate | DHA | - | ↑SDC-1 expression: ↓PDK1/Akt/Bad signaling | [64] |
| PC3, DU145 | Prostate | DHA | - | ↑Mitochondrial ROS: ↓Akt-mTOR signaling | [132] |
| LNCaP, DU145, PC3 | Prostate | DHA | ↑Susceptibility to docetaxel | ↓NF-κB pathway | [135] |
| LNCaP, PacMetUT1 | Prostate | DHA | - | ↓NF-κB pathway: ↓survivin and ↑oxidative stress | [136] |
| PC3 | Prostate | DHA | - | DHA oxidation and 17-HPDHA: binds PPARγ and ↑SDC-1 expression | [163], [164], [165] |
| A549, BEN | Lung | DHA | - | ↑MPK-1:↓ERK1/2 and p38 MAPK phosphorylation | [68] |
| A549, H1299 | Lung | DHA | - | ↑AMPK and ↓PI3K/Akt signaling: ↓mTOR | [133] |
| A549 | Lung | DHA, EPA | - | ↑Oxidative stress: ↑autophagy | [150] |
| AGS | Gastric | DHA | - | ↑ERK and JNK signaling: ↑AP-1, which induces apoptotic genes expression | [52] |
| MGC, SGC | Gastric | EPA, DHA | - | ↑Lipid peroxidation | [147] |
| PaCa-44, MIA-PaCa-2, Capan-2 | Pancreatic | DHA | - | ↑GSH extrusion | [57] |
| MIA-PaCa-2, Capan-2 | Pancreatic | EPA | - | ↑ROS production and caspase-8 activation; ↑autophagy | [58] |
| SW1990, PANC-1 | Pancreatic | DHA, EPA | - | ↑β-catenin/Axin/GSK-3βcomplex-mediated β-catenin degradation | [125] |
| PaCa-44, EJ | Pancreatic, Bladder | DHA | - | Caspase-8 activation | [65] |
| Hep3B, Huh-7, HepG2 | Hepatic | DHA, EPADHA | - | ↑GSK-3β-mediated β-catenin degradation; ↓COX-2/PGE2 signaling | [53] |
| Bel-7402 | Hepatic | DHA | - | Bcl-2 family proteins: ↓Bcl-2 and Bim;↑Bax; caspase-3 activation | [54] |
| HepG2 | Hepatic | EPA | - | ↑ROS-Ca2+-JNK mitochondrial pathway | [55] |
| SCC-13, SCC-25 | Oral squamous cell | EPA | - | ↑EGFR/ERK/p90RSK signaling | [42] |
| CCLP1, HuCCT1, SG231 | Cholangiocarcinoma | DHA, EPA | - | ↓Wnt/β-catenin; ↓COX-2 signaling | [59] |
| SK-N-DZ, SH-SY5Y (chemo-sensitive), SK-N-BE(2) (multi-drug resistant), SK-N-AS, IMR-32 | Neuroblastoma | DHA | ↑Susceptibility to cisplatin, doxorubicin and irinotecan | ↑ROS production and depolarization of mitochondrial membrane potential | [66] |
| SK-N-BE(2) (multi-drug resistant), SH-SY5Y | Neuroblastoma | DHA | ↑Susceptibility to celecoxib | DHA oxidation by 15-LOX to 17-HPDHA; no DHA oxidation by 5-LOX into resolvins and protectins; ↓COX-2/PGE2 signaling | [138] |
| HeLa (expressing HPV-18), SiHa | Cervical | DHA | - | ↑Mitochondrial ROS: ubiquitin-proteasome system activation, leading to E6/E7 onco-proteins degradation | [148] |
| HL-60 | Myeloid leukemia | EPA | - | Caspase-9 and -8 activation | [107] |
| HL-60 (arsenic trioxide resistant), SH-1, Daudi | Myeloid leukemia, Hairy cell leukemia, Burkitt lymphoma | DHA | ↑Susceptibility to arsenic-trioxide | ↑Lipid peroxidation | [144] |
| Ramos | Burkitt’s lymphoma | EPA | - | Caspase-9 and -3 (but not caspase-8) activation | [102] |
| DHL-4 | B cell lymphoma | DHA | - | ↓SOD1 expression | [141] |
| Reh | Acute lymphocytic leukemia | DHA | - | PPARγ activation: ↑p53 protein, activating caspase-9 and -3 | [77] |
| L363, OPM-1, OPM-2, U266 | Multiple myeloma | EPA, DHA | ↑Susceptibility to bortezomib | ↓NF-κB: ↑mytocondrial oxidative stress and caspase-3 activation | [44] |
| SiHa, A549, MCF-7 | Cervical, Lung, Breast | DHA | - | ↓p53/AMPK/mTOR signaling: ↑autophagy | [131] |
| A2780, A2780/CP70, HL-60, Raji, CEM, MCF-7, MM1.S, MM1.R, C8161, HT29, Panc-1 | Ovarian, Leukemia, Breast, Multiple myeloma, Colorectal, Pancreatic | DHA | - | ↓GPx-4 | [142] |
| PA-1, H1299, SiHa, D54MG | Ovarian, Lung, Cervical, Glioblastoma | DHA | - | ↑Mitochondrial ROS: ↑ERK/JNK/p38 signaling | [137] |
| Animal Model | Cancer Type | Diet Fatty Acid | Anti-Cancer Drug | Molecular Targets | Ref. |
|---|---|---|---|---|---|
| Athymic nude mice implanted with human tumor xenograft HCT-15 | Colorectal | FO | - | ↓COX2, HIF-1α/VEGF-A and MMPs signal pathways | [166] |
| Babl/c mice bearing 4T1 mouse breast cancer | Breast | FO | - | ↓Wnt/β-catenin pathway | [126] |
| Athymic nude mice implanted with human tumor xenograft MDA-MB-231 | Breast | FO | - | ↑PTEN expression: ↓PIK3/Akt/NF-κB signaling, ↓transcription of Bcl-2 and Bcl-XL genes, ↑caspase-3 activation | [129] |
| Spontaneous NMU-induced rat mammary tumor | Breast | FO | ↑Susceptibi-lity to epirubicin | ↓GPx-1 response | [143] |
| Athymic nude mice implanted with human tumor xenograft MCF-7 | Breast | FO | - | ↑ROS production and caspase-8 activation | [149] |
| Athymic nude mice implanted with human tumor xenograft MDA-MB-231 | Breast | EPA or DHA ethyl esters | - | ↓PGE2 production | [157] |
| Athymic nude mice implanted with human tumor xenograft DU145 | Prostate | FO | - | ↓PGE2 production | [156] |
| SCID mice implanted with human tumor xenograft LAPC4 | Prostate | FO | - | ↓COX-2/PGE2 pathway | [158] |
| Athymic nude mice implanted with human tumor xenograft A549 | Lung | DHA | - | ↓EGFR onco-protein; ↓EGFR and ERK signaling | [117] |
| Fat-1 transgenic mice implanted with Lewis | Lung | - | - | ↓AMK and PI3K/Akt singnaling: ↑autophagy and apoptosis | [133] |
| Athymic nude mice implanted with human tumor xenograft MIA-PaCa-2 | Pancreatic | FO | - | ↑ROS production; ↑autophagosome formation | [58] |
| Fat-1 transgenic mice implanted with PANC02 | Pancreatic | - | - | ↓Wnt/β-catenin signaling | [125] |
| Athymic nude mice implanted with human tumor xenograft COX-2 negative and positive BxPC-3 | Pancreatic | FO | - | ↓COX-2/PGE2 pathway, ↑PGE3 | [159] |
| Athymic nude rats implanted with human tumor xenograft multi-drug resistant SK-N-BE(2) | Neuroblastoma | DHA | - | ↑lipid peroxidation | [41] |

4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ma, X.; Yu, H. Global burden of cancer. J. Biol. Med. 2006, 79, 85–94. [Google Scholar]
- Basile, K.J.; Aplin, A.E. Resistance to chemotherapy: Short-term drug tolerance and stem cell-like subpopulations. Adv. Pharmacol. 2012, 65, 315–334. [Google Scholar] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Maugeri-Saccà, M.; Vigneri, P.; de Maria, R. Cancer stem cells and chemosensitivity. Clin. Cancer Res. 2011, 17, 4942–4947. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.R.; Bruno, P.M.; Gilbert, L.A.; Capron, K.L.; Lauffenburger, D.A.; Hemann, M.T. Defining principles of combination drug mechanisms of action. Proc. Natl. Acad. Sci. USA 2013, 110, E170–E179. [Google Scholar] [CrossRef] [PubMed]
- Burlingame, B.; Nishida, C.; Uauy, R.; Weisell, R. Fats and fatty acids in human nutrition: Introduction. Ann. Nutr. Metable 2009, 55, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Riediger, N.D.; Othman, R.A.; Suh, M.; Moghadasian, M.H. A systemic review of the roles of n-3 fatty acids in health and disease. J. Am. Diet. Assoc. 2009, 109, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Marine ω-3 Fatty acids and inflammatory processes: Effects, mechanisms and clinical relevance. Biochim. Biophys. Acta 2015, 1851, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Gil, A.; Gil, F. Fish, a Mediterranean source of n-3 PUFA: Benefits do not justify limiting consumption. Br. J. Nutr. 2015, 113, S58–S67. [Google Scholar] [CrossRef] [PubMed]
- Laviano, A.; Rianda, S.; Molfino, A.; Rossi Fanelli, F. ω-3 Fatty acids in cancer. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Bhagat, U.; Das, U.N. Potential role of dietary lipids in the prophylaxis of some clinical conditions. Arch. Med. Sci. 2015, 11, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.; Hraiki, A.; Bebawy, M.; Pazderka, C.; Rawling, T. Anti-tumor activities of lipids and lipid analogues and their development as potential anticancer drugs. Pharmacol. Ther. 2015, 150, 109–128. [Google Scholar] [CrossRef] [PubMed]
- Bang, H.O.; Dyerberg, J.; Nielsen, A.B. Plasma lipid and lipoprotein pattern in Greenlandic West-coast Eskimos. Lancet 1971, 1, 1143–1145. [Google Scholar] [CrossRef]
- Gu, Z.; Shan, K.; Chen, H.; Chen, Y.Q. n-3 Polyunsaturated fatty acids and their role in cancer chemoprevention. Curr. Pharmacol. Rep. 2015, 5, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Serini, S.; Fasano, E.; Piccioni, E.; Cittadini, A.R.; Calviello, G. Dietary n-3 polyunsaturated fatty acids and the paradox of their health benefits and potential harmful effects. Chem. Res. Toxicol. 2011, 24, 2093–2105. [Google Scholar] [CrossRef] [PubMed]
- Chapkin, R.S.; DeClercq, V.; Kim, E.; Fuentes, N.R.; Fan, Y.Y. Mechanisms by which pleiotropic amphiphilic n-3 PUFA reduce colon cancer risk. Curr. Colorectal Cancer Rep. 2014, 10, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Kiyabu, G.Y.; Inoue, M.; Saito, E.; Abe, S.K.; Sawada, N.; Ishihara, J.; Iwasaki, M.; Yamaji, T.; Shimazu, T.; et al. JPHC Study Group. Fish, n-3 polyunsaturated fatty acids and n-6 polyunsaturated fatty acids intake and breast cancer risk: The Japan Public Health Center-based prospective study. Int. J. Cancer. 2015, 137, 2915–2926. [Google Scholar] [CrossRef] [PubMed]
- Brasky, T.M.; Darke, A.K.; Song, X.; Tangen, C.M.; Goodman, P.J.; Thompson, I.M.; Meyskens, F.L., Jr.; Goodman, G.E.; Minasian, L.M.; et al. Plasma phospholipid fatty acids and prostate cancer risk in the SELECT trial. J. Natl. Cancer Inst. 2013, 105, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C.; Deckelbaum, R.J. Dietary fatty acids in health and disease: Greater controversy, greater interest. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Weylandt, K.H.; Serini, S.; Chen, Y.Q.; Su, H.M.; Lim, K.; Cittadini, A.; Calviello, G. ω-3 Polyunsaturated fatty acids: The way forward in times of mixed evidence. Biomed. Res. Int. 2015, 2015, 143109. [Google Scholar] [CrossRef] [PubMed]
- Berquin, I.M.; Edwards, I.J.; Chen, Y.Q. Multi-targeted therapy of cancer by ω-3 Fatty acids. Cancer Lett. 2008, 269, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Serini, S.; Piccioni, E.; Merendino, N.; Calviello, G. Dietary polyunsaturated fatty acids as inducers of apoptosis: Implications for cancer. Apoptosis 2009, 14, 132–152. [Google Scholar] [CrossRef] [PubMed]
- Gleissman, H.; Johnsen, J.I.; Kogner, P. ω-3 Fatty acids in cancer, the protectors of good and the killers of evil? Exp. Cell Res. 2010, 316, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, V.C.; Hassing, M.R.; Lewandowski, P.A. Marine polyunsaturated fatty acids and cancer therapy. Br. J. Cancer 2013, 108, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Biondo, P.D.; Brindley, D.N.; Sawyer, M.B.; Field, C.J. The potential for treatment with dietary long-chain polyunsaturated n-3 fatty acids during chemotherapy. J. Nutr. Biochem. 2008, 19, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, R.A.; Harvey, K.A.; Xu, Z.; Bammerlin, E.M.; Walker, C.; Altenburg, J.D. Docosahexaenoic acid: A natural powerful adjuvant that improves efficacy for anticancer treatment with no adverse effects. Biofactors 2011, 37, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Luo, T.; Li, S.; Zhao, J. The powerful applications of polyunsaturated fatty acids in improving the therapeutic efficacy of anticancer drugs. Expert Opin. Drug Deliv. 2012, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Merendino, N.; Costantini, L.; Manzi, L.; Molinari, R.; D’Eliseo, D.; Velotti, F. Dietary ω -3 polyunsaturated fatty acid DHA: A potential adjuvant in the treatment of cancer. Biomed. Res. Int. 2013, 2013, 310186. [Google Scholar] [CrossRef] [PubMed]
- Hajjaji, N.; Bougnoux, P. Selective sensitization of tumors to chemotherapy by marine-derived lipids: A review. Cancer Treat. Rev. 2013, 39, 473–488. [Google Scholar] [CrossRef] [PubMed]
- de Aguiar Pastore Silva, J.; Emilia de Souza Fabre, M.; Waitzberg, D.L. ω-3 Supplements for patients in chemotherapy and/or radiotherapy: A systematic review. Clin. Nutr. 2015, 34, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N.; Madhavi, N.; Sravan Kumar, G.; Padma, M.; Sangeetha, P. Can tumour cell drug resistance be reversed by essential fatty acids and their metabolites? Prostaglandins Leukot. Essent. Fatty Acids 1998, 58, 39–54. [Google Scholar] [CrossRef]
- Slagsvold, J.E.; Pettersen, C.H.; Størvold, G.L.; Follestad, T.; Krokan, H.E.; Schønberg, S.A. DHA alters expression of target proteins of cancer therapy in chemotherapy resistant SW620 colon cancer cells. Nutr. Cancer 2010, 62, 611–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuan, C.Y.; Walker, T.H.; Luo, P.G.; Chen, C.F. Long-chain polyunsaturated fatty acids promote paclitaxel cytotoxicity via inhibition of the MDR1 gene in the human colon cancer Caco-2 cell line. J. Am. Coll. Nutr. 2011, 30, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Gelsomino, G.; Corsetto, P.A.; Campia, I.; Montorfano, G.; Kopecka, J.; Castella, B.; Gazzano, E.; Ghigo, D.; Rizzo, A.M.; Riganti, C. ω 3 Fatty acids chemosensitize multidrug resistant colon cancer cells by down-regulating cholesterol synthesis and altering detergent resistant membranes composition. Mol. Cancer 2013, 12, 137. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N.; Begin, M.E.; Ells, G.; Huang, Y.S.; Horrobin, D.F. Polyunsaturated fatty acids augment free radical generation in tumor cells in vitro. Biochem. Biophys. Res. Commun. 1987, 145, 15–24. [Google Scholar] [CrossRef]
- Tsai, W.S.; Nagawa, H.; Kaizaki, S.; Tsuruo, T.; Muto, T. Inhibitory effects of n--3 polyunsaturated fatty acids on sigmoid colon cancer transformants. J. Gastroenterol. 1998, 33, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, R.A.; Harvey, K.; Stillwell, W. Anticancer properties of oxidation products of docosahexaenoic acid. Chem. Phys. Lipids 2008, 153, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Giros, A.; Grzybowski, M.; Sohn, V.R.; Pons, E.; Fernandez-Morales, J.; Xicola, R.M.; Sethi, P.; Grzybowski, J.; Goel, A.; et al. Regulation of colorectal cancer cell apoptosis by the n-3 polyunsaturated fatty acids Docosahexaenoic and Eicosapentaenoic. Cancer Prev. Res. 2009, 2, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Toit-Kohn, J.L.; Louw, L.; Engelbrecht, A.M. Docosahexaenoic acid induces apoptosis in colorectal carcinoma cells by modulating the PI3 kinase and p38 MAPK pathways. J. Nutr. Biochem. 2009, 20, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Gleissman, H.; Segerström, L.; Hamberg, M.; Ponthan, F.; Lindskog, M.; Johnsen, J.I.; Kogner, P. ω-3 Fatty acid supplementation delays the progression of neuroblastoma in vivo. Int. J. Cancer 2011, 128, 1703–1711. [Google Scholar] [CrossRef] [PubMed]
- Nikolakopoulou, Z.; Nteliopoulos, G.; Michael-Titus, A.T.; Parkinson, E.K. ω-3 Polyunsaturated fatty acids selectively inhibit growth in neoplastic oral keratinocytes by differentially activating ERK1/2. Carcinogenesis 2013, 34, 2716–2725. [Google Scholar] [CrossRef] [PubMed]
- Ravacci, G.R.; Brentani, M.M.; Tortelli, T.Jr.; Torrinhas, R.S.; Saldanha, T.; Torres, E.A.; Waitzberg, D.L. Lipid raft disruption by docosahexaenoic acid induces apoptosis in transformed human mammary luminal epithelial cells harboring HER-2 overexpression. J. Nutr. Biochem. 2013, 24, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Abdi, J.; Garssen, J.; Faber, J.; Redegeld, F.A. ω-3 Fatty acids, EPA and DHA induce apoptosis and enhance drug sensitivity in multiple myeloma cells but not in normal peripheral mononuclear cells. J. Nutr. Biochem. 2014, 25, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Berstad, P.; Thiis-Evensen, E.; Vatn, M.H.; Almendingen, K. Fatty acids in habitual diet, plasma phospholipids, and tumour and normal colonic biopsies in young colorectal cancer patients. J. Oncol. 2012, 2012, 254801. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.C. Apoptosis and cancer: The genesis of a research field. Nature Rev. Cancer 2009, 9, 501–507. [Google Scholar]
- Logue, S.E.; Gorman, A.M.; Cleary, P.; Keogh, N.; Samali, A. Current concepts in ER stress-induced apoptosis. J. Carcinogene Mutagene 2013. [Google Scholar] [CrossRef]
- Mengeaud, V.; Nano, J.L.; Fournel, S.; Rampal, P. Effects of eicosapentaenoic acid, γ-linolenic acid and prostaglandin E1 on three human colon carcinoma cell lines. Prostaglandins Leukot. Essent. Fatty Acids 1992, 47, 313–319. [Google Scholar] [CrossRef]
- Clarke, R.G.; Lund, E.K.; Latham, P.; Pinder, A.C.; Johnson, I.T. Effect of eicosapentaenoic acid on the proliferation and incidence of apoptosis in the colorectal cell line HT29. Lipids 1999, 34, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; Istfan, N.W. Docosahexaenoic acid is a potent inducer of apoptosis in HT-29 colon cancer cells. Prostaglandins Leukot. Essent. Fatty Acids 2000, 63, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Kubota, H.; Matsumoto, H.; Higashida, M.; Murakami, H.; Nakashima, H.; Oka, Y.; Okumura, H.; Yamamura, M.; Nakamura, M.; Hirai, T. Eicosapentaenoic acid modifies cytokine activity and inhibits cell proliferation in an oesophageal cancer cell line. Anticancer Res. 2013, 33, 4319–4324. [Google Scholar] [PubMed]
- Lee, S.E.; Lim, J.W.; Kim, H. Activator protein-1 mediates docosahexaenoic acid-induced apoptosis of human gastric cancer cells. Ann. N. Y. Acad. Sci. 2009, 1171, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Han, C.; Dai, Y.; Shenm, M.; Wu, T. ω-3 Polyunsaturated fatty acids inhibit hepatocellular carcinoma cell growth through blocking β-catenin and cyclooxygenase-2. Mol. Cancer Ther. 2009, 8, 3046–3055. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.N.; Jia, W.D.; Chen, H.; Ma, J.L.; Ge, Y.S.; Yu, J.H.; Li, J.S. Docosahexaenoic acid (DHA) induces apoptosis in human hepatocellular carcinoma cells. Int. J. of Clin. Exp. Pathol. 2013, 6, 281–289. [Google Scholar]
- Zhang, Y.; Han, L.; Qi, W.; Cheng, D.; Ma, X.; Hou, L.; Cao, X.; Wang, C. Eicosapentaenoic acid (EPA) induced apoptosis in HepG2 cells through ROS-Ca2+-JNK mitochondrial pathways. Biochem. Biophys. Res. Commun. 2015, 456, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, R.A.; Sangster, K.; Arends, M.J. Apoptotic death of pancreatic cancer cells induced by polyunsaturated fatty acids varies with double bond number and involves an oxidative mechanism. J. Pathol. 1998, 185, 61–70. [Google Scholar] [CrossRef]
- Merendino, N.; Loppi, B.; D’Aquino, M.; Molinari, R.; Pessina, G.; Romano, C.; Velotti, F. Docosahexaenoic acid induces apoptosis in the human PaCa-44 pancreatic cancer cell line by active reduced glutathione extrusion and lipid peroxidation. Nutr. Cancer 2005, 52, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Fukui, M.; Kang, K.S.; Okada, K.; Zhu, B.T. EPA, an ω-3 Fatty acid, induces apoptosis in human pancreatic cancer cells: Role of ROS accumulation, caspase-8 activation, and autophagy induction. J. Cell. Biochem. 2013, 114, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Han, C.; Xu, L.; Isse, K.; Demetris, A.J.; Wu, T. Cyclooxygenase-2-derived prostaglandin E2 activates β-catenin in human cholangiocarcinoma cells: Evidence for inhibition of these signaling pathways by ω 3 polyunsaturated fatty acids. Cancer Res. 2008, 68, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Rose, D.P.; Connolly, J.M. Effects of fatty acids and inhibitors of eicosanoid synthesis on the growth of a human breast cancer cell line in culture. Cancer Res. 1990, 50, 7139–7144. [Google Scholar] [PubMed]
- Chamras, H.; Ardashian, A.; Heber, D.; Glaspy, J.A. Fatty acid modulation of MCF-7 human breast cancer cell proliferation, apoptosis and differentiation. J. Nutr. Biochem. 2002, 13, 711–716. [Google Scholar] [CrossRef]
- Sharma, A.; Belna, J.; Logan, J.; Espat, J.; Hurteau, J.A. The effects of ω-3 fatty acids on growth regulation of epithelial ovarian cancer cell lines. Gynecol. Oncol. 2005, 99, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, N.K.; Narayanan, B.A.; Reddy, B.S. A combination of docosahexaenoic acid and celecoxib prevents prostate cancer cell growth in vitro and is associated with modulation of nuclear factor-κB, and steroid hormone receptors. Int. J. Oncol. 2005, 26, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Sun, H.; Owens, R.T.; Gu, Z.; Wu, J.; Chen, Y.Q.; O’Flaherty, J.T.; Edwards, I.J. Syndecan-1-dependent suppression of PDK1/Akt/bad signaling by docosahexaenoic acid induces apoptosis in prostate cancer. Neoplasia 2010, 12, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Molinari, R.; D’Eliseo, D.; Manzi, L.; Zolla, L.; Velotti, F.; Merendino, N. The n3-polyunsaturated fatty acid docosahexaenoic acid induces immunogenic cell death in human cancer cell lines via pre-apoptotic calreticulin exposure. Cancer Immunol. Immunother. 2011, 60, 1503–1507. [Google Scholar] [CrossRef] [PubMed]
- Lindskog, M.; Gleissman, H.; Ponthan, F.; Castro, J.; Kogner, P.; Johnsen, J.I. Neuroblastoma cell death in response to docosahexaenoic acid: Sensitization to chemotherapy and arsenic induced oxidative stress. Int. J. Cancer 2006, 118, 2584–2593. [Google Scholar] [CrossRef] [PubMed]
- Faragó, N.; Fehér, L.Z.; Kitajka, K.; Das, U.N.; Puskás, L.G. MicroRNA profile of polyunsaturated fatty acid treated glioma cells reveal apoptosis-specific expression changes. Lipids Health Dis. 2011, 10, 173. [Google Scholar] [CrossRef] [PubMed]
- Serini, S.; Trombino, S.; Oliva, F.; Piccioni, E.; Monego, G.; Resci, F.; Boninsegna, A.; Picci, N.; Ranelletti, F.O.; Calviello, G. Docosahexaenoic acid induces apoptosis in lung cancer cells by increasing MKP-1 and down-regulating p-ERK1/2 and p-p38 expression. Apoptosis 2008, 13, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.H.; Zhang, X.C.; Fu, T.; Gu, J.Z.; Wang, L.; Wang, Y.; Lai, Y.B.; Wang, Y.Q.; Guo, Y. ω-3 polyunsaturated fatty acids inhibit the proliferation of the lung adenocarcinoma cell line A549 in vitro. Mol. Med. Rep. 2014, 9, 401–406. [Google Scholar] [PubMed]
- Albino, A.P.; Juan, G.; Traganos, F.; Reinhart, L.; Connolly, J.; Rose, D.P.; Darzynkiewicz, Z. Cell cycle arrest and apoptosis of melanoma cells by docosahexaenoic acid: Association with decreased pRb phosphorylation. Cancer Res. 2000, 60, 4139–4145. [Google Scholar] [PubMed]
- Denkins, Y.; Kempf, D.; Ferniz, M.; Nileshwar, S.; Marchetti, D. Role of ω-3 polyunsaturated fatty acids on cyclooxygenase-2 metabolism in brain-metastatic melanoma. J. Lipid Res. 2005, 46, 1278–1284. [Google Scholar] [CrossRef] [PubMed]
- Finstad, H.S.; Myhrstad, M.C.; Heimli, H.; Lømo, J.; Blomhoff, H.K.; Kolset, S.O.; Drevon, C.A. Multiplication and death-type of leukemia cell lines exposed to very long-chain polyunsaturated fatty acids. Leukemia 1998, 12, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Finstad, H.S.; Drevon, C.A.; Kulseth, M.A.; Synstad, A.V.; Knudsen, E.; Kolset, S.O. Cell proliferation, apoptosis and accumulation of lipid droplets in U937-1 cells incubated with eicosapentaenoic acid. Biochem. J. 1998, 336, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Chiu, L.C.M.; Wan, J.M.F. Induction of apoptosis in HL-60 cells by eicosapentaenoic acid (EPA) is associated with downregulation of BCL-2 expression. Cancer Letters 1999, 145, 17–27. [Google Scholar] [CrossRef]
- Chiu, L.C.; Wong, E.Y.; Ooi, V.E. Docosahexaenoic acid modulates different genes in cell cycle and apoptosis to control growth of human leukemia HL-60 cells. Int J Oncol. 2004, 25, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, R.A.; Jenski, L.J.; Neff, K.; Harvey, K.; Kovacs, R.J.; Stillwell, W. Docosahexaenoic acid induces apoptosis in Jurkat cells by a protein phosphatase-mediated process. Biochim. Biophys. Acta 2001, 1499, 265–275. [Google Scholar] [CrossRef]
- Zand, H.; Rhimipour, A.; Bakhshayesh, M.; Shafiee, M.; Nour Mohammadi, I.; Salimi, S. Involvement of PPAR-γ and p53 in DHA-induced apoptosis in Reh cells. Mol. Cell. Biochem. 2007, 304, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Yamagami, T.; Porada, C.D.; Pardini, R.S.; Zanjani, E.D.; Almeida-Porada, G. Docosahexaenoic acid induces dose dependent cell death in an early undifferentiated subtype of acute myeloid leukemia cell line. Cancer Biol. Ther. 2009, 8, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Sravan Kumar, G.; Das, U.N. Cytotoxic action of α-linolenic and eicosapentaenoic acids on myeloma cells in vitro. Prostaglandins Leukot. Essent. Fatty Acids 1997, 56, 285–293. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; de Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Essential fatty acids and their metabolites as modulators of stem cell biology with reference to inflammation, cancer, and metastasis. Cancer Metastasis Rev. 2011, 30, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Fang, S.; Zhang, H.X.; Xu, L.X.; Zhang, Z.Q.; Yuan, K.T.; Xue, C.L.; Yu, H.L.; Zhang, S.; Li, Y.F.; et al. n-3 PUFA shave antiproliferative and apoptotic effects on human colorectal cancer stemlike cells in vitro. J. Nutr. Biochem. 2013, 24, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, A.; Yu, Y.; Banerjee, S.; Woods, J.; Farhana, L.; Rajendra, S.G.; Patel, A.; Dyson, G.; Levi, E.; Maddipati, K.R.; et al. ω-3 Fatty acid is a potential preventive agent for recurrent colon cancer. Cancer Prev. Res. 2014, 7, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- De Carlo, F.; Witte, T.R.; Hardman, W.E.; Claudio, P.P. ω-3 Eicosapentaenoic acid decreases CD133 colon cancer stem-like cell marker expression while increasing sensitivity to chemotherapy. PLoS ONE 2013, 8, e69760. [Google Scholar]
- Xiong, A.; Yu, W.; Liu, Y.; Sanders, B.G.; Kline, K. Elimination of ALDH+ breast tumor initiating cells by docosahexanoic acid and/or γ tocotrienol through SHP-1 inhibition of Stat3 signaling. Mol. Carcinog. 2015. [Google Scholar] [CrossRef] [PubMed]
- Rose, D.P.; Connolly, J.M.; Rayburn, J.; Coleman, M. Influence of diets containing eicosapentaenoic or docosahexaenoic acid on growth and metastasis of breast cancer cells in nude mice. J. Natl. Cancer Inst. 1995, 87, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Yam, D.; Peled, A.; Huszar, M.; Shinitzky, M. Dietary fish oil suppresses tumor growth and metastasis of Lewis lung carcinoma in mice. J. Nutr. Biochem. 1997, 8, 619–622. [Google Scholar] [CrossRef]
- Boudreau, M.D.; Sohn, K.H.; Rhee, S.H.; Lee, S.W.; Hunt, J.D.; Hwang, D.H. Suppression of tumor cell growth both in nude mice and in culture by n-3 polyunsaturated fatty acids: Mediation through cyclooxygenase-independent pathways. Cancer Res. 2001, 61, 1386–1391. [Google Scholar] [PubMed]
- Kato, T.; Hancock, R.L.; Mohammadpour, H.; McGregor, B.; Manalo, P.; Khaiboullina, S.; Hall, M.R.; Pardini, L.; Pardini, R.S. Influence of ω-3 fatty acids on the growth of human colon carcinoma in nude mice. Cancer Lett. 2002, 187, 169–177. [Google Scholar] [CrossRef]
- Camargo, C.Q.; Mocellin, M.C.; Pastore Silva, J.A.; de Souza Fabre, M.E.; Nunes, E.A.; de Moraes Trinidade, E.B. Fish oil supplementation during chemotherapy increases posterior time to tumor progression in colorectal cancer. Nutr. Cancer. (in press). Available online: http://.doi.org/10.1080/01635581.2016.1115097 (accessed on 19 January 2016).
- Bougnoux, P.; Hajjaji, N.; Ferrasson, M.N.; Giraudeau, B.; Couet, C.; le Floch, O. Improving outcome of chemotherapy of metastatic breast cancer by docosahexaenoic acid: A phase II trial. Br. J. Cancer 2009, 1011978–1011985. [Google Scholar] [CrossRef] [PubMed]
- Cockbain, J.; Toogood, G.J.; Hull, M.A. ω-3 Polyunsaturated fatty acids for the treatment and prevention of colorectal cancer. Gut 2012, 61, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.A.; Mourtzakis, M.; Chu, Q.S.; Baracos, V.E.; Reiman, T.; Mazurak, V.C. Supplementation with fish oil increases first-line chemotherapy efficacy in patients with advanced non-small cell lung cancer. Cancer 2011, 117, 3774–3780. [Google Scholar] [CrossRef] [PubMed]
- Patterson, R.E.; Flatt, S.W.; Newman, V.A.; Natarajan, L.; Rock, C.L.; Thomson, C.A.; Caan, B.J.; Parker, B.A.; Pierce, J.P. Marine fatty acid intake is associated with breast cancer prognosis. J. Nutr. 2011, 141, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Cockbain, A.J.; Volpato, M.; Race, A.D.; Munarini, A.; Fazio, C.; Belluzzi, A.; Loadman, P.M.; Toogood, G.J.; Hull, M.A. Anticolorectal cancer activity of the ω-3 polyunsaturated fatty acid eicosapentaenoic acid. Gut 2014, 63, 1760–1768. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Lara, K.; Turcott, J.G.; Juárez-Hernández, E.; Nuñez-Valencia, C.; Villanueva, G.; Guevara, P.; de la Torre-Vallejo, M.; Mohar, A.; Arrieta, O. Effects of an oral nutritional supplement containing eicosapentaenoic acid on nutritional and clinical outcomes in patients with advanced non-small cell lung cancer: Randomised trial. Clin. Nutr. 2014, 33, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Arshad, A.; Chung, W.Y.; Isherwood, J.; Mann, C.D.; Al-Leswas, D.; Steward, W.P.; Metcalfe, M.S.; Dennison, A.R. Cellular and plasma uptake of parenteral ω-3 rich lipid emulsion fatty acids in patients with advanced pancreatic cancer. Clin. Nutr. 2014, 33, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.J.; Yu, J.; Xiao, J.; Cao, B.W. The consumption of ω-3 polyunsaturated fatty acids improves clinical outcomes and prognosis in pancreatic cancer patients: A systematic evaluation. Nutr. Cancer 2015, 67, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.F.; Bilottom, S.; Russom, G.L.; Orhan, I.E.; Habtemariam, S.; Daglia, M.; Devi, K.P.; Loizzo, M.R.; Tundis, R.; Nabavi, S.M. ω-3 polyunsaturated fatty acids and cancer: Lessons learned from clinical trials. Cancer Metastasis Rev. 2015, 34, 359–380. [Google Scholar] [CrossRef] [PubMed]
- Khankari, N.K.; Bradshaw, P.T.; Steck, S.E.; He, K.; Olshan, A.F.; Shen, J.; Ahn, J.; Chen, Y.; Ahsan, H.; Terry, M.B.; et al. Dietary intake of fish, polyunsaturated fatty acids, and survival after breast cancer: A population-based follow-up study on Long Island, New York. Cancer 2015. [Google Scholar] [CrossRef] [PubMed]
- Mocellin, M.C.; Camargo, C.Q.; Nunes, E.A.; Fiates, G.M.; Trindade, E.B. A systematic review and meta-analysis of the n-3 polyunsaturated fatty acids effects on inflammatory markers in colorectal cancer. Clin. Nutr. 2015. [Google Scholar] [CrossRef] [PubMed]
- Heimli, H.; Giske, C.; Naderi, S.; Drevon, C.A.; Hollung, K. Eicosapentaenoic acid promotes apoptosis in Ramos cells via activation of caspase-3 and -9. Lipids 2002, 37, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Llor, X.; Pons, E.; Roca, A.; Alvarez, M.; Mañé, J.; Fernández-Bañares, F.; Gassull, M.A. The effects of fish oil, olive oil, oleic acid and linoleic acid on colorectal neoplastic processes. Clin. Nutr. 2003, 22, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Danbara, N.; Yuri, T.; Tsujita-Kyutoku, M.; Sato, M.; Senzaki, H.; Takada, H.; Hada, T.; Miyazawa, T.; Okazaki, K.; Tsubura, A. Conjugated docosahexaenoic acid is a potent inducer of cell cycle arrest and apoptosis and inhibits growth of colo 201 human colon cancer cells. Nutr. Cancer 2004, 50, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Calviello, G.; Di Nicuolo, F.; Serini, S.; Piccioni, E.; Boninsegna, A.; Maggiano, N.; Ranelletti, F.O.; Palozza, P. Docosahexaenoic acid enhances the susceptibility of human colorectal cancer cells to 5-fluorouracil. Cancer Chemother. Pharmacol. 2005, 55, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, C.H.; Størvold, G.L.; Bremseth, H.; Follestad, T.; Sand, K.; Mack, M.; Olsen, K.S.; Lundemo, A.G.; Iversen, J.G.; Krokan, H.E.; et al. DHA induces ER stress and growth arrest in human colon cancer cells: Associations with cholesterol and calcium homeostasis. J. Lipid. Res. 2008, 49, 2089–2100. [Google Scholar] [CrossRef] [PubMed]
- Arita, K.; Kobuchi, H.; Utsumi, T.; Takehara, Y.; Akiyama, J.; Horton, A.A.; Utsumi, K. Mechanism of apoptosis in HL-60 cells induced by n-3 and n-6 polyunsaturated fatty acids. Biochem. Pharmacol. 2001, 62, 821–828. [Google Scholar] [CrossRef]
- Narayanan, B.A.; Narayanan, N.K.; Reddy, B.S. Docosahexaenoic acid regulated genes and transcription factors inducing apoptosis in human colon cancer cells. Int. J. Oncol. 2001, 19, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Kolch, W.; Halasz, M.; Granovskaya, M.; Kholodenko, B.N. The dynamic control of signal transduction networks in cancer cells. Nat. Rev. Cancer 2015, 15, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Yu, L.C. Pathophysiological mechanisms of death resistance in colorectal carcinoma. World J. Gastroenterol. 2015, 21, 11777–11792. [Google Scholar] [CrossRef] [PubMed]
- Glatz, J.F.; Luiken, J.J.; van Nieuwenhoven, F.A.; van der Vusse, G.J. Molecular mechanism of cellular uptake and intracellular translocation of fatty acids. Prostaglandins Leukot. Essent. Fatty Acids 1997, 57, 3–9. [Google Scholar] [CrossRef]
- Wassall, S.R.; Stillwell, W. Polyunsaturated fatty acid-cholesterol interactions: Domain formation in membranes. Biochim. Biophys. Acta 2009, 1788, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yu, H.; Ni, X.; Shen, S.; Das, U.N. Growth inhibitory effect of polyunsaturated fatty acids (PUFAs) on colon cancer cells via their growth inhibitory metabolites and fatty acid composition changes. PLoS ONE 2015, 10, e0123256. [Google Scholar]
- Ibarguren, M.; López, D.J.; Escribá, P.V. The effect of natural and synthetic fatty acids on membrane structure, microdomain organization, cellular functions and human health. Biochim. Biophys. Acta 2014, 1838, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Corsetto, P.A.; Cremona, A.; Montorfano, G.; Jovenitti, I.E.; Orsini, F.; Arosio, P.; Rizzo, A.M. Chemical-physical changes in cell membrane microdomains of breast cancer cells after ω-3 PUFA incorporation. Cell Biochem. Biophys. 2012, 64, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Schley, P.D.; Brindley, D.N.; Field, C.J. (n-3) PUFA alter raft lipid composition and decrease epidermal growth factor receptor levels in lipid rafts of human breast cancer cells. J. Nutr. 2007, 137, 548–553. [Google Scholar] [PubMed]
- Rogers, K.R.; Kikawa, K.D.; Mouradian, M.; Hernandez, K.; McKinnon, K.M.; Ahwah, S.M.; Pardini, R.S. Docosahexaenoic acid alters epidermal growth factor receptor-related signaling by disrupting its lipid raft association. Carcinogenesis 2010, 31, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Corsetto, P.A.; Montorfano, G.; Zava, S.; Jovenitti, I.E.; Cremona, A.; Berra, B.; Rizzo, A.M. Effects of n-3 PUFAs on breast cancer cells through their incorporation in plasma membrane. Lipids Health Dis. 2011, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Yun, U.J.; Koo, K.H.; Sung, J.Y.; Shim, J.; Ye, S.K.; Hong, K.M.; Kim, Y.N. Down-regulation of lipid raft-associated onco-proteins via cholesterol-dependent lipid raft internalization in docosahexaenoic acid-induced apoptosis. Biochim. Biophys. Acta 2014, 1841, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.K.; Klaire, S.; Kharotia, S.; Wiggins, A.K.; Thompson, L.U. α-linolenic acid and docosahexaenoic acid, alone and combined with trastuzumab, reduce HER2-overexpressing breast cancer cell growth but differentially regulate HER2 signaling pathways. Lipids Health Dis. 2015, 14, 91. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Ma, Z.; Rasenick, M.M.; Yeh, S.; Yu, J. n-3 poly-unsaturated fatty acids shift estrogen signaling to inhibit human breast cancer cell growth. PLoS ONE 2012, 7, e52838. [Google Scholar] [CrossRef] [PubMed]
- Ewaschuk, J.B.; Newell, M.; Field, C.J. Docosahexanoic acid improves chemotherapy efficacy by inducing CD95 translocation to lipid rafts in ER− breast cancer cells. Lipids 2012, 47, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Wu, J.; Wang, S.; Suburu, J.; Chen, H.; Thomas, M.J.; Shi, L.; Edwards, I.J.; Berquin, I.M.; Chen, Y.Q. Polyunsaturated fatty acids affect the localization and signaling of PIP3/AKT in prostate cancer cells. Carcinogenesis 2013, 34, 1968–1975. [Google Scholar] [CrossRef] [PubMed]
- Calviello, G.; Resci, F.; Serini, S.; Piccioni, E.; Toesca, A.; Boninsegna, A.; Monego, G.; Ranelletti, F.O.; Palozza, P. Docosahexaenoic acid induces proteasome-dependent degradation of β-catenin, down-regulation of survivin and apoptosis in human colorectal cancer cells not expressing COX-2. Carcinogenesis 2007, 28, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Song, K.S.; Jing, K.; Kim, J.S.; Yun, E.J.; Shin, S.; Seo, K.S.; Park, J.H.; Heo, J.Y.; Kang, J.X.; Suh, K.S.; et al. ω-3-Polyunsaturated fatty acids suppress pancreatic cancer cell growth in vitro and in vivo via downregulation of Wnt/β-catenin signaling. Pancreatology 2011, 11, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Wang, Q.; Zhao, J.; Dong, L.; Ge, Y.; Hou, L.; Liu, Y.; Zheng, Z. Docosahexaenoic acid inhibited the Wnt/β-catenin pathway and suppressed breast cancer cells in vitro and in vivo. J. Nutr. Biochem. 2014, 25, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Hu, Y.; Gu, Z.; Owens, R.T.; Chen, Y.Q.; Edwards, I.J. ω-3 Fatty acids induce apoptosis in human breast cancer cells and mouse mammary tissue through syndecan-1 inhibition of the MEK-Erk pathway. Carcinogenesis 2011, 32, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Schley, P.D.; Jijon, H.B.; Robinson, L.E.; Field, C.J. Mechanisms of ω-3 fatty acid-induced growth inhibition in MDAMB-231 human breast cancer cells. Breast Cancer Res. Treat. 2005, 92, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Ghosh-Choudhury, T.; Mandal, C.C.; Woodruff, K.; St Clair, P.; Fernandes, G.; Choudhury, G.G.; Ghosh-Choudhury, N. Fish oil targets PTEN to regulate NFκB for downregulation of anti-apoptotic genes in breast tumor growth. Breast Cancer Res. Treat. 2009, 118, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Engelbrecht, A.M.; Toit-Kohn, J.L.; Ellis, B.; Thomas, M.; Nell, T.; Smith, R. Differential induction of apoptosis and inhibition of the PI3-kinase pathway by saturated, monounsaturated and polyunsaturated fatty acids in a colon cancer cell model. Apoptosis 2008, 13, 1368–1377. [Google Scholar] [CrossRef] [PubMed]
- Jing, K.; Song, K.S.; Shin, S.; Kim, N.; Jeong, S.; Oh, H.R.; Park, J.H.; Seo, K.S.; Heo, J.Y.; Han, J.; et al. Docosahexaenoic acid induces autophagy through p53/AMPK/mTOR signaling and promotes apoptosis in human cancer cells harboring wild-type p53. Autophagy 2011, 7, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Jing, K.; Jeong, S.; Kim, N.; Song, K.S.; Heo, J.Y.; Park, J.H.; Seo, K.S.; Han, J.; Park, J.I.; et al. The ω-3 polyunsaturated fatty acid DHA induces simultaneous apoptosis and autophagy via mitochondrial ROS-mediated Akt-mTOR signaling in prostate cancer cells expressing mutant p53. Biomed. Res. Int. 2013, 2013, 568671. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Jeong, S.; Jing, K.; Shin, S.; Kim, S.; Heo, J.Y.; Kweon, G.R.; Park, S.K.; Wu, T.; Park, J.I.; et al. Docosahexaenoic acid induces cell death in human non-small cell Lung cancer cells by repressing mTOR via AMPK activation and PI3K/Akt inhibition. Biomed. Res. Int. 2015, 2015, 239764. [Google Scholar] [CrossRef] [PubMed]
- Rescigno, T.; Capasso, A.; Tecce, M.F. Effect of Docosahexaenoic acid on cell cycle pathways in Breast cell lines with different transformation degree. J. Cell. Physiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, I.A.; Brown, I.; Schofield, A.C.; Wahle, K.W.; Heys, S.D. Docosahexaenoic acid enhances the efficacy of docetaxel in prostate cancer cells by modulation of apoptosis: The role of genes associated with the NF-κB pathway. Prostate 2008, 68, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Cavazos, D.A.; Price, R.S.; Apte, S.S.; de Graffenried, L.A. Docosahexaenoic acid selectively induces human prostate cancer cell sensitivity to oxidative stress through modulation of NF-κB. Prostate 2011, 71, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Jing, K.; Kim, N.; Shin, S.; Kim, S.; Song, K.S.; Heo, J.Y.; Park, J.H.; Seo, K.S.; Han, J.; et al. Docosahexaenoic acid-induced apoptosis is mediated by activation of mitogen-activated protein kinases in human cancer cells. BMC Cancer 2014, 14, 481. [Google Scholar] [CrossRef] [PubMed]
- Gleissman, H.; Yang, R.; Martinod, K.; Lindskog, M.; Serhan, C.N.; Johnsen, J.I.; Kogner, P. Docosahexaenoic acid metabolome in neural tumors: Identification of cytotoxic intermediates. FASEB J. 2010, 24, 906–915. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Arita, M.; Hong, S.; Gotlinger, K. Resolvins, docosatrienes, and neuroprotectins, novel ω-3-derived mediators, and their endogenous aspirin-triggered epimers. Lipids 2004, 39, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Lu, Y.; Yang, R.; Gotlinger, K.H.; Petasis, N.A.; Serhan, C.N. Resolvin D1, protectin D1, and related docosahexaenoic acid-derived products: Analysis via electrospray/low energy tandem mass spectrometry based on spectra and fragmentation mechanisms. J. Am. Soc. Mass Spectrom. 2007, 18, 128–144. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.Q.; Vaught, J.L.; Yamauchi, H.; Lind, S.E. Differential sensitivity of cancer cells to docosahexaenoic acid-induced cytotoxicity: The potential importance of down-regulation of superoxide dismutase 1 expression. Mol. Cancer Ther. 2004, 3, 1109–1117. [Google Scholar] [PubMed]
- Ding, W.Q.; Lind, S.E. Phospholipid hydroperoxide glutathione peroxidase plays a role in protecting cancer cells from docosahexaenoic acid-induced cytotoxicity. Mol. Cancer Ther. 2007, 6, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Vibet, S.; Goupille, C.; Bougnoux, P.; Steghens, J.P.; Goré, J.; Mahéo, K. Sensitization by docosahexaenoic acid (DHA) of breast cancer cells to anthracyclines through loss of glutathione peroxidase (GPx1) response. Free Radic. Biol. Med. 2008, 44, 1483–1491. [Google Scholar] [CrossRef] [PubMed]
- Sturlan, S.; Baumgartner, M.; Roth, E.; Bachleitner-Hofmann, T. Docosahexaenoic acid enhances arsenic trioxidemediated apoptosis in arsenic trioxide-resistant HL-60 cells. Blood 2003, 101, 4990–4997. [Google Scholar] [CrossRef] [PubMed]
- Granci, V.; Cai, F.; Lecumberri, E.; Clerc, A.; Dupertuis, Y.M.; Pichar, C. Colon cancer cell chemosensitisation by fish oil emulsion involves apoptotic mitochondria pathway. Br. J. Nutr. 2013, 109, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Hossain, Z.; Hosokawa, M.; Takahashi, K. Growth inhibition and induction of apoptosis of colon cancer cell lines by applying marine phospholipid. Nutrition and Cancer 2009, 61, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Shen, J.; Pan, W.; Shen, S.; Das, U.N. Effects of polyunsaturated fatty acids on the growth of gastric cancer cells in vitro. Lipids Health Dis. 2013, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Jing, K.; Shin, S.; Jeong, S.; Kim, S.; Song, K.S.; Park, J.H.; Heo, J.Y.; Seo, K.S.; Park, S.K.; Kweon, G.R.; et al. Docosahexaenoic acid induces the degradation of HPV E6/E7 oncoproteins by activating the ubiquitin-proteasome system. Cell Death Dis. 2014, 5, e1524. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.S.; Wang, P.; Yamabe, N.; Fukui, M.; Jay, T.; Zhu, B.T. Docosahexaenoic acid induces apoptosis in MCF-7 cells in vitro and in vivo via reactive oxygen species formation and caspase 8 activation. PLoS ONE 2010, 5, e10296. [Google Scholar] [CrossRef] [PubMed]
- Zajdel, A.; Wilczok, A.; Tarkowski, M. Toxic effects of n-3 polyunsaturated fatty acids in human lung A549 cells. Toxicol. Vitro 2015. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Hong, S.; Gronert, K.; Colgan, S.P.; Devchand, P.R.; Mirick, G.; Moussignac, R.L. Resolvins: A family of bioactive products of ω-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 2002, 196, 1025–1037. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, M.; Xu, H.; Yang, X. Anti-inflammatory and Pro-resolving effects of n-3 PUFA in Cancers: Structures and Mechanisms. Curr. Top Med. Chem. 2016, 16, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Hawcroft, G.; Loadman, P.M.; Belluzzi, A.; Hull, M.A. Effect of eicosapentaenoic acid on E-type prostaglandin synthesis and EP4 receptor signaling in human colorectal cancer cells. Neoplasia 2010, 12, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Poorani, R.; Bhatt, A.N.; Dwarakanath, B.S.; Das, U.N. COX-2, aspirin and metabolism of arachidonic, eicosapentaenoic and docosahexaenoic acids and their physiological and clinical significance. Eur. J. Pharmacol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Karmali, R.A.; Reichel, P.; Cohen, L.A.; Terano, T.; Hirai, A.; Tamura, Y.; Yoshida, S. The effects of dietary ω−3 fatty acids on the DU-145 transplantable human prostatic tumor. Anticancer Res. 1987, 17, 1173–1180. [Google Scholar]
- Rose, D.P.; Cohen, L.A. Effects of dietary menhaden oil and retinyl acetate on the growth of DU145 human prostatic adenocarcinoma cells transplanted into athymic nude mice. Carcinogenesis 1988, 9, 603–605. [Google Scholar] [CrossRef] [PubMed]
- Rose, D.P.; Connolly, J.M. Dietary fat and breast cancer metastasis by human tumor xenografts. Breast Cancer Res. Treat. 1997, 46, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Barnard, R.J.; Henning, S.M.; Elashoff, D.; Reddy, S.T.; Cohen, P.; Leung, P.; Hong-Gonzalez, J.; Freedland, S.J.; Said, J.; et al. Effect of altering dietary ω-6/ω-3 fatty acid ratios on prostate cancer membrane composition, cyclooxygenase-2, and prostaglandin E2. Clin. Cancer Res. 2006, 12, 4662–4670. [Google Scholar] [CrossRef] [PubMed]
- Funahashi, H.; Satake, M.; Hasan, S.; Sawai, H.; Newman, R.A.; Reber, H.A.; Hines, O.J.; Eibl, G. Opposing effects of n-6 and n-3 polyunsaturated fatty acids on pancreatic cancer growth. Pancreas 2008, 36, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, B.A.; Narayanan, N.K.; Desai, D.; Pittman, B.; Reddy, B.S. Effects of a combination of docosahexaenoic acid and 1,4-phenylene bis(methylene) selenocyanate on cyclooxygenase 2, inducible nitric oxide synthase and β-catenin pathways in colon cancer cells. Carcinogenesis 2004, 25, 2443–2449. [Google Scholar] [CrossRef] [PubMed]
- Calviello, G.; Serini, S.; Piccioni, E. n-3 polyunsaturated fatty acids and the prevention of colorectal cancer: Molecular mechanisms involved. Curr. Med. Chem. 2007, 14, 3059–3069. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Berquin, I.M.; Owens, R.T.; O’Flaherty, J.T.; Edwards, I.J. Peroxisome proliferator-activated receptor γ-mediated up-regulation of syndecan-1 by n-3 fatty acids promotes apoptosis of human breast cancer cells. Cancer Res. 2008, 68, 2912–2919. [Google Scholar] [CrossRef] [PubMed]
- Edwards, I.J.; Sun, H.; Hu, Y.; Berquin, I.M.; O’Flaherty, J.T.; Cline, J.M.; Rudel, L.L.; Chen, Y.Q. In vivo and in vitro regulation of syndecan 1 in prostate cells by n-3 polyunsaturated fatty acids. J. Biol. Chem. 2008, 283, 18441–18449. [Google Scholar] [CrossRef] [PubMed]
- O’Flaherty, J.T.; Hu, Y.; Wooten, R.E.; Horita, D.A.; Samuel, M.P.; Thomas, M.J.; Sun, H.; Edwards, I.J. 15-lipoxygenase metabolites of docosahexaenoic acid inhibit prostate cancer cell proliferation and survival. PLoS ONE 2012, 7, e45480. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Sun, H.; O’Flaherty, J.T.; Edwards, I.J. 15-Lipoxygenase-1-mediated metabolism of docosahexaenoic acid is required for syndecan-1 signaling and apoptosis in prostate cancer cells. Carcinogenesis 2013, 34, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Meng, X.; Meng, Y.; Liu, J.; Liu, B.; Zhang, S.; Ding, W.; Wu, J.; Zhou, J. Microarray analysis of anti-cancer effects of docosahexaenoic acid on human colon cancer model in nude mice. Int. J. Clin. Exp. Med. 2015, 8, 5075–5084. [Google Scholar] [PubMed]
- Sheng, H.; Li, P.; Chen, X.; Liu, B.; Zhu, Z.; Cao, W. ω-3 PUFAs induce apoptosis of gastric cancer cells via ADORA1. Front. Biosci. 2014, 19, 854–861. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Eliseo, D.; Velotti, F. Omega-3 Fatty Acids and Cancer Cell Cytotoxicity: Implications for Multi-Targeted Cancer Therapy. J. Clin. Med. 2016, 5, 15. https://doi.org/10.3390/jcm5020015
D’Eliseo D, Velotti F. Omega-3 Fatty Acids and Cancer Cell Cytotoxicity: Implications for Multi-Targeted Cancer Therapy. Journal of Clinical Medicine. 2016; 5(2):15. https://doi.org/10.3390/jcm5020015
Chicago/Turabian StyleD’Eliseo, Donatella, and Francesca Velotti. 2016. "Omega-3 Fatty Acids and Cancer Cell Cytotoxicity: Implications for Multi-Targeted Cancer Therapy" Journal of Clinical Medicine 5, no. 2: 15. https://doi.org/10.3390/jcm5020015
APA StyleD’Eliseo, D., & Velotti, F. (2016). Omega-3 Fatty Acids and Cancer Cell Cytotoxicity: Implications for Multi-Targeted Cancer Therapy. Journal of Clinical Medicine, 5(2), 15. https://doi.org/10.3390/jcm5020015